

Abstract
Objectives
Progesterone treatment can effectively increase levels of circulating endothelial progenitor cells (EPCs) and improve neurological functional outcome in a traumatic brain injury (TBI) rat model. However, the mechanisms of progesterone's effects on EPC viability remain elusive. The CXCL12/CXCR4 (CXC chemokine ligand 12/CXC chemokine receptor 4) signalling pathway regulates cell proliferation; we hypothesize that it mediates progesterone‐induced EPC viability.
Materials and methods
EPCs were isolated from bone marrow‐derived mononuclear cells (BM‐MNCs) and treated with progesterone (5, 10 and 100 nm). MTS assay was used to investigate EPC viability. Protein expression was examined by Western blotting, ELISA assay and flow cytometry. Cell membrane and cytoplasm proteins were extracted with membrane and cytoplasm protein extraction kits. CXCR4 antagonist (AMD3100) and phosphatidylinositol 3‐kinases (PI3K) antagonist (LY294002) were used to characterize underlying mechanisms.
Results
Progesterone‐induced EPC viability was time‐ and dose‐dependent. Administration of progesterone facilitated EPC viability and increased expression of CXCL12 and phosphorylated Akt (also known as protein kinase B, pAkt) activity (P < 0.05). Progesterone did not regulate CXCR4 protein expression in cultured EPC membranes or cytoplasm. However, progesterone‐induced EPC viability was significantly attenuated by AMD3100 or LY294002. Inhibition of the signalling pathway with AMD3100 and LY294002 subsequently reduced progesterone‐induced CXCL12/CXCR4/PI3K/pAkt signalling activity.
Conclusions
The CXCL12/CXCR4/PI3K/pAkt signalling pathway increased progesterone‐induced EPC viability.
Introduction
Progesterone is a steroid hormone synthesized in both males and females. Though its function is related to sex and reproduction, especially in females, there is growing evidence that progesterone is also neuroprotective and has angiogenic properties in traumatic brain injury (TBI) 1. Some studies have revealed that progesterone could enhance ovarian cancer cell proliferation through progesterone receptor membrane component‐1 2 and regulate viability of granulosa cells 3. Our previous studies have found that progesterone treatment increases circulating levels of endothelial progenitor cells (EPCs) and improves neurological functional outcome in a rat model of traumatic brain injury (TBI) 4. However, the mechanisms of progesterone's effects on EPC proliferation have remained elusive.
Circulating EPCs have been shown to supplement damaged endothelium and augment neovascularization in a number of animal models 5, 6. Under stress, EPCs migrate to damage sites to replenish injured endothelium and enhance neovascularization 7. Increasing EPC levels play a key role in the process of angiogenesis, as well as being involved in neuroprotection, after brain injury. However, there are very few peripheral EPCs, and they do not proliferate readily 8, 9, 10. Progesterone may increase numbers of circulating EPCs and reinforce vascular remodelling, which could improve neurological outcome in TBI rats 4. Based on these findings, one can assume that enhancing EPC viability may benefit patient recovery 11. In order to develop more efficient therapeutic regimens for TBI, it is important to explore the possible benefits of expanding EPC levels in vitro. In this study we tested the effects of progesterone treatment on the viability of EPCs in vitro, as well as the molecular signalling pathway possibly responsible for enhancing EPC viability.
Chemokines are cytokines and signalling proteins, responsible for proliferation and viability of numerous types of cells 12, 13, 14. The CXCL12/CXCR4 signalling pathway has been extensively investigated and has been shown to play a key role in angiogenesis 15. CXCL12 (also called stromal cell‐derived factor 1 (SDF‐1)), is a small, secreted α chemokine protein. After combining with CXCR4, CXCL12 mediates functional changes by inducing activation of GTP‐binding protein G inhibitory (Gi) and activating other signalling pathways in several types of cells including neural progenitor cells (NPCs) 16, 17. The CXCL12/CXCR4 pathway enhances chemotaxis, cell survival and proliferation. CXCL12/CXCR4 also stimulates phosphatidylinositol 3‐kinase (PI3K)/Akt signalling activity, thereby promoting NPC proliferation and neuronal survival 17, 18. Akt, a downstream target of PI3K, regulates cell proliferation, metabolism and cell size 19, 20. However, potential effects of CXCL12/CXCR4 in EPC viability and its connection with progesterone‐induced EPC viability have remained unclear. Accordingly, we hypothesized that progesterone might enhance EPC viability through the CXCL12/CXCR4/PI3K/Akt signalling pathway. By employing a well‐established in vitro culture system, we studied the effects of progesterone on EPC viability, and whether the CXCL12/CXCR4/PI3K/Akt signalling pathway plays a key role in this process.
Materials and methods
EPC culture
All animal procedures were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University General Hospital, and in accord with the US National Institutes of Health's Guide for the Care and Use of Laboratory Animals (Bethesda, MD, USA). Bone marrow‐derived mononuclear cells (BM‐MNCs) were isolated from 14‐day‐old male Wistar rats by the density gradient centrifugation method, using Histopaque‐1083 (Sigma‐Aldrich, St. Louis, MO, USA). BM‐MNC were seeded on six‐well plates (5 × 106 cells/well) pre‐coated with human fibronectin (1 μg/ml; Millipore Bedford Mass, USA) and incubated at 37 °C in a humidified 5% CO2 atmosphere. First medium change was performed after 24 h in culture, and non‐adherent cells were removed; adherent cells were continuously cultured in fresh EGM‐2 media (Lonza, Walkersville, MD, USA) 21 and medium was changed every 2 days for 14 days. After 14 days culture, cells were identified by a combination of specific surface antigen expression and functional properties 22.
EPC characterization
To confirm their endothelial phenotype, uptake of DiI‐acetylated low‐density lipoprotein (DiI‐acLDL) (Molecular Probe, Eugene, OR, USA) was performed. EPCs were washed in PBS (phosphate‐buffered saline) and stained with DiI‐labelled acetylated low‐density lipoprotein (acLDL) (2.5 mg/ml; Molecular Probe) for 30 min at 37 °C and then stained with fluorescein isothiocyanate (FITC)‐labelled Ulex europaeus agglutinin (UEA) (Vector Laboratories, Burlingame, CA, USA). Endothelial cells markers stained for were, von Willebrand factor (vWF, 1:200, rabbit polyclonal; Santa Cruz Biotechnology, Shanghai, China), and kinase domain receptor (KDR, 1:200, mouse monoclonal; Santa Cruz Biotechnology). CD34 is expressed on hematopoietic stem and progenitor cells, thus, CD34 status was examined by using anti‐CD34 antibody (1:200, mouse monoclonal, Santa Cruz Biotechnology). Positive staining was detected by fluorescent microscopy after incubation with TRITC (tetramethyl rhodamine isothiocynate)‐ or FITC (fluorescein isothiocyanate)‐conjugated secondary antibodies. Nuclear counterstaining was performed with 4′, 6‐diamidino‐2‐phenylindole (DAPI) (1:1000; Sigma‐Aldrich). All processes were visualized using an inverted phase‐contrast microscope (IX81; Olympus, Tokyo, Japan).
Matrigel assay
To analyse capillary‐like tube formation of EPCs, a Matrigel assay was performed as previously described 23. Briefly, growth factor‐reduced Matrigel (BD Biosciences, Bedford, MA, USA) was thawed overnight at 4 °C. 50 μl/well of chilled Matrigel matrix was added to ice‐cold 96‐well plates. Plates were then incubated for 60 min at 37 °C to allow the Matrigel to solidify. After gel layer formation, 100 μl late outgrowth EPCs (1 × 104 cells) in EGM‐2 were seeded in each well and incubated for 6 h. Microscope images of tube formation were captured using inverted phase‐contrast microscopy (IX81; Olympus).
EPC viability assay
To test whether progesterone treatment would regulate EPC viability, three different concentrations of progesterone were employed. In order to address whether CXCL12 directly impacts EPC viability, we also stimulated EPCs with different concentrations of CXCL12 (CXCL12 dissolved in culture medium). Selection of concentrations of progesterone and CXCL12 were based on previous studies 17, 24. Progesterone is liposoluble and was dissolved in a 0.16% ethanol solution. EPC viability was measured by MTS [colorimetric3‐(4,5‐dimethylthia‐zol‐2‐yl)‐5‐(3‐carboxy‐methoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetra‐zolium] assay (Cell Titer 96 AQ, Promega, Madison, WI, USA). After 14 days culture, EPCs were cultured in the presence of different doses of progesterone (0, 5, 10 and 100 nm) (Sigma‐Aldrich) or vehicle (0.016% ethanol) for 3, 6, 12 and 24 h respectively. We also treated EPCs with different concentrations of CXCL12 (0, 10, 20, 60, 80 and 120 ng/ml; PEPROTECH, Rocky Hill, NJ, USA) for 6 h. After treatment with progesterone and CXCL12, EPCs were harvested and reseeded on 96‐well plates (1 × 104 cells/well, n = 6 wells/group) in 100 μl culture medium. Then, 20 μl MTS reagent were pipetted into each well and plates were incubated for 1 h at 37 °C in a humidified, 5% CO2 atmosphere. Absorbance was recorded at 490 nm using an ELISA plate reader (Bionetics Laboratory, Kensington, CA, USA).
CXCL12 ELISA assay
CXCL12 secreted by EPCs in the supernatant was measured using a CXCL12 ELISA kit (Biovol, Shanghai, China) according to the manufacturer's instructions. Briefly, cells were pre‐treated with or without 5 nm progesterone for 6 h. Samples of cell culture supernatant were incubated in plates with CXCL12 antibody for 1 h at 37 °C (n = 6 wells/group). After washing, 100 μl horseradish peroxidase‐labelled CXCL12 antibody solution was added to each well and incubated for 30 min at 37 °C. After washing, tetramethyl benzidine chromogenic reagent was added to each well. When the liquid in each well turned blue, plates were read on a plate reader (Bionetics Laboratory), colour intensity of samples being proportional to quantity of CXCL12. Concentration of CXCR12 in the samples was determined by comparing optical density (OD) of samples to the standard curve.
Flow cytometry assay for membrane protein of CXCR4
To detect CXCR4 on cell membranes, flow cytometric analysis was preformed. EPCs were treated with or without progesterone (5 nm) for 6 h.Cells were collected and washed twice in PBS. All cells were resuspended in 100 μl binding buffer and incubated with 5 μl CXCR4 antibody (ab2074; Abcam, Cambridge Cambridgeshire, UK) for 30 min at 4 °C in the dark, and then washed twice in PBS. Next, FITC‐conjugated goat anti‐rabbit secondary antibody (1:50; ZSGO‐BIO, Beijing, China) was used, and cells were washed twice in PBS. Finally, cells were analysed on a flow cytometer (Becton Dickinson and Company, Franklin Lakes, NJ, USA).
Cytomembrane and cytoplasmic protein extraction
Cytomembrane and cytoplasmic proteins were extracted using the Membrane and Cytosol Protein Extraction Kit (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer's instructions. Briefly, EPCs were treated with or without progesterone (5 nm) for 6 h. They were then pelleted and resuspended without pancreatin in solution A provided by the kit. Cells were then lysed and centrifuged at 14000 × g for 30 min at 4 °C. Cytoplasmic proteins were extracted from the supernatant, and the precipitate was treated with solution B provided by the kit, then centrifuged at 14000 × g for 5 min at 4 °C. Membrane proteins were extracted from the supernatant. Protein concentrations of cytomembrane and cytoplasmic proteins were determined with a BCA (Bicinchoninic acid) protein assay kit (Solarbio, Beijing, China) and proteins were stored at −80 °C. Cytomembrane and cytoplasmic differential protein expressions were analysed using Western blotting.
Western Blotting assay
To explore the mechanisms of progesterone‐mediated EPC viability, CXCR4 inhibitor AMD3100 and the PI3K inhibitor LY294002 were used. EPCs were pre‐treated with AMD3100 (20 μm, 60 μm, Sigma, St. Louis, MO, USA), LY294002 (20 μm, Selleckchem, Shanghai, China) for 2 h followed by stimulation with 5 nm progesterone for 6 h. Choice of inhibitor concentrations was based on previous assays 17, 25, 26. Western blotting assay was performed to detect CXCL12, CXCR4 and pAkt. Cell proteins were isolated using RIPA (Radio Immunoprecipitation Assay) lysis buffer (Solarbio), containing phosphatase inhibitor (1:100, Solarbio) and PMSF (phenylmethanesulfonyl fluoride) (1:1000, Solarbio). The BCA protein kit (Solarbio) was used to determine protein concentrations. Proteins were separated using SDS‐polyacrylamide gel then transferred to polyvinylidene fluoride (PVDF) membranes (Bio‐Rad, Hercules, CA, USA). Primary antibodies against pAkt (1:1000, 9271S; Cell Signaling Technologies, Danvers, MA, USA), CXCL12 (1:1000, 3530S, Cell Signaling Technologies), CXCR4 (1:1000, ab2074; Abcam) and total Akt (1:1000, 4685, Cell Signaling Technologies) were added and incubated overnight at 4 °C. Membranes were then washed and horseradish peroxidase‐conjugated secondary antibodies (1:5000, Solarbio) were added. Membranes were washed and then treated with chemiluminescent (ECL) solution (Millipore, Shanghai, China) and signal bands were visualized using an appropriate scanner (BioTek, Hercules, CA, USA).
Statistical analysis
All data are shown as mean ± standard deviation (SD) and at least three independent experiments were performed for all analyses. In cell experiments, six wells were performed in each group. Data were analysed using SPSS software (version 10.0). After confirming normal distribution of all variables, statistical analysis of differences between two groups was performed using Student's t‐test. Two‐way ANOVA assessed the effects of treatments and time on change in EPC viability and were followed by Bonferroni post hoc test. Also, differences within and among groups were analysed using two‐way ANOVA for EPC viability with progesterone and antagonists as factors, and followed by the Bonferroni post hoc test. All data were considered statistically significant at P < 0.05. ELISA data were analysed by curve expert (version 1.4). P < 0.05 was considered statistically significant.
Results
EPC characterization
As shown in Fig. 1, total BM‐MNCs were cultured for 14 days and all cells appeared to be homogeneous and cobblestone‐like (Fig. 1a). Together with tubular formation capacity (Fig. 1b), EPCs were identified by their expression of progenitor or stem cell marker CD34 (Fig. 1c), and EC markers KDR (Fig. 1d) and vWF (Fig. 1e), binding with UEA‐I‐FITC and up take of DiI‐acLDL (Fig. 1f), a typical feature in characterization of endothelial lineage cells. This information indicated that the cultured cells were EPCs.
Figure 1.
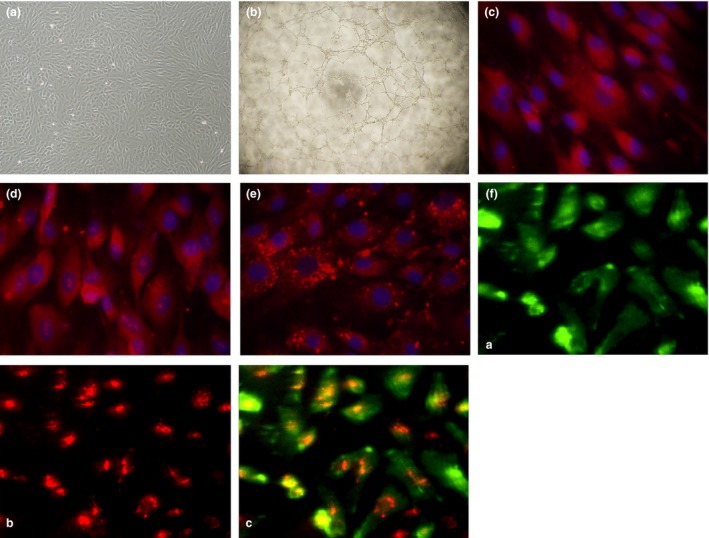
(a) Typical morphological appearance of EPC s in culture (10× objective lens). Typical homogeneous and cobblestone‐like morphology of cells after 14 days culture. (b) Tube formation capacity on Matrigel matrix (10 × 4 objective lens). (c–f) Representative immunofluorescence staining of endothelial cell markers including stem cell marker CD34 (c, red), KDR (d, red), vWF (e, red). Cell nuclei were counterstained with DAPI in blue (40x). (f) The cultured cells were able to bind UEA‐1‐FITC (f‐(a), green) and take up DiI‐acLDL (f‐(b), red), (f‐(c), two‐colour overlay)
Progesterone‐induced EPC viability was time‐ and dose‐dependent
To clarify whether progesterone enhanced EPC viability, MTS assay was performed. As shown in (Fig. 2), EPCs were treated with low concentrations of progesterone (5 and 10 nm) and showed progressive increase in viability starting at 6 h and reaching a peak by 12 h. Notably, the 5 nm progesterone treatment significantly increased the viability at 12 h, compared to control, 10 nm or 100 nm progesterone treatment groups respectively. High‐dose progesterone treatment (100 nm) dramatically impaired EPC viability compared to control (P < 0.05). There was no statistical difference in OD values between vehicle and control groups. The data indicated that 5 nm progesterone was the optimal dose to promote EPC viability. Therefore, this concentration (5 nm) was used in the following studies.
Figure 2.
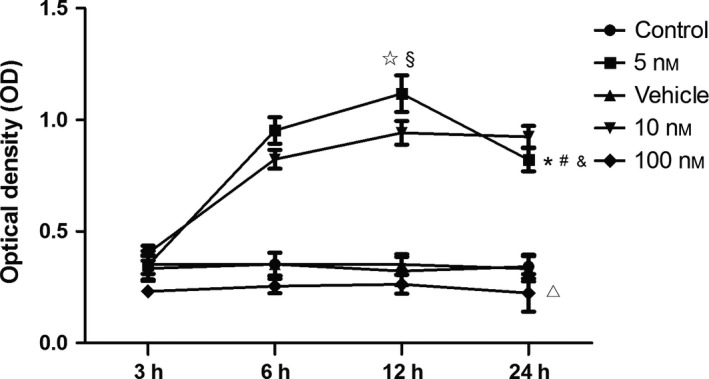
Concentration‐ and time‐dependent effects of progesterone on EPC viability. Low concentration of progesterone (5 nm) enhanced EPC viability to the greatest extent (*P < 0.05 versus control, vehicle, 10 and 100 nm at 12 h) when incubation was sustained for a short term (no more than 12 h, § P < 0.05 versus 3, 6 and 24 h at 5 nm concentration). High‐dose progesterone treatment (100 nm) dramatically impaired viability of EPCs compared to control. There was no statistical difference in OD values between vehicle and control groups.*Two‐way ANOVA: Significant effect among 3, 6, 12 and 24 h in 5 nm group (P < 0.01) #Two‐way ANOVA: Significant treatment effect between 5 nm group and 10 nm group (P < 0.05), &Two‐way ANOVA: treatment effect between 5 nm group and control (P < 0.05), ▵Two‐way ANOVA: Impaired treatment effect between 100 nm group and control (P < 0.05).
Progesterone increased CXCL12 rather than CXCR4 in EPCs: increasing CXCL12 mediated progesterone‐induced EPC viability
To further explore which intracellular pathways were responsible for progesterone‐mediated EPC viability, we investigated effects of progesterone on expression of CXCL12 and CXCR4. By using western Blot assay, we found that progesterone significantly increased CXCL12 expression instead of that of CXCR4(Fig. 3a). As the data shows, 5 nm progesterone was the optimal dose to promote EPC viability. After EPCs were treated with this, cell culture supernatant was found to contain significantly higher levels of CXCL12 (Fig. 3b).
Figure 3.
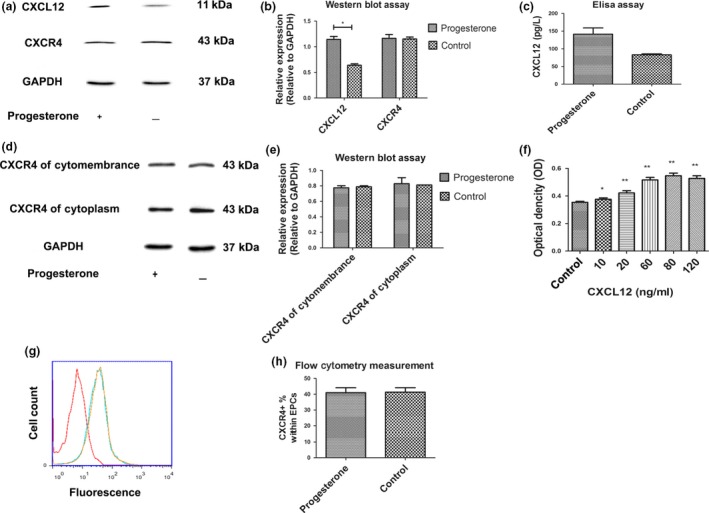
(a) Treating EPC s with progesterone significantly increased levels of CXCL 12 but not CXCR 4 expression. (b) Quantitative analysis of the relative expression of CXCL12 and CXCR4 according to expression of the GAPDH. *P < 0.05 versus control. (c) CXCL12 expression in cultured EPC supernatant.**P < 0.01 versus control. (d) Western blot analysis of CXCR4 protein expressed in cytoplasm and cytomembranes, suggesting that progesterone treatment did not change CXCR4 levels in either. (e) Relative expression of CXCR4 proteinexpressed in cytoplasm and cytomembranes was analysed on the basis of CXCR4/GAPDH ratio. (f) Effect of CXCL12 on EPC viability was measured by MTS. *P < 0.05 versus control, **P < 0.01 versus control. (g) Flow cytometric measurement: progesterone treatment did not increase CXCR4 expression (blue parabola) compared to the non‐treatment group (yellow parabola). Red parabola is the control group without CXCR4 antibody. (H)The percentage of CXCR4+ EPCs in total EPCs after progesterone treatment was measured by flow cytometry.
To further confirm that progesterone administration did not have any impact on compartment protein expression of CXCR4, its levels in membrane proteins were examined by flow cytometry. Furthermore, cytomembrane and cytoplasmic proteins were separately isolated from whole EPCs. Western blotting was performed to confirm that progesterone did not induce differential expression of CXCR4 in either cytomembrane or cytoplasmic proteins (Fig. 3c). Flow cytometry indicated that expression of CXCR4 in membrane proteins was not changed by progesterone treatment (Fig. 3d). This seems to indicate that progesterone did not induce differential expression of CXCR4.
To address whether CXCL12 had any direct impact on EPC viability, we employed the MTS viability assay. EPCs were treated with or without appropriate concentrations of CXCL12 (10–120 ng/ml) for 6 h, then EPC viability was measured. Results indicated that CXCL12 induced a dose‐dependent increase in EPC viability (Fig. 3e).
CXCL12/CXCR4/PI3K/Akt signalling pathways mediated progesterone‐induced EPC viability
To further clarify the role of the PI3K/Akt pathway in progesterone‐induced EPC viability, EPCs were treated with or without 20 μm of specific PI3K inhibitor, LY294002, for 2 h before being treated with progesterone. PI3K/Akt signalling activity was measured. We found that progesterone significantly increased pAkt levels as well as EPC viability compared to the non‐treatment control group. Inhibition of PI3K/Akt activity by LY294002 treatment significantly attenuated progesterone‐induced EPC viability and increased expression of pAkt when compared to the progesterone alone treatment group (Fig. 4a,b).
Figure 4.
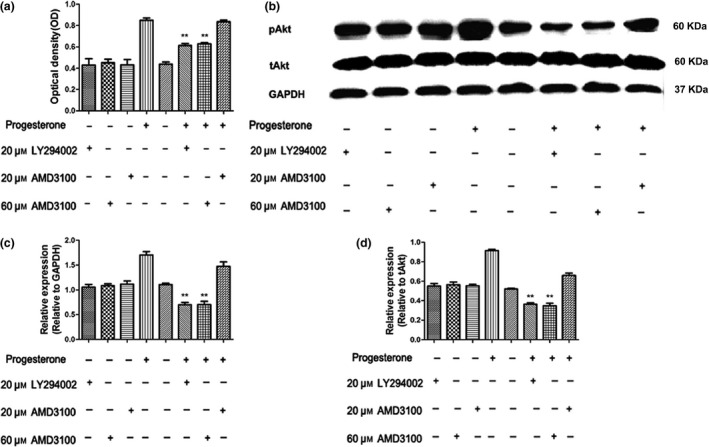
(a) Progesterone treatment increased EPC viability, while AMD 3100 (60 μ m ) or LY 294002 (20 μ m ) treatment significantly attenuated progesterone‐induced EPC viability. Inhibitors alone had no potential effect on EPC viability. **Two‐way ANOVA: P < 0.01 versus the progesterone group. (b) Western blot analysis indicated that progesterone treatment increased pAkt activity while AMD3100 or LY294002 treatment reduced progesterone‐induced pAkt activity. Treating with inhibitors alone did not change pAkt. (c) Quantitative analysis of the relative expression of pAkt according to the expression of the GAPDH. **Two‐way ANOVA: P < 0.01 versus progesterone group. (d) Relative expression of pAkt was analysed on the basis of pAkt/total Akt ratio. **Two‐way ANOVA: P < 0.01 versus progesterone group.
To test whether PI3K/Akt was downstream of CXCL12/CXCR4 axis and mediated progesterone‐induced EPC viability, CXCR4 antagonist, AMD3100 (20 and 60 μm) was employed. The results showed that inhibition of the CXCL12/CXCR4 signalling pathway by high dose AMD3100 (60 μm) significantly attenuated progesterone‐induced EPC viability (Fig. 4a) and reduced the level of p‐Akt compared to the progesterone treatment alone group (Fig. 4b). Low dose of ADM3100 (20 μm) did not reduce EPC viability or pAkt expression (Fig. 4). There was no potential effect of the inhibitors alone in EPC viability and pAkt. Thus, the data suggest that the CXCL12/CXCR4/PI3K/Akt pathway was involved in progesterone‐mediated EPC viability.
Discussion
It is well established that male and female patients suffering from severe TBI have different outcomes,possibly due to different levels of sex hormones. We postulated that progesterone might play a key role in TBI recovery, as diverse levels of progesterone have been found between TBI patients of different genders 27, 28. In addition to its multifarious functions, such as reducing free radicals, apoptosis and oedema, progesterone could also be responsible for EPC‐induced angiogenesis following brain injury 4. It has also been shown that EPCs are rapidly mobilized from the bone marrow in response to brain injury and then play an essential role in neurorestoration 29. EPCs might mediate vasculogenesis in the neurovascular repair process, which is known to be an alternative source of ECs after vascular injury 30, 31. Thus, increasing EPC viability and homing by progesterone treatment may play an important role in brain injury after TBI. Our study was designed to determine whether treatment with physiological concentrations of progesterone (5, 10 and 100 nm) could alter EPC viability. At physiological concentrations, we found that low concentrations of progesterone (5 nm) had the best viability enhancing effect on EPCs when incubation was sustained for a short term (no more than 12 h). However, high concentrations (100 nm) of progesterone and prolonged stimulation (24 h) with it impaired EPC viability. The data indicate that enhancing EPC viability by progesterone may be involved in the improved functional outcome observed following TBI. But, the mechanisms underlying the biphasic response of progesterone on EPCs viability is not clear. Consistent with our results, Okada et al. have demonstrated that high concentrations of progesterone (100 nm) reduce estradiol‐induced vascular endothelial growth factor (VEGF) and CXCL12 levels in human endometrial stromal cells 32. Progesterone also inhibits cAMP‐stimulated chloride transport in 100 nm progesterone 24, through which progesterone may reduce cell viability 33. VEGF, CXCL12 and chloride transport have connections with cell viability 33, 34, 35, thus, decreasing them with high concentrations of progesterone may cause the observed decreased EPC viability. As progesterone‐induced EPC viability rise is dose‐dependent, lower doses of progesterone treatment in TBI should be considered in the clinic. There is significant evidence that progesterone might improve patients’ functional outcome in TBI 1, 36, but, recent studies have indicated that progesterone was not associated with any benefit over placebo 37, 38. Skolnick and colleagues infused progesterone intravenously with loading dose of 0.50 mg/kg/h for 119 h, which lead to high concentrations of progesterone in patients. However, our data showed that high‐dose progesterone treatment (100 nm) dramatically impaired EPC viability, which may result in failure of progesterone treatment. In Wright and colleagues study, progesterone infusion was maintained for 71 h. However, our results showed that low‐level progesterone treatment significantly enhanced EPC viability, but it was restricted by time, with long‐term treatment leading to unsatisfactory consequences. There are fewer studies that focus on changes in the progesterone‐induced signalling pathways during TBI, which may become more complex and play critical roles in the outcome of TBI patients undergoing progesterone treatment. These findings imply that to help develop novel therapies for TBI, optimal treatment time and dose should be identified for progesterone, as well as fully understanding the underlying mechanisms.
The mechanism responsible for progesterone‐induced EPC viability is not known. CXCL12 is a pleiotropic cytokine that binds to and signals through the G protein‐coupled receptor, CXCR4. The CXCL12/CXCR4 signalling pathway plays a critical role in angiogenesis, as well as metastasis, cell proliferation and survival 39. The main functions of CXCL12 are regulating trafficking and homing of stem and progenitor cells, as well as blood vessel formation and angiogenesis 40, 41, 42. The CXCL12/CXCR4 axis is fully active during cell proliferation and viability 35, 43. In tumours, CXCL12/CXCR4 signalling has been shown to regulate vascularization of tumours and foster tumour growth 44, 45. Stimulation of cell growth by CXCL12/CXCR4 has been documented in human breast cancer cells, due to overexpression of CXCL12 46. In addition, CXCL12 has also been shown to increase rat NPC proliferation and cardiomyocyte viability 47, 48. Overexpression of CXCL12 overlaps with strong expression of the progesterone receptor (PR) and when localized with PR‐positive cells has indicated that there is a potential relationship between progesterone and CXCL12 49. However, it is not clear whether progesterone acts through a CXCL12‐dependent or ‐independent manner. Our study shows that CXCL12 expression is tightly associated with progesterone administration. Progesterone or CXCL12 treatment, both significantly increase EPC viability. In addition to regulating CXCL12, progesterone also increases the phosphatidylinositol 3‐kinase (PI3K)/Akt pathway 50, extracellular signal‐related kinase (ERK) pathway 51 and Ca2+ influx/PKC activation 52. Progesterone also interacts with neurotransmitter receptors, including gamma‐aminobutyric acid A (GABA‐A) receptor 53, and Sigma‐1/2 54 receptors to exert its protective effects. Through these signalling pathways, progesterone plays an important role in regulation of cell viability 53, 55 and cell proliferation 56, in a CXCL12‐independent manner. Furthermore, our data also show that induction of proliferation by CXCL12 appears to be more modest compared to progesterone treatment. The antagonist of CXCL12 receptor‐CXCR4 failed to entirely abolish progesterone‐induced viability. Hence, progesterone‐induced EPC viability may have CXCL12‐dependent and ‐independent pathways. The underlying mechanisms by which progesterone induces viability warrant further study. Meanwhile, we found that progesterone treatment did not regulate CXCR4 expression in cultured EPCs. In addition, there was no differential expression between the membrane compartment and the cytosolic compartment of CXCR4 proteins. Progesterone‐induced EPC viability was abolished by a CXCR4 inhibitor. Thus, it is reasonable to infer that CXCL12 plays a key role in progesterone‐induced EPC viability and the function relies on its combination with CXCR4. CXCR4 expression in EPCs is necessary for progesterone‐induced increase in EPC viability. Thus, the CXCL12/CXCR4 signalling pathway appears to be involved in progesterone‐inducing EPC viability.
The PI3K/Akt pathway plays a key role in the CXCL12/CXCR4 pathway's control of survival and migration of haematopoietic cells 57. Study of NPCs has shown that activation of PI3K/Akt stimulated by CXCL12 through CXCR4 affects cell proliferation 17. Akt plays an important role in cell proliferation, differentiation and survival, and it also regulates many genes as a downstream target of PI3K 19, 58, 59. It is accepted that the PI3K/Akt/mTOR/p70S6K pathway plays a role in cell viability in colorectal cancer cells 60. Survival of both normal and malignant cells relies on PI3K/Akt signalling. PI3K and its downstream factors have a central role in regulating muliple functions of mammalian cells, as well as cell proliferation and viability 61, 62, 63. In this study, we found that progesterone significantly increased EPC viability and pAkt expression. Furthermore, inhibition of the PI3K/Akt signalling pathway by LY294002 abolished progesterone‐induced pAkt activity and progesterone‐mediated EPC viability. The role of the PI3K/Akt signalling pathway was then suggested to be involved in progesterone‐induced EPC viability. We also found that inhibition of CXCL12/CXCR4 by ADM3100 significantly attenuated both progesterone‐inducing EPC viability and pAkt activity. Thus, CXCL12/CXCR4/PI3K/Akt pathway seems to play a key role in progesterone‐induced EPC viability.
In conclusion, we found that progesterone does dependently augmented EPC viability and increased CXCL12 expression and PI3K/Akt signalling pathway activity. The CXCL12/CXCR4/PI3K/Akt signalling pathway contributed to progesterone‐induced EPC viability.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant 81271359), The Ontario‐China Research and Innovation Fund (OCRIF, 2011DFG33430) and Tianjin Research Program of Application Foundation and Advanced Technology (grant 11JCZDJC18100).
References
- 1. Hu Z, Li Y, Fang M, Wai MS, Yew DT (2009) Exogenous progesterone: a potential therapeutic candidate in CNS injury and neurodegeneration. Curr. Med. Chem. 16, 1418–1425. [DOI] [PubMed] [Google Scholar]
- 2. Peluso JJ, Liu X, Saunders MM, Claffey KP, Phoenix K (2008) Regulation of ovarian cancer cell viability and sensitivity to cisplatin by progesterone receptor membrane component‐1. J. Clin. Endocrinol. Metab. 93, 1592–1599. [DOI] [PubMed] [Google Scholar]
- 3. Peluso JJ, Yuan A, Liu X, Lodde V (2013) Plasminogen activator inhibitor 1 RNA‐binding protein interacts with progesterone receptor membrane component 1 to regulate progesterone's ability to maintain the viability of spontaneously immortalized granulosa cells and rat granulosa cells. Biol. Reprod. 88, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Z, Wang B, Kan Z, Zhang B, Yang Z, Chen J et al (2012) Progesterone increases circulating endothelial progenitor cells and induces neural regeneration after traumatic brain injury in aged rats. J. Neurotrauma 29, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xue S, Zhang HT, Zhang P, Luo J, Chen ZZ, Jang XD et al (2010) Functional endothelial progenitor cells derived from adipose tissue show beneficial effect on cell therapy of traumatic brain injury. Neurosci. Lett. 473, 186–191. [DOI] [PubMed] [Google Scholar]
- 6. Fan Y, Shen F, Frenzel T, Zhu W, Ye J, Liu J et al (2010) Endothelial progenitor cell transplantation improves long‐term stroke outcome in mice. Ann. Neurol. 67, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Briasoulis A, Tousoulis D, Antoniades C, Papageorgiou N, Stefanadis C (2011) The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc. Ther. 29, 125–139. [DOI] [PubMed] [Google Scholar]
- 8. Heeschen C, Lehmann R, Honold J, Assmus B, Aicher A, Walter DH et al (2004) Profoundly reduced neovascularization capacity of bone marrow mononuclear cells derived from patients with chronic ischemic heart disease. Circulation 109, 1615–1622. [DOI] [PubMed] [Google Scholar]
- 9. Herbrig K, Haensel S, Oelschlaegel U, Pistrosch F, Foerster S, Passauer J (2006) Endothelial dysfunction in patients with rheumatoid arthritis is associated with a reduced number and impaired function of endothelial progenitor cells. Ann. Rheum. Dis. 65, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC et al (2004) Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 53, 195–199. [DOI] [PubMed] [Google Scholar]
- 11. Zhao LR, Du YJ, Chen L, Liu ZG, Pan YH, Liu JF et al (2014) Quercetin protects against high glucose‐induced damage in bone marrow‐derived endothelial progenitor cells. Int. J. Mol. Med. 34, 1025–1031. [DOI] [PubMed] [Google Scholar]
- 12. Bolitho C, Hahn MA, Baxter RC, Marsh DJ (2010) The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr. Relat. Cancer 17, 929–940. [DOI] [PubMed] [Google Scholar]
- 13. Basso FG, Soares DG, Pansani TN, Turrioni AP, Scheffel DL, de Souza Costa CA et al (2015) Effect of LPS treatment on the viability and chemokine synthesis by epithelial cells and gingival fibroblasts. Arch. Oral Biol. 60, 1117–1121. [DOI] [PubMed] [Google Scholar]
- 14. Spratte J, Schonborn M, Treder N, Bornkessel F, Zygmunt M, Fluhr H (2015) Heparin modulates chemokines in human endometrial stromal cells by interaction with tumor necrosis factor alpha and thrombin. Fertil. Steril. 103, 1363–1369. [DOI] [PubMed] [Google Scholar]
- 15. Li B, Bai W, Sun P, Zhou B, Hu B, Ying J (2014) The effect of CXCL12 on endothelial progenitor cells: potential target for angiogenesis in intracerebral hemorrhage. J. Interferon Cytokine Res. 35, 23–31. [DOI] [PubMed] [Google Scholar]
- 16. Vlahakis SR, Villasis‐Keever A, Gomez T, Vanegas M, Vlahakis N, Paya CV (2002) G protein‐coupled chemokine receptors induce both survival and apoptotic signaling pathways. J. Immunol. 169, 5546–5554. [DOI] [PubMed] [Google Scholar]
- 17. Wu Y, Peng H, Cui M, Whitney NP, Huang Y, Zheng JC (2009) CXCL12 increases human neural progenitor cell proliferation through Akt‐1/FOXO3a signaling pathway. J. Neurochem. 109, 1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khan MZ, Brandimarti R, Musser BJ, Resue DM, Fatatis A, Meucci O (2003) The chemokine receptor CXCR4 regulates cell‐cycle proteins in neurons. J. Neurovirol. 9, 300–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plas DR, Thompson CB (2005) Akt‐dependent transformation: there is more to growth than just surviving. Oncogene 24, 7435–7442. [DOI] [PubMed] [Google Scholar]
- 20. Jiang J, Zhang Y, Guo Y, Yu C, Chen M, Li Z et al (2015) MicroRNA‐3127 promotes cell proliferation and tumorigenicity in hepatocellular carcinoma by disrupting of PI3K/AKT negative regulation. Oncotarget 6, 6359–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colombo E, Calcaterra F, Cappelletti M, Mavilio D, Della Bella S (2013) Comparison of fibronectin and collagen in supporting the isolation and expansion of endothelial progenitor cells from human adult peripheral blood. PLoS ONE 8, e66734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brandl A, Yuan Q, Boos AM, Beier JP, Arkudas A, Kneser U et al (2014) A novel early precursor cell population from rat bone marrow promotes angiogenesis in vitro. BMC Cell Biol. 15, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsukada S, Kwon SM, Matsuda T, Jung SY, Lee JH, Lee SH et al (2013) Identification of mouse colony‐forming endothelial progenitor cells for postnatal neovascularization: a novel insight highlighted by new mouse colony‐forming assay. Stem Cell Res. Ther. 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayol JM, Arbeo‐Escolar A, Alarma‐Estrany P, Adame‐Navarrete Y, Fernandez‐Represa JA (2002) Progesterone inhibits chloride transport in human intestinal epithelial cells. World J. Surg. 26, 652–656. [DOI] [PubMed] [Google Scholar]
- 25. Zhou Z, Deng H, Yan W, Luo M, Tu W, Xia Y et al (2014) AEG‐1 promotes anoikis resistance and orientation chemotaxis in hepatocellular carcinoma cells. PLoS ONE 9, e100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carr AN, Howard BW, Yang HT, Eby‐Wilkens E, Loos P, Varbanov A et al (2006) Efficacy of systemic administration of SDF‐1 in a model of vascular insufficiency: support for an endothelium‐dependent mechanism. Cardiovasc. Res. 69, 925–935. [DOI] [PubMed] [Google Scholar]
- 27. Bounds TA, Schopp L, Johnstone B, Unger C, Goldman H (2003) Gender differences in a sample of vocational rehabilitation clients with TBI. NeuroRehabilitation 18, 189–196. [PubMed] [Google Scholar]
- 28. Stein DG (2011) Is progesterone a worthy candidate as a novel therapy for traumatic brain injury? Dialogues Clin. Neurosci. 13, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu L, Liu H, Jiao J, Liu H, Bergeron A, Dong JF et al (2007) Changes in circulating human endothelial progenitor cells after brain injury. J. Neurotrauma 24, 936–943. [DOI] [PubMed] [Google Scholar]
- 30. Tepper OM, Capla JM, Galiano RD, Ceradini DJ, Callaghan MJ, Kleinman ME et al (2005) Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow‐derived cells. Blood 105, 1068–1077. [DOI] [PubMed] [Google Scholar]
- 31. Frontczak‐Baniewicz M, Walski M (2003) New vessel formation after surgical brain injury in the rat's cerebral cortex I. Formation of the blood vessels proximally to the surgical injury. Acta Neurobiol. Exp. (Wars). 63, 65–75. [DOI] [PubMed] [Google Scholar]
- 32. Okada H, Okamoto R, Tsuzuki T, Tsuji S, Yasuda K, Kanzaki H (2011) Progestins inhibit estradiol‐induced vascular endothelial growth factor and stromal cell‐derived factor 1 in human endometrial stromal cells. Fertil. Steril. 96, 786–791. [DOI] [PubMed] [Google Scholar]
- 33. Suh KS, Yuspa SH (2005) Intracellular chloride channels: critical mediators of cell viability and potential targets for cancer therapy. Curr. Pharm. Des. 11, 2753–2764. [DOI] [PubMed] [Google Scholar]
- 34. Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M et al (2012) Autocrine VEGF‐VEGFR2‐Neuropilin‐1 signaling promotes glioma stem‐like cell viability and tumor growth. J. Exp. Med. 209, 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gatti M, Pattarozzi A, Bajetto A, Wurth R, Daga A, Fiaschi P et al (2013) Inhibition of CXCL12/CXCR4 autocrine/paracrine loop reduces viability of human glioblastoma stem‐like cells affecting self‐renewal activity. Toxicology 314, 209–220. [DOI] [PubMed] [Google Scholar]
- 36. Xiao G, Wei J, Yan W, Wang W, Lu Z (2008) Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit. Care 12, R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M et al (2014) Very early administration of progesterone for acute traumatic brain injury. N. Engl. J. Med. 371, 2457–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD et al (2014) A clinical trial of progesterone for severe traumatic brain injury. N. Engl. J. Med. 371, 2467–2476. [DOI] [PubMed] [Google Scholar]
- 39. Teicher BA, Fricker SP (2010) CXCL12 (SDF‐1)/CXCR4 pathway in cancer. Clin. Cancer Res. 16, 2927–2931. [DOI] [PubMed] [Google Scholar]
- 40. Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ et al (2004) CXCR4‐SDF‐1 signalling, locomotion, chemotaxis and adhesion. J. Mol. Histol. 35, 233–245. [DOI] [PubMed] [Google Scholar]
- 41. Juarez J, Bendall L (2004) SDF‐1 and CXCR4 in normal and malignant hematopoiesis. Histol. Histopathol. 19, 299–309. [DOI] [PubMed] [Google Scholar]
- 42. Wang L, Li X, Zhao Y, Fang C, Lian Y, Gou W et al (2015) Insights into the mechanism of CXCL12‐mediated signaling in trophoblast functions and placental angiogenesis. Acta Biochim. Biophys. Sin. (Shanghai) 47, 663–672. [DOI] [PubMed] [Google Scholar]
- 43. Wei L, Zhang B, Cao W, Xing H, Yu X, Zhu D (2014) Inhibition of CXCL12/CXCR4 suppresses pulmonary arterial smooth muscle cell proliferation and cell cycle progression via PI3K/Akt pathway under hypoxia. J. Recept. Signal Transduct. Res. 35, 329–339. [DOI] [PubMed] [Google Scholar]
- 44. Orimo A, Gupta PB, Sgroi DC, Arenzana‐Seisdedos F, Delaunay T, Naeem R et al (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF‐1/CXCL12 secretion. Cell 121, 335–348. [DOI] [PubMed] [Google Scholar]
- 45. Ao M, Franco OE, Park D, Raman D, Williams K, Hayward SW (2007) Cross‐talk between paracrine‐acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 67, 4244–4253. [DOI] [PubMed] [Google Scholar]
- 46. Kang H, Mansel RE, Jiang WG (2005) Genetic manipulation of stromal cell‐derived factor‐1 attests the pivotal role of the autocrine SDF‐1‐CXCR4 pathway in the aggressiveness of breast cancer cells. Int. J. Oncol. 26, 1429–1434. [PubMed] [Google Scholar]
- 47. Gong X, He X, Qi L, Zuo H, Xie Z (2006) Stromal cell derived factor‐1 acutely promotes neural progenitor cell proliferation in vitro by a mechanism involving the ERK1/2 and PI‐3K signal pathways. Cell Biol. Int. 30, 466–471. [DOI] [PubMed] [Google Scholar]
- 48. Atluri P, Liao GP, Panlilio CM, Hsu VM, Leskowitz MJ, Morine KJ et al (2006) Neovasculogenic therapy to augment perfusion and preserve viability in ischemic cardiomyopathy. Ann. Thorac. Surg. 81, 1728–1736. [DOI] [PubMed] [Google Scholar]
- 49. Shiah YJ, Tharmapalan P, Casey AE, Joshi PA, McKee TD, Jackson HW et al (2015) A progesterone‐CXCR4 axis controls mammary progenitor cell fate in the adult gland. Stem Cell Rep. 4, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng S, Huang J, Zhou K, Xiang Q, Zhang Y, Tan Z et al (2012) Progesterone enhances vascular endothelial cell migration via activation of focal adhesion kinase. J. Cell Mol. Med. 16, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Su C, Cunningham RL, Rybalchenko N, Singh M (2012) Progesterone increases the release of brain‐derived neurotrophic factor from glia via progesterone receptor membrane component 1 (Pgrmc1)‐dependent ERK5 signaling. Endocrinology 153, 4389–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swiatek‐De Lange M, Stampfl A, Hauck SM, Zischka H, Gloeckner CJ, Deeg CA et al (2007) Membrane‐initiated effects of progesterone on calcium dependent signaling and activation of VEGF gene expression in retinal glial cells. Glia 55, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 53. Ishihara Y, Kawami T, Ishida A, Yamazaki T (2013) Allopregnanolone‐mediated protective effects of progesterone on tributyltin‐induced neuronal injury in rat hippocampal slices. J. Steroid Biochem. Mol. Biol. 135, 1–6. [DOI] [PubMed] [Google Scholar]
- 54. Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F et al (2011) Identification of the PGRMC1 protein complex as the putative sigma‐2 receptor binding site. Nat. Commun. 2, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaur P, Jodhka PK, Underwood WA, Bowles CA, de Fiebre NC, de Fiebre CM et al (2007) Progesterone increases brain‐derived neuroptrophic factor expression and protects against glutamate toxicity in a mitogen‐activated protein kinase‐ and phosphoinositide‐3 kinase‐dependent manner in cerebral cortical explants. J. Neurosci. Res. 85, 2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu B, Arbogast LA (2009) Gene expression profiles of intracellular and membrane progesterone receptor isoforms in the mediobasal hypothalamus during pro‐oestrus. J. Neuroendocrinol. 21, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moll NM, Ransohoff RM (2010) CXCL12 and CXCR4 in bone marrow physiology. Expert Rev. Hematol. 3, 315–322. [DOI] [PubMed] [Google Scholar]
- 58. Scioli MG, Bielli A, Gentile P, Mazzaglia D, Cervelli V, Orlandi A (2014) The biomolecular basis of adipogenic differentiation of adipose‐derived stem cells. Int. J. Mol. Sci. 15, 6517–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ahn JY (2014) Neuroprotection signaling of nuclear akt in neuronal cells. Exp. Neurobiol. 23, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang H, Duan L, Zou Z, Li H, Yuan S, Chen X et al (2014) Activation of the PI3K/Akt/mTOR/p70S6K pathway is involved in S100A4‐induced viability and migration in colorectal cancer cells. Int. J. Med. Sci. 11, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Engelman JA (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9, 550–562. [DOI] [PubMed] [Google Scholar]
- 62. Vanhaesebroeck B, Stephens L, Hawkins P (2012) PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 13, 195–203. [DOI] [PubMed] [Google Scholar]
- 63. Xu Q, Ma J, Lei J, Duan W, Sheng L, Chen X et al (2014) alpha‐Mangostin suppresses the viability and epithelial‐mesenchymal transition of pancreatic cancer cells by downregulating the PI3K/Akt pathway. Biomed. Res. Int. 2014, 546353. [DOI] [PMC free article] [PubMed] [Google Scholar]