

Abstract
We report the photoredox alkylation of halopyridines using functionalized alkene and alkyne building blocks. Selective single-electron reduction of the halogenated pyridines provides the corresponding heteroaryl radicals, which undergo anti-Markovnikov addition to the alkene substrates. The system is shown to be mild and tolerant of a variety of alkene and alkyne subtypes. A combination of computational and experimental studies support a mechanism involving proton-coupled electron transfer followed by medium-dependent alkene addition and rapid hydrogen atom transfer mediated by a polarity-reversal catalyst.
Graphical Abstract
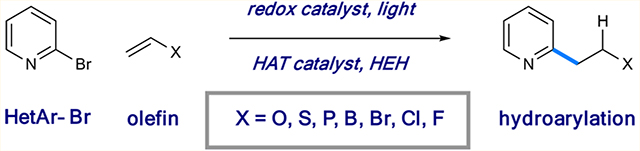
INTRODUCTION
Functionalized pyridines are ubiquitous structural elements in bioactive small molecules that span a wide range of applications.1 Accordingly, a number of powerful synthetic strategies have been developed to effectively access this heterocyclic family.2 In addition to classical methods for pyridine construction using acyclic precursors,3 diversification of pyridine units using transition-metal-based cross-coupling4 and C–H functionalization5 strategies plays key roles in modern drug discovery, as do nucleophilic aromatic substitution reactions.6 In part because of the orthogonal reactivity of radical intermediates to acidic (i.e., X–H) or Lewis basic functional groups,7 Minisci radical addition to pyridines remains a tremendously impactful retrosynthetic tool in complex pyridine synthesis.8 However, the typical requirement for strong oxidants and the pronounced substrate-dependent regiocontrol limit the applicability of this strategy in many cases.9
Among others, we have utilized an alternative approach to complex (hetero)arene synthesis that operates through single-electron reduction of halogenated aryl units to give rise to aryl radicals in a regiospecific manner.10,11 In contrast to arenediazonium salts, this approach draws from a vast collection of stable, often commercially available substrates as radical precursors.12 While arene activation in this manner requires the action of strong reductants, the introduction of photoredox catalysis has enabled the formation of highly reactive aryl radicals under mild conditions.13 Our studies in this area have centered on the intermolecular reaction of pyridyl radicals with an array of olefin subtypes. Interestingly, we have developed conditions that enforce divergent reactivity profiles of these typically ambiphilic radicals based solely on the reaction solvent (Figure 1). For example, in aqueous dimethyl sulfoxide (DMSO), pyridyl radicals selectively engage electron-poor alkenes through a radical conjugate addition (RCA) mechanism.10a However, the use of 2,2,2-trifluoroethanol (TFE) as the solvent enables highly selective hydroarylation with simple electron-neutral olefins.11 Importantly, this protocol delivers the desired pyridyl products with complete regiocontrol with respect to both the heterocyclic and olefinic subunits (anti- Markovnikov addition) as a complement to recent radical hydroarylation systems reported by Herzon14 and Shenvi15 (Markovnikov addition). Here we describe the development of a system based on this mechanistic blueprint that allows for radical hydroarylation of a diverse collection of olefinic substrate classes. Many of these are shown for the first time as substrates under this synthetic disconnection, giving rise to densely functionalized alkylpyridines. In addition, we offer a mechanistic evaluation of these complementary pathways.
Figure 1.

Catalytic intermolecular reactions of pyridyl radical intermediates.
RESULTS AND DISCUSSION
Radical Hydroarylation of Olefinic Substrates
At the outset of this investigation, we treated the feedstock reagent vinyl acetate with 4-bromopyridine (1) under conditions that we recently described for anti-Markovnikov hydroarylation of aliphatic olefins.11 More specifically, we employed the commercial iridium-based photoredox catalyst [Ir(ppy)2 dtbbpy]PF6 (1 mol %) and Hantzsch ester (HEH) (1.3 equiv) as a stoichiometric reductant using TFE as the reaction solvent. We found that irradiation of this mixture for 16 h with blue light furnished the desired product 2-(4-pyridyl)ethyl acetate (2) in low yield (17%) and that the use of TFE was crucial for product formation. However, significant amounts of oligomeric byproducts where pyridyl units coupled with multiple vinyl acetate equivalents (pyridine:olefin ratios of 1:2, 1:3, etc.) were formed. We reasoned that increasing the rate of hydrogen atom transfer (HAT) to the nucleophilic α-oxy radical intermediate—resulting from radical addition to vinyl acetate—would preclude further olefin incorporation.16 Therefore, we surveyed a number of electrophilic polarity-reversal catalysts (Scheme 1).
Scheme 1. Anti-Markovnikov Vinyl Acetate Hydroarylation Using HAT Catalystsa.

aPerformed with [Ir(ppy)2(dtbbpy)]PF6 (1 mol %), Hantzsch ester (1.3 equiv), halopyridine (1 equiv), vinyl acetate (3 equiv) in 2,2,2-trifluoroethanol (0.1 M) at 23 °C for 16 h. Yields were determined by 1H NMR analysis. b4-Bromopyridine hydrochloride was used as the substrate.
While the use of the nitroxyl-based HAT catalysts NHPI17 and HOBt18 did not significantly alter the reaction outcome, employing cyclohexanethiol (5 mol % loading) resulted in significantly improved production of 2 (63% yield).19 Here a much cleaner reaction profile was observed, with pyridine production (via radical hydrodehalogenation) being the major alternative pathway. The beneficial effect of the thiol catalyst was also observed using the isomeric pyridyl radical precursor 3 as the limiting reagent, thus delivering the desired adduct 4 in 65% yield. A range of thiol HAT catalysts were surveyed, although no clear trend was observed between thiol electronics and reaction efficiency. Control reactions revealed that both the photo-catalyst and light were necessary for effective conversion, and in line with our previous results, acidic additives such as AcOH or NH4Cl increased the yield (see the Supporting Information (SI) for details).
We then applied our optimized conditions to a variety of functionalized alkenes (containing heteroatom substitution) that were absent from the current hydroarylation literature as well as common polymer feedstocks (Table 1).20 Halogenated alkenes were particularly appealing because of the broad utility of the products for further functionalization. Vinyl bromide was an effective coupling partner under the reaction conditions, affording alkyl bromide 6 in 78% yield with no further reduction to the corresponding ethylpyridine. Exposure of the crude mixture to silica gel resulted in rapid quantitative conversion to the corresponding vinylpyridine, a product typically accessed through Stille cross-coupling with vinylstannane.21 2-Chloropropene was similarly effective, providing secondary alkyl chloride 7 in 70% yield, again without undesired reduction of the resulting chloroalkane, and elimination during purification was not observed in this case. The reaction conditions were also tolerant of alkenyl fluorides, producing alkyl fluoride 8 in good yield. Robust methods for the synthesis of alkyl fluorides are highly prized, particularly where SN2-type approaches are competitive with elimination byproducts.22
Table 1.
Anti-Markovnikov Hydroarylation with Pyridyl Radicals: Scope of the Functionalized Olefinaa
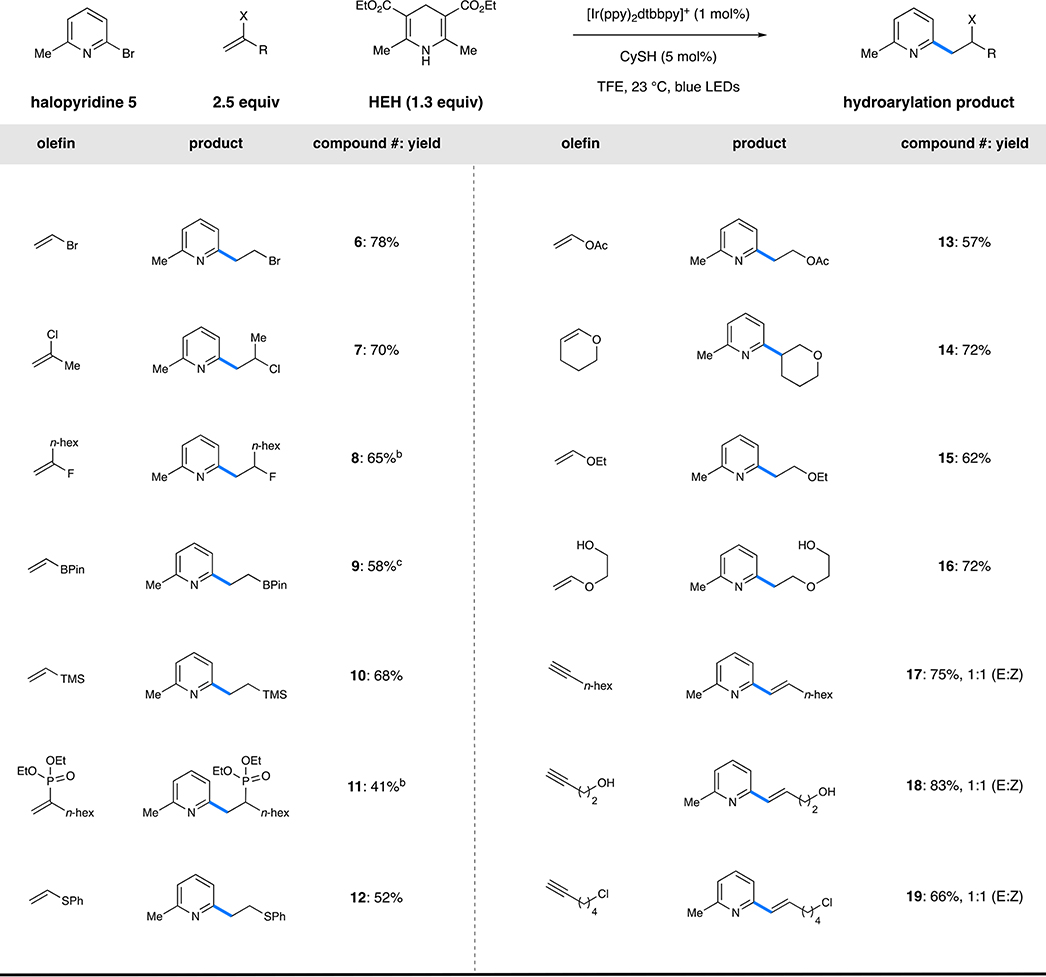 |
Performed with [Ir(ppy)2 dtbbpy]PF6 (1 mol %), Hantzsch ester (1.3 mmol), 2-bromo-6-methylpyridine (1 mmol), alkene (3 mmol), and CySH (5 mol %) in 2,2,2-trifluoroethanol (0.1 M) at 23 °C for 16 h. Isolated yields are shown.
The reaction was performed on a 0.1 mmol scale.
The reaction was performed on a 0.25 mmol scale.
In a process complementary to the recently described work of Aggarwal,23 alkenylboronic esters also underwent the reaction smoothly under the reaction conditions. Homobenzylic alkylboronic ester 9 was effectively synthesized through this method (58%) without any undesired protodeboronation or oxidation. Alkenes substituted with main-group elements were also readily incorporated, despite relatively scarce examples of the hydroarylation of these moieties and a dearth of knowledge concerning the reactivity of the resulting α-heteroatomic radicals.2 Alkenes substituted with Si, P, and S were all tolerated, delivering 10, 11, and 12, respectively, in good yields (41–68%). We envision this method to be applicable to the synthesis of novel bidentate P,N or S,N ligands that are nontrivial to access through other methods.25
The polymer feedstocks vinyl acetate and ethyl vinyl ether both combined with bromopyridine 5 in good yields (57% and 62% yield, respectively), where the remainder of the mass balance was hydrodehalogenated 5 and not higher-order oligomers. Alkyl enol ethers smoothly participated in the reaction to give 14 and 16, both in 72% isolated yield. In addition to heteroatom-substituted alkenes, a number of alkynes were smoothly converted by this system to provide the corresponding vinylpyridines 17–19 as 1:1 mixtures of geometrical isomers (66%−83% yield). In particular, these transformations were unaffected by unprotected alcohols and primary alkyl chlorides. This method provides an alternative to classical Mizoroki–Heck chemistry, which is typically intolerant of such sensitive functionality.26
We next explored the scope of the halopyridine component using isopropenyl acetate as the olefinic coupling partner (as indicated in Table 2). The conditions were tolerant of alkyl substituents on the radical precursor (20, 24–25) in addition to other electron-donating groups, including alkoxy (23) and carbamate (27) functions, with only minor deviations in yield (42–57%). Alkyne-containing pyridine 30 could also be synthesized under the reaction conditions (26%), providing exclusive chemoselectivity for alkene addition, leaving a useful functional handle for further manipulation. Fused heterocycles were likewise tolerated, with pyrrolopyrimidine 26 and azaindole 31 being formed in moderate yields (38–55%). Moreover, 3,4-dibromopyridine underwent chemoselective reaction at the more electron-poor 4-position to provide alkylated bromopyridine 28 without subsequent reductive dehalogenation (38%). Here, radical anion fragmentation occurs selectively through cleavage of the weaker C–Br bond (at the 4-position), and 3-bromopyridine (formed via hydro-debromination) constituted the mass balance. To further illustrate the generality of the process for all pyridine regioisomers, representative members of the alkene scope were coupled to 3- and 4-halopyridines (Table 2, 32–43). To further demonstrate the scope of the alkene-coupling partner, the antidiabetic pioglitazone27 was prepared in a short synthetic sequence (Table 2, 44, 64%) that utilized an aryl vinyl ether as olefinic partner. The unique disconnection afforded by this hydroarylation protocol allows for a highly modular synthesis that would be appealing for early-stage discovery programs.28
Table 2.
Scope of the Halogenated Pyridineaa
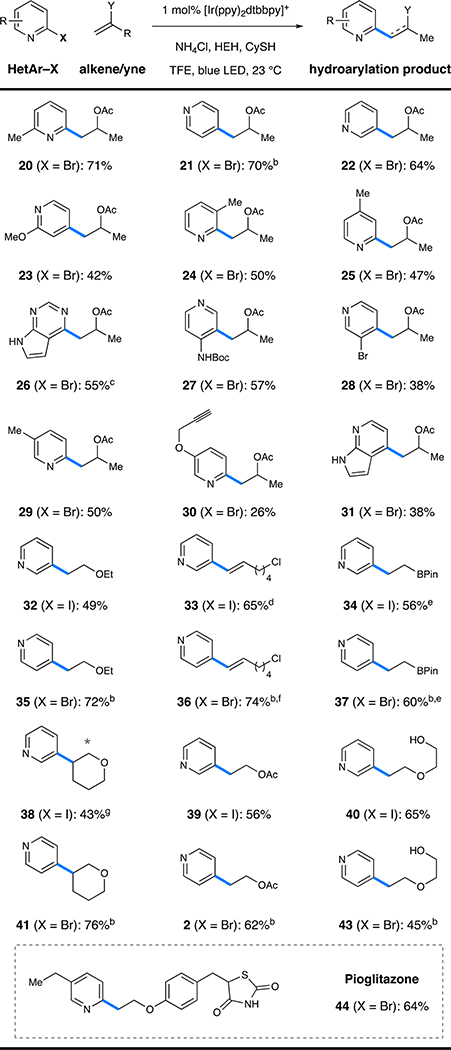 |
The reactions were conducted as in Table 1. Isolated yields are shown.
4-Bromopyridine⋅HCl was used as the starting material.
The reaction was performed on a 0.5 mmol scale.
Formed as a 1.7:1 mixture of olefin isomers.
The reaction was performed on a 0.25 mmol scale.
Formed as a 3:1 mixture of olefin isomers.
Isolated as a 1.2:1 mixture of regioisomers. The gray asterisk indicates the connectivity of the minor regioisomer.
This photoredox process was amenable to gram-scale synthesis without erosion of the isolated yield (71 vs 76%). Compound 20 was synthesized on a 10 mmol scale without any specialized equipment using only 0.1 mol % Ir catalyst (Figure 2). It has been well-demonstrated that scale-up of photocatalytic processes can be limited by light penetration;29 a common solution to this issue is the use of continuous flow methods,30 which have facilitated the large-scale production of active pharmaceutical ingredients.31 To demonstrate that our method is amenable to such scale-up procedures, we performed the reaction in a custom-built flow reactor. By the use of a plug-flow regime with a residence time of 20 min, the alkylated product was formed in 75% yield, corresponding to a continuous flow production of 4.5 mmol/h. Further optimization of the flow reactor setup is ongoing within our laboratory.
Figure 2.

Enol acetate hydroarylation on a gram scale.
Mechanistic Investigation
Having established the utility of pyridyl radical intermediates in the hydroarylation of functionalized olefins, we were interested in gaining a deeper mechanistic understanding of this system. Of particular interest were the following two elements: (i) reductive activation of halopyridine substrates is efficient even in cases where single electron transfer (SET) would appear to be endergonic (as indicated by thermodynamic reduction potentials), and (ii) pyridyl radical species display remarkable chemoselectivity for different olefin types depending on the reaction solvent.
Shown in Figure 3 is a proposed mechanistic scenario for the dual-catalytic hydroarylation process introduced above. Specifically, reductive quenching of the photoexcited iridium catalyst (*[Ir]III) would give rise to the reducing [Ir]II species ( V vs SCE). This mechanism would likely be initiated by oxidation of a sacrificial amount of HEH, as supported by Stern–Volmer quenching studies, in agreement with previous studies by Knowles.32 Reduction of bromopyridine substrate 3 via proton-coupled electron transfer (PCET) (involving TFE as the proton source) followed by rapid mesolytic cleavage would provide protonated pyridine radical 44 and an equivalent of bromide. Regioselective radical addition to the terminal carbon of vinyl acetate (like other nucleophilic olefins) would furnish radical species 45, which would undergo polarity-matched HAT from the thiol catalyst, concurrently producing the hydroarylation product and thiyl species. Regeneration of the HAT catalyst with HEH would deliver the corresponding dihydropyridine radical (HEH⋅), a mild reductant that would close the photoredox catalytic cycle, delivering the corresponding pyridinium (HP).
Figure 3.

Mechanistic proposal: the solvent dictates the nature of the pyridyl radical, imparting divergent chemoselectivity.
Key to this electrophilic radical reactivity is the notorious H-bond-donating (but poorly H-bond-accepting) solvent TFE.33 Indeed, when the diene compound vinyl crotonate (46) was reacted in trifluoroethanol, the pyridyl radical derived from bromopyridine 5 engaged the nucleophilic alkene, giving rise to hydroarylation product 47 as a single regioisomer (72% yield). In contrast, activation of the same radical precursor with the same catalyst in aqueous DMSO as the solvent (25% v/v H2O/DMSO) resulted in exclusive radical conjugate addition to the Michael acceptor, affording 48 as a single regioisomer (56% yield). Regiochemical analysis in these experiments was conducted by GC and, in both cases, <1% yield of the alternate regioisomer was observed. Here, picoline production was the major alternative pathway. These results, which are completely consistent with a previously reported set of competition experiments, indicate that pyridyl radical addition to olefins operates through different reactive intermediates, as dictated by the solvent.11 Further evaluation of these scenarios is presented in the following sections.
Proton-Coupled Electron Transfer
As indicated above, we propose that pyridyl radical formation occurs primarily through a reductive quenching pathway of the iridium photocatalyst. However, analysis of the salient reduction potentials in this system is complicated. Accurate measurement of the reduction potentials of halogenated pyridines remains a challenge because fragmentation of the resulting radical anions is rapid and pyridyl radicals readily undergo reduction to the corresponding anions. However, the reduction potential of 2-bromopyridine has been reported to be between −1.8034 and −2.29 V vs SCE,35 which is significantly beyond the reducing ability of the Ir catalyst ( V vs SCE). Although protonation of the halopyridine substrate would significantly decrease the reduction potential ( V vs SCE), a cursory analysis of the pKa values of TFE (pKa = 12 in DMSO) and 2-bromopyridinium (pKa = 0.5 in DMSO) does not support the formation of a discrete pyridinium salt. Thus, we reason that reductive activation of pyridine substrates in TFE occurs through a PCET mechanism.36
The effect of pyridine protonation on the energetics of electron transfer has been well-studied, particularly within the context of CO2 reduction.37 However, reports detailing the exact impact of protonation on the pyridine reduction potential remain disparate. Because the complications associated with halopyridine reduction potential measurements (vide supra) are compounded by the fact that TFE has a narrow usable electrochemical window, we turned to density functional theory (DFT) calculations to further interrogate this step (Scheme 2A). At the uB3LYP level of theory, the reduction potential of 2-bromopyridine was calculated to be −2.61 V vs SCE,38 and the potentials of H-bonded substrate 49 ( V vs SCE; N–H–O bond length = 2 Å) and fully protonated substrate 50 ( V vs SCE; bond length = 1.1 Å) were also computed. Indeed, the measured reduction potential of the [Ir]II catalytic species ( V vs SCE) lies squarely between these values.13 As shown in Scheme 2B, protonation of the neutral species with TFE is endergonic by 27 kcal/mol but is thermoneutral after reduction. Similarly, reduction of bromopyridine at 2 Å is endergonic by 18.1 kcal/mol but exergonic by 9.4 kcal/mol after protonation. The lowest energetic barrier (where the two curves intersect) was calculated to be 13 kcal/mol at a N–H bond length of 1.55 Å, a reasonable energetic barrier for the proposed PCET event. An endergonic electron transfer of this is type is feasible from a ground-state reductant because of the highly exergonic heterolytic cleavage event that occurs immediately after reduction (i.e., the Curtin–Hammett principle). While this principle could account for a decoupled protonation/electron transfer sequence, we posit that a concerted mechanism is operative.
Scheme 2. Proposed Mechanism of Reduction: PCETa.

aDFT calculations were performed at the uB3LYP/6–311+G(d,p)/CPCM=DMSO level of theory.
Chemoselective C—C Bond Formation
Among the most interesting features of this pyridyl-radical-based strategy is the highly pronounced chemoselectivity that is imparted by the chosen reaction solvent. Shown in Scheme 3 are two electronically diverse olefins along with the optimal reaction solvents for their intermolecular couplings with pyridyl radicals. Because high chemoselectivity for either electron-rich or -poor olefins has been observed (vide supra), we hypothesize that these complementary systems differ in the identity of the pertinent pyridyl radical species. More specifically, we propose that protonated, electrophilic radical species (e.g., 44) are active in TFE, while proton dissociation in polar, H-bond-accepting solvents favors neutral, nucleophilc aryl radicals (e.g., 51).39
Scheme 3. Proposal for the Observed Solvent-Dependent Chemoselectivity of Pyridyl Radicalsa.

aThe competition experiments were performed with [Ir(ppy)2(dtbbpy)]PF6 (1 mol %), Hantzsch ester (1.3 equiv), 2-bromo-6-methylpyridine (1 equiv), dimethylethylidene malonate (3 equiv), and isopropenyl acetate (3 equiv) in 2,2,2-trifluoroethanol (0.1 M) at 23 °C for 16 h.
As an excellent H-bond donor and a weak H-bond acceptor, TFE has the ability to form strong H-bonding networks that effectively stabilize anions or protonate very weak bases.33 As a result, the use of TFE as the reaction solvent has led to divergent selectivity profiles in other systems that are reminiscent of our findings.33 To investigate the relevance of this solvent property in our systems, we attempted to overturn the observed pyridyl radical chemoselectivity through a series of competition experiments. As indicated in Scheme 3, treatment of 5 with both electron-rich isopropenyl acetate and electron-poor dimethylethylidene malonate (3 equiv each) under standard conditions in TFE led to the exclusive production of 20. However, addition of various amounts of water was accompanied by increasingly significant formation of the radical conjugate addition product 52. With 150 equiv of H2O, the two products were formed in nearly equal amounts, presumably resulting from increased dissociation of the proton from the pyridyl radical species by the H-bond acceptor H2O.40 Addition of acids to the same mixtures in DMSO did not have an impact on the chemoselectivity, which is also consistent with the suggestion that protons are highly dissociated from the pyridyl radical species in polar (aqueous) solvents.
An alternative set of reactive intermediates that could potentially account for the observed solvent-based complementary selectivity of this method could arise from differential rates of radical anion fragmentation depending on the solvent. In this scenario, an α-bromo, α-amino radical (resulting directly from PCET) would undergo C–C bond formation prior to bromide dissociation, presumably as an intermediate in radical conjugate addition (i.e., aqueous DMSO). Indeed, Weaver has proposed that the rate of radical anion fragmentation is responsible for altered selectivity in reductive photoredox processes of 2-haloazoles (where differential reactivity resulted from different halogens).41 Along the same lines, it has been shown that the rate of 2-halopyridine (2-X-pyr) radical anion fragmentation decreases significantly in the series X = I > Br > Cl ≫F.35a While the fluoropyridine-based radical anion persists long enough to observe reversible redox by CV, bromopyridine radical anion fragmentation is exceedingly rapid, as measured by Savéant and co-workers.42 Solvent polarity has also been shown to alter the fragmentation rate to a lesser extent, but unimolecular fragmentation rates remain significantly higher than the relevant bimolecular addition rates for aryl radical addition to olefins.43
Radical Termination after Alkene Addition
During our investigations of the addition of pyridyl radicals to olefins, we have observed different radical termination mechanisms that are highly dependent on the employed alkene subtype. While different reaction modes often lead to identical outcomes (e.g., Giese termination via HAT44 or a reduction/enolate protonation sequence45), they could be expected to have discrete implications for photocatalyst turnover or, more importantly, the structural nature of the product. Using combinations of deuterium-labeled HEH and solvents, we have shown that dehydroalanine-derived 53 undergoes essentially exclusive reduction and enolate protonation to give the desired amino acid products.10b In contrast, radicals 54 and 55 receive α-protons or deuterons primarily from HEH, indicating HAT as the primary termination events (>80% incorporation of the label from HEH rather than the solvent).10a,11 When coupling to more nucleophilic vinyl heteroatoms is considered, both oxidation and HAT processes of the intermediate radical (e.g., 56 derived from vinyl acetate) are relevant, as they would likely be product-determining. For example, in a method described by Buchwald, a similar α-oxy radical generated by Meerwein addition to an enol acetate is efficiently oxidized by ferrocinium to provide an aldehyde product.46 However, the conditions that are introduced here appear to enforce termination via HAT, even though single-electron oxidation of 56 would be facile ( V vs SCE).
Evidence for product formation via terminal HAT is offered in Figure 4, where the two potentially competing pathways involving radical intermediate 57 are illustrated. Work by Moeller47 and Horner and Newcomb48 indicates that oxidation to the corresponding cation would be followed by exceedingly rapid cyclization to form dioxolane 58. However, hydroarylation product 19 was delivered in 72% yield without any indication of 58 by NMR or GC–MS analysis. Furthermore, isotopic labeling experiments involving deuterated HEH and/or solvent were consistent with this proposal (see the SI for details). The discrepancy between these results and the system reported by Buchwald46 arises from effective concentration of the putative oxidant: 10 mol % Cp2Fe+ (generated through highly exergonic reduction of arenediazonium salts) versus the minute concentration of photooxidant.
Figure 4.

Mechanisms of radical termination.
Alternative Redox Pathways
Although the mechanistic analyses that we have presented are consistent with experimental data, the observed results likely arise from an ensemble of related mechanistic pathways. For example, the proposed role of the stoichiometric reductant (HEH) is highly consistent with previous reports, facilitating product formation through sequential HAT and SET steps. However, the order of these events is experimentally ambiguous, as HEH consumption via the same steps in reverse order (SET then HAT) would reasonably lead to the same experimental results.
To further evaluate the validity of the proposed photoredox pathway, we conducted a series of temporal studies. Under these conditions, hydroarylation of isopropenyl acetate (to give 20) exhibited zeroth-order kinetics with respect to the starting materials and catalyst in a manner that was highly dependent on the light source,49 as shown in Figure 5. In conjunction with the finding that the benchmark reaction reached completion within 10 min, these results led us to question the degree of radical chain character of this process.50 However, the relatively low quantum yield of this process (Φ = 0.31) indicates that photosensitized processes are dominant. Control experiments showed that formation of 20 can occur in the absence of the iridium catalyst, but the reaction rate is considerably lower (~10% yield after 16 h vs 72% yield after 10 min; see the SI for details). We suggest that the catalyst-free reaction arises from reduction of the halopyridine by the HEH excited state.51 Charge transfer complexes between HEH and bromopyridine were not observed in either the ground state via 1H NMR analysis or the excited state via UV/vis spectroscopy (see the SI for further details). Moreover, alteration of the catalyst properties had a noticeable impact on the overall reaction efficiency (see the SI for full details).50 Taken in aggregate, these findings implicate the outlined catalytic manifold as the major contributor to product formation in this system.50
Figure 5.

Temporal evaluation of the reaction profile.
CONCLUSIONS
A general and efficient method for the hydroarylation of electron-rich olefins has been developed. Addition of a thiol polarity-reversal catalyst promotes a rapid intermolecular HAT step that prevents side reactions such as olefin polymerization and single-electron oxidation to provide the anti-Markovnikov products exclusively. Investigation of the reaction mechanism revealed a solvent-dependent mechanistic divergence. Use of the highly coordinating solvent TFE leads to a protonated, electrophilic pyridyl radical that is polarity-matched for alkene addition with electron-rich olefins. Use of a H-bond-accepting medium such as aqueous DMSO leads to a dissociated, neutral pyridyl radical that is polarity-matched for electron-poor olefins. We believe that these data will provide a greater rationale for chemistries utilizing heteroaryl radicals and expand the scope and understanding of related radical–olefin couplings.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by funds from the National Institutes of Health (R01 GM129495) and NMR spectra were obtained under the support of the National Science Foundation (CHE-1531620). Z. Xu acknowledges financial support by the U.S. Department of Energy, Solar Photochemistry Program (DE-FG02–07ER-15906). The authors thank Savannah Post (Emory University) for preliminary experiments, Professors R. Brian Dyer and Tianquan Lian (Emory University) for generous access to instrumentation, and Professor Zachary Wickens (University of Wisconsin) for helpful discussions.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10238.
Experimental procedures and spectral data (PDF)
REFERENCES
- (1).(a) Guan A-Y; Liu C-L; Sun X-F; Xie Y; Wang M-A Discovery of pyridine-based agrochemicals by using intermediate derivatization methods. Bioorg. Med. Chem 2016, 24, 342–353. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (c) Bakhite EA; Abd-Ella AA; El-Sayed MEA; Abdel-Raheem SAA Pyridine Derivatives as Insecticides. Part 1: Synthesis and Toxicity of Some Pyridine Derivatives Against Cowpea Aphid, Aphis craccivora Koch (Homoptera: Aphididae). J. Agric. Food Chem 2014, 62, 9982–9986. [DOI] [PubMed] [Google Scholar]
- (2).Eicher T; Hauptmann S The Chemistry of Heterocycles: Structures, Reactions, Synthesis and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]; (b) Joule JA; Mills K Heterocyclic Chemistry, 4th ed.; Blackwell: Malden, MA, 2000. [Google Scholar]
- (3).(a) Hill MD Recent Strategies for the Synthesis of Pyridine Derivatives. Chem. - Eur. J 2010, 16, 12052–12062. [DOI] [PubMed] [Google Scholar]; (b) Boger DL Diels-Alder reactions of heterocyclic aza dienes. Scope and applications. Chem. Rev 1986, 86, 781–793. [Google Scholar]; (c) Kröhnke F The Specific Synthesis of Pyridines and Oligopyridines. Synthesis 1976, 1976, 1–24. [Google Scholar]
- (4).Schäfer P; Palacin V; Sidera M; Fletcher SP Asymmetric Suzuki-Miyaura coupling of heterocycles via Rhodium-catalysed allylic arylation of racemates. Nat. Commun 2017, 8, 15762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dick GR; Woerly EM; Burke MD A General Solution for the 2-Pyridyl Problem. Angew. Chem., Int. Ed 2012, 51, 2667–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Billingsley KL; Anderson KW; Buchwald SL A Highly Active Catalyst for Suzuki-Miyaura Cross-Coupling Reactions of Heteroaryl Compounds. Angew. Chem., Int. Ed 2006, 45, 3484–3488. [DOI] [PubMed] [Google Scholar]
- (5).(a) Bull JA; Mousseau JJ; Pelletier G; Charette AB Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev 2012, 112, 2642–2713. [DOI] [PubMed] [Google Scholar]; (b) Nakao Y; Kanyiva KS; Hiyama T A Strategy for C—H Activation of Pyridines: Direct C-2 Selective Alkenylation of Pyridines by Nickel/Lewis Acid Catalysis. J. Am. Chem. Soc 2008, 130, 2448–2449. [DOI] [PubMed] [Google Scholar]; (c) Cho SH; Hwang SJ; Chang S Palladium-Catalyzed C—H Functionalization of Pyridine N-Oxides: Highly Selective Alkenylation and Direct Arylation with Unactivated Arenes. J. Am. Chem. Soc 2008, 130, 9254–9256. [DOI] [PubMed] [Google Scholar]
- (6).Bunnett JF; Zahler RE Aromatic Nucleophilic Substitution Reactions. Chem. Rev 1951, 49, 273–412. [Google Scholar]
- (7).Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Punta C; Minisci F Minisci reaction: a Friedel—Crafts type process with opposite reactivity and selectivity. Selective homolytic alkylation, acylation, carboxylation and carbamoylation of heterocyclic aromatic bases. Trends Heterocycl. Chem 2008, 13, 1–68. [Google Scholar]
- (9).(a) Duncton MAJ Minisci reactions: Versatile CH-functionalizations for medicinal chemists. MedChemComm 2011, 2, 1135–1161. [Google Scholar]; (b) Minisci F; Citterio A; Giordano C Electron-transfer processes: peroxydisulfate, a useful and versatile reagent in organic chemistry. Acc. Chem. Res 1983, 16, 27–32. [Google Scholar]
- (10).(a) Aycock RA; Wang H; Jui NT A mild catalytic system for radical conjugate addition of nitrogen heterocycles. Chem. Sci 2017, 8, 3121–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aycock RA; Vogt DB; Jui NT A practical and scalable system for heteroaryl amino acid synthesis. Chem. Sci 2017, 8, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Boyington AJ; Riu M-L; Jui NT Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates. J. Am. Chem. Soc 2017, 139, 6582–6585. [DOI] [PubMed] [Google Scholar]
- (12).Heinrich MR Intermolecular Olefin Functionalisation Involving Aryl Radicals Generated from Arenediazonium Salts. Chem. - Eur. J 2009, 15, 820–833. [DOI] [PubMed] [Google Scholar]
- (13).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ma X; Herzon SB Intermolecular Hydropyridylation of Unactivated Alkenes. J. Am. Chem. Soc 2016, 138, 8718–8721. [DOI] [PubMed] [Google Scholar]; (b) Ma X; Dang H; Rose JA; Rablen P; Herzon SB Hydro-heteroarylation of Unactivated Alkenes Using N-Methoxyheteroarenium Salts. J. Am. Chem. Soc 2017, 139, 5998–6007. [DOI] [PubMed] [Google Scholar]
- (15).(a) Green SA; Matos JLM; Yagi A; Shenvi R A. Branch- Selective Hydroarylation: Iodoarene-Olefin Cross-Coupling. J. Am. Chem. Soc 2016, 138, 12779–12782. [DOI] [PubMed] [Google Scholar]; (b) Green SA; Vásquez-Céspedes S; Shenvi RA Iron-Nickel Dual-Catalysis: A New Engine for Olefin Functionalization and the Formation of Quaternary Centers. J. Am. Chem. Soc 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Roberts BP Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem. Soc. Rev 1999, 28, 25–35. [Google Scholar]
- (17).Recupero F; Punta C Free Radical Functionalization of Organic Compounds Catalyzed by N-Hydroxyphthalimide. Chem. Rev 2007, 107, 3800–3842. [DOI] [PubMed] [Google Scholar]
- (18).Cooper JC; Luo C; Kameyama R; Van Humbeck JF Combined Iron/Hydroxytriazole Dual Catalytic System for Site Selective Oxidation Adjacent to Azaheterocycles. J. Am. Chem. Soc 2018, 140, 1243–1246. [DOI] [PubMed] [Google Scholar]
- (19).Denes F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (20).(a) Debuigne A; Caille J-R; Jérôme R Highly Efficient Cobalt-Mediated Radical Polymerization of Vinyl Acetate. Angew. Chem., Int. Ed 2005, 44, 1101–1104. [DOI] [PubMed] [Google Scholar]; (b) Percec V; Popov AV; Ramirez-Castillo E; Monteiro M; Barboiu B; Weichold O; Asandei AD; Mitchell CM Aqueous Room Temperature Metal-Catalyzed Living Radical Polymerization of Vinyl Chloride. J. Am. Chem. Soc 2002, 124, 4940–4941. [DOI] [PubMed] [Google Scholar]
- (21).Over half of the reports on vinylpyridine synthesis from halopyridines come from stannanes. The remaining examples use mostly boron reagents (Scifinder 2018). For an example of Stille coupling to form vinylpyridines, see: Nuñez A; Abarca B; Cuadro AM; Alvarez-Builla J; Vaquero JJ Ring-Closing Metathesis Reactions on Azinium Salts: Straightforward Access to Quinolizinium Cations and Their Dihydro Derivatives. J. Org Chem 2009, 74, 4166–4176. [DOI] [PubMed] [Google Scholar]
- (22).Nielsen MK; Ugaz CR; Li W; Doyle AG PyFluor: A Low-Cost, Stable, and Selective Deoxyfluorination Reagent. J. Am. Chem. Soc 2015, 137, 9571–9574. [DOI] [PubMed] [Google Scholar]
- (23).Noble A; Mega RS; Pflasterer D; Myers EL; Aggarwal VK Visible-Light-Mediated Decarboxylative Radical Additions to Vinyl Boronic Esters: Rapid Access to γ-Amino Boronic Esters. Angew. Chem., Int. Ed 2018, 57, 2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) The reactivity of α-oxy and α-amino radicals is well-studied. For examples of the reactivity of α-oxy radicals, see: Tang X; Studer A. Sodium-Ketyl Radical Anions by Reverse Pinacol Reaction and Their Coupling with Iodoarenes. Org. Lett 2016, 18, 4448–4450. [DOI] [PubMed] [Google Scholar]; (b) Zhang W-C; Li C-J Magnesium-Mediated Carbon-Carbon Bond Formation in Aqueous Media: Barbier-Grignard Allylation and Pinacol Coupling of Aldehydes. J. Org. Chem 1999, 64, 3230–3236. [DOI] [PubMed] [Google Scholar]; (c) Jeffrey JL; Terrett JA; MacMillan DWC O-H hydrogen bonding promotes H-atom transfer from α C-H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. For examples of the reactivity of α-amino radicals, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native functionality in triple catalytic cross-coupling: sp3 C-H bonds as latent nucleophiles. Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ruiz Espelt L; McPherson IS; Wiensch EM; Yoon TP Enantioselective Conjugate Additions of α-Amino Radicals via Cooperative Photoredox and Lewis Acid Catalysis. J. Am. Chem. Soc 2015, 137, 2452–2455. For a recent example of degradation of α-halo radicals to aldehydes, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ma X; Herzon SB Synthesis of Ketones and Esters from Heteroatom-Functionalized Alkenes by Cobalt-Mediated Hydrogen Atom Transfer. J. Org. Chem 2016, 81, 8673–8695. [DOI] [PubMed] [Google Scholar]
- (25).Carroll MP; Guiry PJP,N ligands in asymmetric catalysis. Chem. Soc. Rev 2014, 43, 819–833. [DOI] [PubMed] [Google Scholar]
- (26).Primary alcohols can be oxidized under Heck conditions. For an example, see: Kim BH; Lee JG; Yim T; Kim H-J; Lee HY; Kim YG Highly efficient two-step selective synthesis of 2,6-dimethylnaphthalene. Tetrahedron Lett. 2006, 47, 7727–7730. [Google Scholar]
- (27).Sohda T; Ikeda H; Meguro K Studies on Antidiabetic Agents. XII. Synthesis and Activity of the Metabolites of (±)-5-[p-[2-(5-Ethyl-2-pyridyl)ethoxy]benzyl]-2,4-thiazolidinedione (Pioglitazone). Chem. Pharm. Bull 1995, 43, 2168–2172. [DOI] [PubMed] [Google Scholar]
- (28).Beeler AB; Schaus SE Jr; Porco JA Chemical library synthesis using convergent approaches. Curr. Opin. Chem. Biol 2005, 9, 277–284. [DOI] [PubMed] [Google Scholar]
- (29).Tucker JW; Zhang Y; Jamison TF; Stephenson CRJ Visible-Light Photoredox Catalysis in Flow. Angew. Chem., Int. Ed 2012, 51, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Plutschack MB; Pieber B; Gilmore K; Seeberger PH The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev 2017, 117, 11796–11893. [DOI] [PubMed] [Google Scholar]; (b) Lima F; Kabeshov MA; Tran DN; Battilocchio C; Sedelmeier J; Sedelmeier G; Schenkel B; Ley SV Visible Light Activation of Boronic Esters Enables Efficient Photoredox C(sp2)-C(sp3) Cross-Couplings in Flow. Angew. Chem., Int. Ed 2016, 55, 14085–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).(a) Beatty JW; Douglas JJ; Miller R; McAtee RC; Cole KP; Stephenson CRJ Photochemical Perfluoroalkylation with Pyridine N-Oxides: Mechanistic Insights and Performance on a Kilogram Scale. Chem. 2016, 1, 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Caron A; Hernandez-Perez AC; Collins SK Synthesis of a Carprofen Analogue Using a Continuous Flow UV-Reactor. Org. Process Res. Dev 2014, 18, 1571–1574. [Google Scholar]; (c) Šterk D; Jukič M; Časar Z Application of Flow Photochemical Bromination in the Synthesis of a 5-Bromomethylpyrimidine Precursor of Rosuvastatin: Improvement of Productivity and Product Purity. Org. Process Res. Dev 2013, 17, 145–151. [Google Scholar]; (d) Lévesque F; Seeberger PH Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem., Int. Ed 2012, 51, 1706–1709. [DOI] [PubMed] [Google Scholar]
- (32).Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]
- (33).Bégué J-P; Bonnet-Delpon D; Crousse B Fluorinated Alcohols: A New Medium for Selective and Clean Reaction. Synlett 2004, 1, 18–29. [Google Scholar]
- (34).Holubek J; Volke J Polarography of heterocyclic aromatic compounds. XIII. Polarographic fission of carbon-halogen bonds in monohalogenopyridines. Collect. Czech. Chem. Commun 1962, 27, 680–692. [Google Scholar]
- (35).(a) Enemӕrke RJ; Christensen TB;Jensen H; Daasbjerg K Application of a new kinetic method in the investigation of cleavage reactions of haloaromatic radical anions. J. Chem. Soc., Perkin Trans 2001, 2, 1620–1630. [Google Scholar]; (b) Andrieux CP; Blocman C; Dumas-Bouchiat J-M; Saveant J-M Heterogeneous and homogeneous electron transfers to aromatic halides. An electrochemical redox catalysis study in the halobenzene and halopyridine series. J. Am. Chem. Soc 1979, 101, 3431–3441. [Google Scholar]
- (36).(a) Tarantino KT; Liu P; Knowles RR Catalytic Ketyl-Olefin Cyclizations Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2013, 135, 10022–10025. [DOI] [PubMed] [Google Scholar]; (b) Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- (37).(a) Yan Y; Zeitler EL; Gu J; Hu Y; Bocarsly AB Electrochemistry of Aqueous Pyridinium: Exploration of a Key Aspect of Electrocatalytic Reduction of CO2 to Methanol. J. Am. Chem. Soc 2013, 135, 14020–14023. [DOI] [PubMed] [Google Scholar]; (b) Keith JA; Carter EA Theoretical Insights into Pyridinium-Based Photoelectrocatalytic Reduction of CO2. J. Am. Chem. Soc 2012, 134, 7580–7583. [DOI] [PubMed] [Google Scholar]
- (38).DFT calculations were also performed at different levels of theory with different solvation parameters, giving a range of values for pyridine reduction potentials (see the SI for full details).
- (39).Joerg S; Drago RS; Adams J Donor-acceptor and polarity parameters for hydrogen bonding solvents. J. Chem. Soc., Perkin Trans. 2 1997, 2, 2431–2438. [Google Scholar]
- (40).Krishnan R; Fillingim TG; Lee J; Robinson GW Solvent structural effects on proton dissociation. J. Am. Chem. Soc 1990, 112, 1353–1357. [Google Scholar]
- (41).(a) Arora A; Teegardin K; Weaver JD Reductive Alkylation of 2-Bromoazoles via Photoinduced Electron Transfer: A Versatile Strategy to Csp2-Csp3 Coupled Products. Org. Lett 2015, 17, 3722–3725. [DOI] [PubMed] [Google Scholar]; (b) Singh A; Arora A; Weaver JD Photoredox-Mediated C–H Functionalization and Coupling of Tertiary Aliphatic Amines with 2-Chloroazoles. Org. Lett 2013, 15, 5390–5393. [DOI] [PubMed] [Google Scholar]; (c) Arora A; Weaver JD Photocatalytic Generation of 2-Azolyl Radicals: Intermediates for the Azolylation of Arenes and Heteroarenes via C-H Functionalization. Org. Lett 2016, 18, 3996–3999. [DOI] [PubMed] [Google Scholar]
- (42).Andrieux CP; Robert M; Saveant J-M Role of Environmental Factors in the Dynamics of Intramolecular Dissociative Electron Transfer. Effect of Solvation and Ion-Pairing on Cleavage Rates of Anion Radicals. J. Am. Chem. Soc 1995, 117, 9340–9346. [Google Scholar]
- (43).Giese B Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew. Chem., Int. Ed. Engl 1983, 22, 753–764. [Google Scholar]
- (44).Giese B; González-Gómez JA; Witzel T The Scope of Radical CC-Coupling by the “Tin Method. Angew. Chem., Int. Ed. Engl 1984, 23, 69–70. [Google Scholar]
- (45).Yin Y; Dai Y; Jia H; Li J; Bu L; Qiao B; Zhao X; Jiang Z Conjugate Addition-Enantioselective Protonation of N-Aryl Glycines to α-Branched 2-Vinylazaarenes via Cooperative Photoredox and Asymmetric Catalysis. J. Am. Chem. Soc 2018, 140, 6083–6087. [DOI] [PubMed] [Google Scholar]
- (46).Chernyak N; Buchwald SL Continuous-Flow Synthesis of Monoarylated Acetaldehydes Using Aryldiazonium Salts. J. Am. Chem. Soc 2012, 134, 12466–12469. [DOI] [PubMed] [Google Scholar]
- (47).Campbell JM; Xu H-C; Moeller KD Investigating the Reactivity of Radical Cations: Experimental and Computational Insights into the Reactions of Radical Cations with Alcohol and p-Toluene Sulfonamide Nucleophiles. J. Am. Chem. Soc 2012, 134, 18338–18344. [DOI] [PubMed] [Google Scholar]
- (48).Horner JH; Taxil E; Newcomb M Laser Flash Photolysis Kinetic Studies of Enol Ether Radical Cations. Rate Constants for Heterolysis of α-Methoxy-β-phosphatoxyalkyl Radicals and for Cyclizations of Enol Ether Radical Cations. J. Am. Chem. Soc 2002, 124, 5402–5410. [DOI] [PubMed] [Google Scholar]
- (49).Le C; Wismer MK; Shi Z-C; Zhang R; Conway DV; Li G; Vachal P; Davies IW; MacMillan DWC A General Small-Scale Reactor To Enable Standardization and Acceleration of Photocatalytic Reactions. ACS Cent. Sci 2017, 3, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cismesia MA; Yoon TP Characterizing chain processes in visible light photoredox catalysis. Chem. Sci 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Huang W; Cheng X Hantzsch Esters as Multifunctional Reagents in Visible-Light Photoredox Catalysis. Synlett 2017, 28, 148–158. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.