

Abstract
Abstract. Heat shock proteins (HSPs) are involved in a variety of intracellular processes and can have both pro‐ and anti‐apoptotic action. However, little is known about the role of HSPs in the progression of apoptosis. Translocation of HSPs to the surface of apoptotic cells is a previously observed phenomenon demonstrating participation of these proteins in execution of the terminal stages of apoptosis. In a previous study we showed that development of EL‐4 lymphoma cell apoptosis in vitro is accompanied by elevation of surface HSP expression. In this study we used this model to analyse the relationship of HSP70 expression and its translocation to the cell surface during apoptosis with some key intracellular events. Our data demonstrate a synchronization of surface and intracellular HSP70 expression with bcl‐2 expression, intracellular reactive oxygen species (ROS) concentration and caspase‐3 activity. A maximum level of surface and intracellular HSP70 expression was observed at an irreversible phase of EL‐4 cell apoptosis after mitochondrial potential depolarization. In addition, an enhancement of the relative level of cytoplasmic HSP70 translocation to the cell surface was concomitant with EL‐4 cell apoptosis. However, the size of surface and intracellular pools of HSP70, increasing for initial and intermediate stages of cell death, decreased at the terminal phase of apoptosis. Western blot analysis of HSP70 in conditioned supernatant obtained from EL‐4 cell tissue showed that the observed decrease of HSP70 cell content might be due to surface HSP70 shedding into the intercellular space.
Introduction
The superfamily of heat shock proteins (HSPs) is characterized both by heterogeneity of physicochemical properties and variety of functions. The chaperone functions of intracellular HSPs connected with protein folding, transport and reparation and their protective functions increasing cell resistance to stressful conditions have been extensively studied (Schlesinger 1990; Parsell & Lindquist 1993; Craig et al. 1994; Morimoto et al. 1994; Hartl 1996). HSPs have also been shown to be involved in intracellular signal transduction (Pratt & Toft 1997). In comparison with intracellular HSPs, the functions of HSPs expressed on the surface of normal (Erkeller‐Yeksel et al. 1992; Ishiyama et al. 1996), infected (Di Cesare et al. 1992), transformed (Ferrarini et al. 1992; Altmeyer et al. 1996; Multhoff & Hightower 1996; Multhoff et al. 1997; Rogias et al. 1998; Sapozhnikov et al. 1999; Hantschel et al. 2000) and apoptotic (Poccia et al. 1996) cells are poorly understood. Some data indicated that surface HSPs might serve as markers of cells subject to elimination by the innate immune system (Multhoff & Hightower 1996; Multhoff et al. 1997; Rogias et al. 1998). However, such a role for surface HSPs might be a consequence of their surface localization but not an underlying cause of HSP translocation onto the plasma membrane. One reason for translocation might be connected to the known capacity of HSPs to stabilize plasma membranes (Tõrõk et al. 1997). This property of HSPs may be used by cells to reinforce the membranes in the case of their destabilization (due to cell stress, cell transformation, apoptosis, etc.). However, at present there is no direct evidence for the role of surface HSP participation in cell protection.
The functions of HSP translocated to the cell surface may not be restricted to membrane and protein stabilization. Surface HSP localization can be considered as an intermediate stage of the process of HSP production by cells into the extracellular space. Indeed, HSPs have been detected in the culture medium of some cell lines (Multhoff & Hightower 1996). The mechanisms of HSP secretion and the functions of the extracellular pool of HSPs have not been studied. Some authors believe that the presence of HSPs in the extracellular milieu is a result of cell destruction (necrosis) followed by intracellular HSP release (Basu et al. 2000). Alternatively, cells may actively secrete cytoplasmic HSPs (Hightower & Guidon 1989; Multhoff & Hightower 1996). In favour of the latter mechanism, recent data have suggested that exogenous HSPs may possess cytokine properties (Asea et al. 2000), and certainly, all cytokines are secreted factors. Collectively, the above data hint at some functional significance for extracellular HSPs but have not elucidated any clear role in cell physiology. In addition, mechanisms of HSP secretion into the extracellular milieu are largely unclear and need to be determined.
Previously, we demonstrated that in vitro culture of EL‐4 lymphoma cells is characterized by spontaneous apoptosis and concurrent expression of surface HSPs (Sapozhnikov et al. 1999). In this model, the initiation of apoptosis was accompanied by elevated levels of surface HSPs and a subsequent decrease of their expression at the terminal phase of cell death. The registered variation of surface HSP expression during apoptosis suggested a possibility of using this model for studying the process of cytoplasmic HSP translocation onto the plasma membrane. In our present work, we have used this model to identify several successive stages of EL‐4 cell‐programmed death and have analysed the relationship of surface and intracellular HSP70 expression with some key apoptotic events, including bcl‐2 and p53 expression, caspase‐3 activity, intracellular levels of reactive oxygen species (ROS) and mitochondrial transmembrane potential (ΔΨm). We have also measured the secretion/shedding of HSPs during apoptosis and have confirmed that this in fact does occur.
Materials and methods
Cell culture
Mouse EL‐4 lymphoma cells were grown in RPMI‐1640 medium supplemented with 10% heat‐inactivated fetal bovine serum (FBS), 4 mm l‐glutamine, 20 mm HEPES and 50 µg/ml gentamicin (complete medium; all reagents from Sigma, St Louis, MO, USA). Cells were passaged every 3 days. Cell number was determined using a haemocytometer and viability was assessed by their ability to exclude trypan blue.
Flow cytometric analysis
All flow cytometric analysis was performed using an EPICS ELITE fluorescence‐activated cell sorter (Coulter Corporation, Miami, FL, USA) equipped with a water‐cooled argon‐ion laser emitting at 488 nm.
Live cell assays
For immunolabelling, cells were transferred from culture medium into phosphate‐buffered saline (PBS) supplemented with 1% FBS and 0.05% NaN3 and were incubated with an antibody specific for HSP70 (clone BRM‐22, Sigma) or for bcl‐2 (clone 3F11, BD Pharmingen, San Diego, CA, USA) for 45 min at 4 °C. An isotype‐matched control antibody was used in each experiment. The cells were then washed and incubated under the same conditions with a secondary anti‐immunoglobulin antibody coupled to fluorescein (FITC). Labelled cells were washed twice and measured immediately. To assess ΔΨm (mitochondrial membrane potential), rhodamine 123 (Rh123, Sigma) and 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolocarbocyanine iodide (JC‐1, Sigma) were used. EL‐4 cells were incubated in 50 ng/ml Rh123 or with 1 µm JC‐1 for 30 min at 37 °C followed by flow cytometric analysis. For samples with Rh123, the level of green fluorescence (proportional to both ΔΨm and the total mitochondria volume) was measured in cell subpopulations being at different stages of apoptosis. In the case of JC‐1, the ratio of the red fluorescence level to green fluorescence level was measured. This ratio was mitochondrial potential‐dependent and mitochondrial volume‐independent (Reers et al. 1995).
To evaluate caspase‐3 activity at different stages of EL‐4 cell apoptosis by flow cytometry, the protease‐specific fluorogenic substrate PhiPhiLux‐G1D2 (OncoImmunin Inc., Gaithersburg, MD, USA) was used (Hirata et al. 1998). Tubes containing about 106 cells were centrifuged and 50 µl (10 µm stock concentration) substrate solution was added to the centrifuged cell pellets. After 30–60 min incubation at 37 °C in CO2‐incubator, samples were washed with cold dilution buffer and resuspended in 1 ml of fresh dilution buffer. The samples were subsequently kept at 4 °C and analysed within 60–90 min. Intracellular ROS concentration during EL‐4 cell apoptosis was determined by incubation of the cells in the membrane‐permeant H2O2 probe 10 µm 2′7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA, Molecular Probes, Eugene, OR) for 30 min at 37 °C, followed by flow cytometric analysis for FITC fluorescence (Rothe & Valet 1990).
The mitochondrial membrane potential, caspase‐3 activity and ROS concentration analyses were performed together with the propidium iodide (PI, Sigma) exclusion analysis to determine viability (and terminal stage of apoptosis) in measured cell suspension.
Fixed and permeabilized cell assays
To simultaneously detect surface and intracellular HSP70 and for evaluation of bcl‐2 and p53 expression during apoptosis, a series of EL‐4 cell samples were first immunolabelled with the antibody directed to these proteins as described above, or with antip53 antibody (clone PAb 246, PharMingen), then fixed with 0.5% paraformaldehyde for 30 min. Cells were then permeabilized with 0.1% Triton X‐100 in PBS at 4 °C for 3 min followed by washing and standard staining with indirect immunofluorescence using the same secondary antibody described above. Thus, these samples were labelled for both surface and cytoplasmic proteins. Another series of samples of the same cells were labelled only for surface protein expression as previously described for viable cells. For discrimination of different stages of apoptosis 20 µg/ml PI and 1 mg/ml RNase A were added to every sample 10 min prior to analysis.
In all experiments, more than 5–10 × 103 cells were analysed for each sample. For evaluation of the level of HSP70, bcl‐2, p53 expression, caspase‐3 activity, ROS concentration and mitochondrial potential, the arithmetic mean level of cell fluorescence was used. The relative level of protein expression was presented as: [mean fluorescence intensity (experiment) – mean fluorescence intensity (control)].
Detection of HSP70 shedding or secretion
To detect possible shedding of surface HSP70, we prepared the samples of conditioned supernatant obtained from EL‐4 cell culture for immunoblot analysis of HSP70 content. For this purpose complete medium in EL‐4 culture was gently changed to serum‐free medium at the termination of exponential phase cell growth (2 days after passing, for cell density about 106 cells/ml). Two hours later the medium was changed once more to remove all traces of serum. The cells were cultivated 1 day before the supernatant was harvested. Adherent (live cells) and non‐adherent (apoptotic cells) fractions of EL‐4 cell culture were separated [as described in Sapozhnikov et al. (1999)] and washed at the same phase of cell growth, and supernatant was collected after 24 h incubation of these different cell populations in serum‐free medium. The supernatant obtained was dialysed against distilled water, lyophilized and dissolved in a volume of PBS to a final protein concentration approximately 20‐fold higher than the initial concentration. These samples were measured for protein concentration by a standard method (Bradford 1976) and were analysed by immunoblotting for HSP70 content.
Immunoblotting analysis
The samples of conditioned supernatant were subjected to SDS‐10% PAGE (approximately 50 µg of protein was loaded in each line) under reducing (100 mmβ‐mercaptoethanol + 2% SDS) conditions and then transferred to nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany) by semidry electroblotting (Bio‐Rad, Hercules, CA). To check equal protein loading, the membranes were stained with Ponceau Red (Sigma). For blocking nonspecific binding, the nitrocellulose membrane strips were incubated in PBS containing 5% skim milk and 0.1% Tween‐20 for 2 h at room temperature. The strips were then washed in PBS containing 0.1% Tween 20, and incubated with anti‐HSP70 (BRM22, 1 : 500) for 1 h at room temperature followed by washing and incubation with peroxidase‐conjugated goat antimouse IgG (Sigma). After washing with blocking buffer, the blots were developed with 4‐chloronaphthol (1 mg/ml) and hydrogen peroxide (0.003%).
Results
Relationship of surface HSP70 expression with ΔΨm, caspase‐3 activity and intracellular ROS activity during EL‐4 cell apoptosis
Previously we demonstrated that cell light scatter properties detected by flow cytometry allow us to identify two successive stages of programmed death of EL‐4 cells in vitro (Sapozhnikov et al. 1999). The first, earliest stage of cell death detected in this model can be identified as the phase of initial decreasing of forward cell light scatter (FS), reflecting cell size, and increasing of side cell light scatter (SS), reflecting cell granularity. This first pool of cells can be identified on a FS/SS dot plot as a region of ‘neck’ cells positioned between the regions of live and apoptotic cells (Fig. 1a). A second region of apoptotic cells contains two subpopulations – stained and unstained by PI (Fig. 1b). These features of EL‐4 cells measured by flow cytometric analysis therefore allow identification of three successive stages of apoptosis: first, ‘neck’ cells, the earliest detected apoptotic cells in this model; second, cells from apoptotic region detected by light scattering that are negative for PI staining (representing an intermediate stage of apoptosis); and third, cells from the same apoptotic region positively stained by PI (representing the terminal phase of apoptosis).
Figure 1.
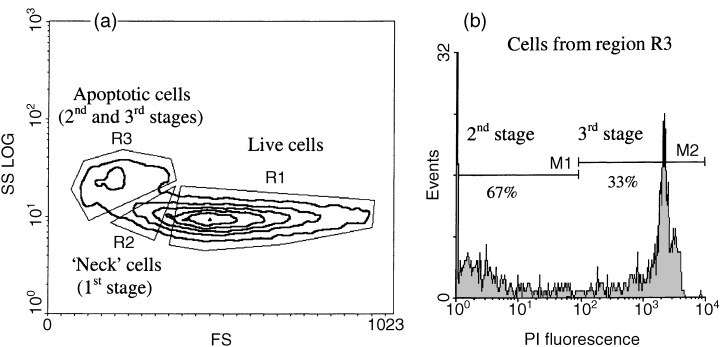
Different stages of apoptosis detected by flow cytometry in in vitro culture of EL‐4 cells. (a) Regions of live and apoptotic cells selected by their morphological features (forward and side light scatter) . (b) Second and third stages of apoptosis selected by the level of PI staining cells from apoptotic region identified by morphological features (region R3, second and third stages).
This flow cytometric assay for these three stages of apoptosis allowed simultaneous analysis of surface HSP expression, mitochondrial membrane potential using the fluorescent probes rhodamine 123 and JC‐1, and caspase‐3 activity using a fluorogenic substrate. Measurement of surface HSP70 expression in EL‐4 cell culture for the three stages of apoptosis confirmed our previous data, demonstrating an increase during first and second stages, followed by a decrease in the third, terminal phase of apoptosis (2, 3). The analysis of the relative mitochondrial membrane potential ΔΨm for these successive phases of programmed death of EL‐4 cells showed that it was significantly elevated during the earliest stage of apoptosic neck cells (2, 3). After this stage, the progress of EL‐4 cell apoptosis was characterized by depolarization of mitochondria (2, 3, stages 2 and 3). The elevation of ΔΨm at the earliest phase of EL‐4 cell apoptosis was not an artifact connected with a rise of the mitochondria volume because the ΔΨm potential was detected using both rhodamine123, a probe dependent on the potential and volume of mitochondria, and JC‐1, a ratiometric probe reflecting transmembrane mitochondrial potential independently of the volume of mitochondria (2, 3). In addition, a separate series of experiments using NAO, a fluorescence probe sensitive to mitochondrial volume and insensitive to transmembrane potential, showed that in our model the development of apoptosis is not accompanied by significant elevation of mitochondrial volume (data not shown). These data show that the increase in surface HSP expression occurs after the alteration in mitochondrial membrane potential known to be an early step in the apoptotic cascade.
Figure 2.
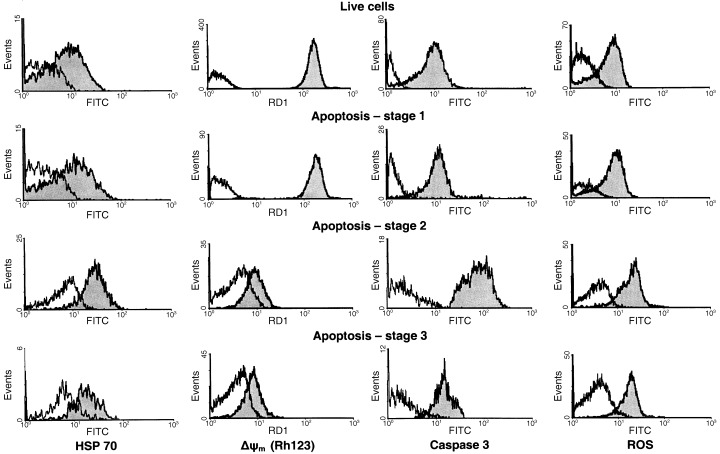
Flow cytometric histograms of surface HSP70 expression, mitochondrial membrane potential, caspase‐3 and ROS activity at progressive stages of EL‐4 cell death.
Figure 3.
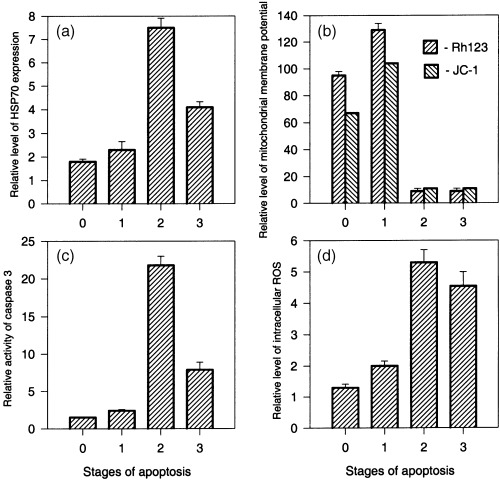
Surface HSP70 expression, mitochondrial membrane potential, caspase‐3 and ROS activity at progressive stages of EL‐4 cell death. (a) HSP70 expression; (b) mitochondrial transmembrane potential; (c) caspase‐3 activity, and (d) intracellular concentration of ROS during the progress of programmed EL‐4 cell death. Results are presented as the mean of fluorescence intensity ± SEM. Data are representative of three comparable experiments.
An increase of caspase‐3 activity using a fluorogenic caspase substrate (PhiPhiLux G1D2) was also observed at the earliest phase of apoptosis in our EL‐4 cell model (2, 3, stage 1). Transition of cells from the first to the second phase of apoptosis was accompanied by a strong elevation (about 10‐fold) in caspase‐3 activity (2, 3, stage 2) followed by a marked decrease at the terminal phase of cell death (2, 3, stage 3). Caspase‐3 activation and surface HSP elevation therefore occurred more or less simultaneously in EL‐4 cell death. The synchronization may reflect dependence of the level of HSP70 expression and activity of caspase‐3 on the same regulatory signals, in accordance with our supposition that HSP can participate in execution of the programme of apoptosis at its intermediate and later stages.
Also coincident with surface HSP70 expression was intracellular ROS, similarly measured by flow cytometry. In particular, entry of EL‐4 cells into apoptosis was accompanied by a small elevation of intracellular ROS concentration (2, 3, stage 1). This concentration was maximum at the second phase of cell death (2, 3, stage 2) followed by a decrease at the terminal phase of apoptosis (2, 3, stage 3). The registered correlation between the level of surface HSP70 and intracellular ROS concentration suggested that, in our model, ROS may be similarly associated with HSP expression in some functional way.
Analysis of HSP70, bcl‐2 and p53 expression during EL‐4 cell apoptosis
The experiments described required the analysis of live EL‐4 cells; as a result, the HSP expression levels measured were limited to surface expression. In this series of experiments, we analysed the redistribution of HSP70 between the cytoplasm and plasma membrane during EL‐4 cell apoptosis, and analysed the relationship between HSP expression and expression of cell death‐associated proteins bcl‐2 and p53. We first immunolabelled live cells for surface proteins following cell fixation, permeabilization and immunolabelling for intracellular proteins. Isotype control samples were similarly prepared for calculating the ratio of surface to intracellular pools, being labelled for only surface or intracellular proteins with an accompanying isotype‐matched control antibody. For identification of successive phases of apoptosis in these experiments, we also used cell light scatter properties and PI labelling. As previously done for live cells, the first phase of apoptosis was gated at the region of neck cells (Fig. 4a) because cell fixation and permeabilization did not markedly change cell morphology. However, there was a difference in identification of the intermediate and terminal phases of cell death. This was based on the presence of two cell subpopulations within the region of apoptotic cells based on light scatter (Fig. 4a), with differing levels of hypodiploid DNA content (Fig. 4d). We have identified the cell subpopulation with higher PI fluorescence as an intermediate phase of apoptosis and the cell subpopulation with lower PI fluorescence as the terminal phase of apoptosis (Fig. 4d).
Figure 4.
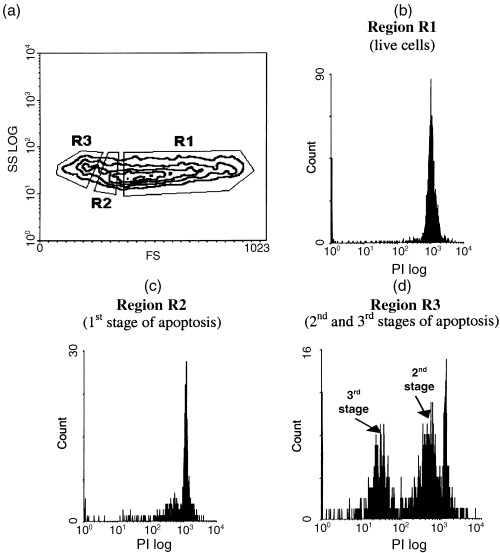
Identification of different stages of apoptosis in EL‐4 cells prepared for measuring intracellular proteins by fixation and permeabilization (see Materials and Methods ) and stained with PI. (a) Regions of live and apoptotic cells selected by their morphological features (forward and side scatter); (b) measurement of PI fluorescence in region R1 demonstrating that the majority of cells within this region are live cells; (c) PI staining cells from region R2 showing that this region contains cells at an early phase of apoptosis (first stage for our model); (d) PI staining showed the presence within region R3 of two hypodiploid peaks with differing intensity of fluorescence, allowing a distinction to be made between this region in the intermediate (second stage) and terminal (third stage) phases of apoptosis.
Using this approach we revealed that in live EL‐4 cells the majority of HSP70 was localized to the cytoplasm (Fig. 5a, stage 0). Triggering of programmed death induced an elevation of both cytoplasmic and surface HSP70 (Fig. 5a, stage 1). Entry of cells into the intermediate phase of apoptosis was accompanied by a sharp increase in surface HSP70 expression and a marginal decrease in intracellular levels (Fig. 5a, stage 2). This stage of programmed death was characterized by the maximum observed level of total HSP70 expression. At the terminal stage of EL‐4 cell apoptosis, a significant drop in total HSP70 cell content was observed; the major part of this decrease was due to exhaustion of intracellular HSP70 (Fig. 5a, stage 3). These results showed that the surface HSP70 as a function of total cell concentration of this protein increased during development of EL‐4 cell apoptosis (Fig. 5b). This in turn indicated that programmed cell death was accompanied by an elevation of HSP70 translocation on the plasma membrane.
Figure 5.
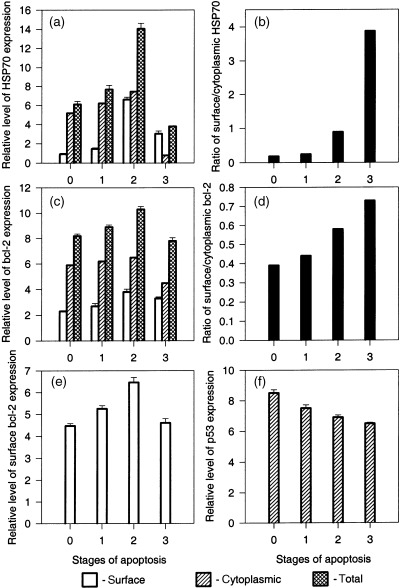
Flow cytometric analysis of surface and intracellular expression of HSP70 (a) , bcl‐2 (c, e) and p53 (f) for successive stages of EL‐4 cell apoptosis. (b) Variation of the ration surface/cytoplasmic HSP70 during the process of EL‐4 cell apoptosis. (d) Variation of the ratio surface/cytoplasmic bcl‐2 during the process of EL‐4 cell apoptosis. For (a), (c) , (e) and (f), results are presented as the mean of fluorescence intensity ± SEM. Data are representative of three comparable experiments.
Measurement of p53 expression in EL‐4 cells showed that intracellular p53 concentration slowly decreased during the progress of apoptosis (Fig. 5f). This might be explained by degradation of this protein as it has a short half‐life (Monney et al. 1998). As was expected, this protein was strictly intracellular and was not observed on the cell surface. In contrast to p53, cytometric analysis of bcl‐2 expression in our model showed localization of this intracellular protein, not only in the cytoplasm, but also on the surface of EL‐4 cells. Patterns of surface and intracellular bcl‐2 expression during apoptosis were similar to that for HSP70; some increase in bcl‐2 expression was observed at the earliest phase of apoptosis. The size of surface and cytoplasmic bcl‐2 pools were maximal at the second phase of apoptosis. Transition to the terminal phase of programmed death was accompanied by a significant decrease of both the intracellular and surface pools of bcl‐2 (Fig. 5c). Overall, however, progress of apoptosis was accompanied by an elevation in the ratio of surface to intracellular bcl‐2 (Fig. 5d). Unusual surface localization of bcl‐2 in our model was confirmed in experiments with immunophenotyping live EL‐4 cells (Fig. 5e). These experiments, in addition to those described above, demonstrated synchronization of surface bcl‐2 and HSP70 variations during EL‐4 cell apoptosis (3, 5). Additional experiments with normal mouse thymocytes and mouse cytotoxic lymphocyte line CTLL‐2 for surface bcl‐2 immunophenotyping did not show localization of this protein on the cell surface (data not shown). These data provide evidence that development of EL‐4 cell apoptosis is accompanied by increased translocation on the cell surface of both protective HSPs and anti‐apoptotic bcl‐2.
Extracellular pool of HSP70 in EL‐4 cell culture in vitro
The above results indicate significant elevation of the relative portion of surface HSP70 in the total pool of this protein during EL‐4 cell apoptosis (Fig. 5b), implying increased translocation of HSP70 to the cell surface during this process. At the same time, total HSP70 cell content is decreased at the terminal stage of programmed EL‐4 cell death, as compared with the middle phase of apoptosis (Fig. 5a). It is unlikely that a decrease in the total HSP70 pool is connected with proteolysis of these proteins. It was previously shown that in lymphoid cells the level of intracellular HSP70 after stress remained elevated for several days longer than the typical duration of apoptosis (Guzhova et al. 1997). Taking into consideration our data showing that increasing translocation of HSP70 to the cell surface is concomitant with the decrease of HSP70 cell content, we proposed that surface HSP70 could be shed from the cell into the extracellular space This in turn would cause a loss of this protein at the terminal phase of apoptosis. To examine this proposition we tested the presence of HSP70 in conditioned serum‐free supernatant of EL‐4 cell culture.
Immunoblotting of EL‐4 tissue culture supernatants showed the presence of HSP70 in the conditioned culture medium (Fig. 6a). The nature of this extracellular fraction of HSP70 was not connected with the protein released from necrotic cells because, as was expected, the control supernatant obtained from EL‐4 cells destroyed by freezing/thawing contained different protein fractions (Fig. 6d) as compared with the samples of medium from conventional conditions, which contained only one major and a few minor bands (Fig. 6a). The supernatant obtained after permeabilization of plasma membrane of EL‐4 cells by detergent also differed from the normal samples by the presence of a large spectrum of protein fractions (data not shown). The comparison of samples of supernatant obtained after incubation of adherent (live cells; Fig. 6c) and non‐adherent (apoptotic cells; Fig. 6b) EL‐4 cell fractions showed that the latter had higher levels of soluble HSP70. These results confirmed our proposition that the decrease in HSP70 cell content during the terminal stage of EL‐4 cell apoptosis could be connected with shedding of surface HSP70 into the tissue culture milieu.
Figure 6.

Immunoblotting of conditioned supernatants obtained from conventional in vitro culture of (a) viable EL‐4 cells, (b) an apoptotic (non‐adherent) subpopulation of the EL‐4 cell culture, (c) a subpopulation of live (adherent) EL‐4 cells, and (d) EL‐4 cells destroyed by freezing/thawing. An electophoretogram and a blot developed using anti‐HSP70 antibodies are presented for each sample.
Discussion
The phenomenon of translocation of cytoplasmic HSPs onto the cell surface, previously described for normal (Erkeller‐Yeksel et al. 1992; Ishiyama et al. 1996), virus‐infected (Di Cesare et al. 1992), transformed (Ferrarini et al. 1992; Altmeyer et al. 1996; Multhoff & Hightower 1996; Multhoff et al. 1997; Rogias et al. 1998; Sapozhnikov et al. 1999; Hantschel et al. 2000) and apoptotic (Possia et al. 1996) cells, has until now had no convincing explanation. To study this phenomenon we used an in vitro model of spontaneous apoptosis of mouse lymphoma EL‐4 characterized by constitutive expression of a spectrum of surface HSPs (Sapozhnikov et al. 1999). In our previous work we used flow cytometry to demonstrate variation in the level of surface HSPs during EL‐4 cell apoptosis. In the present work we analysed the relationship between HSP70 expression and some key events for programmed cell death, namely transmembrane potential of mitochondria (ΔΨm), caspase‐3 activity, intracellular ROS concentration, bcl‐2 and p53 expression.
The approach based on multiparametric cytometry used in our experiments allowed us to select three successive stages of EL‐4 cell apoptosis and simultaneously detect HSP and other protein expression (both intracellular and cell surface), caspase‐3 activation, mitochondrial transmembrane potential and ROS activity. We found transmembrane mitochondrial potential hyperpolarization at the earliest stage of apoptosis detected in our model, followed by mitochondrial depolarization at the subsequent stage of EL‐4 cell death (Fig. 3b). Similar elevation of ΔΨm during apoptosis has been previously demonstrated in another model of T‐lymphocyte apoptosis (Banki et al. 1999). Our data demonstrate that the mitochondrial membrane hyperpolarization is concomitant with the earliest morphological change in dying cells and with the beginning of increased surface HSP70 expression (Fig. 3a), caspase‐3 activity (Fig. 3c) and intracellular ROS concentration (Fig. 3d). It is important to note that maximum HSP70 expression was displayed after mitochondria depolarization (Fig. 3a, stage 2 and Fig. 5a, stage 2), i.e. at an irreversible phase of apoptosis. It has been previously demonstrated that increased expression of HSP70 protects cells from apoptosis. However, in our cell death model elevation of HSP70 expression after the ‘point of decision to die’ is not likely to be connected with the anti‐apoptotic functions of this protein, but might instead reflect a participation of HSP70 in the execution of the programme of apoptosis at its effector and terminal stages, as we have previously proposed (Sapozhnikov et al. 1999). In favour of this proposition are our data showing an association between HSP70 expression and activity of caspase‐3 (Fig. 3a and c), a late‐stage caspase triggering the process of proteolysis in dying cells. This correlation suggests that HSP70 may participate in the process of degradation and utilization of intracellular proteins during apoptosis. Moreover, it may be speculated that the chaperonin functions of HSP70 are directed toward protection of effector proteases executing the programme of cell death. Such a contribution of HSP70 in realizing the programme of apoptosis can explain the synchronization of this protein expression with concentration of ROS (Fig. 3a and d) damaging intracellular structures including caspases (Hampton & Orrenius 1997). This synchronization may also be due to the ability of ROS to induce HSPs expression (Gorman et al. 1999). In addition, ROS can influence bcl‐2 expression (Katon et al. 1999) and this may be reflected in the similar kinetics of bcl‐2 and ROS in our model (2, 4). It is well known that bcl‐2 possesses antioxidant properties and is able to protect cell membranes from the action of ROS (Hockenbery et al. 1993). The latter may be a cause of the bcl‐2 localization on the plasma membrane being registered in our model.
Our data also showed that EL‐4 cell apoptosis is accompanied by an increase in the relative level of cytoplasmic HSP70 translocation on the cell surface (Fig. 5a and b). These results support our supposition that elevated surface HSP expression during apoptosis is due to the ability of these proteins to stabilize plasma membranes (Török et al. 1997) and is directed to prevent dying cell rupture during this stage of cytoskeleton degradation. HSP70 may be translated onto the cell surface concomitantly with cytoskeleton degradation as a temporary ‘reinforcing’ element to protect the cell membrane before it is ‘sewed up’ by transglutaminase (Knight et al. 1991).
Unexpected results were obtained in our experiments with bcl‐2 expression in EL‐4 cells. In these experiments we used the same protocol of cell staining as for HSP70, which allowed us to distinguish surface and cytoplasmic fractions of the proteins (see Materials and methods). It was revealed that bcl‐2 was present on the surface of EL‐4 cells (Fig. 5c). Additional experiments with direct immunophenotyping for surface bcl‐2 using live EL‐4 cells confirmed the unusual surface localization of this protein in our model (Fig. 5e). To our knowledge these data are the first direct evidence for cell surface bcl‐2 expression, although some authors have demonstrated that the cell plasma membrane fraction can contain this protein (Chen‐Levy et al. 1989; Bruce‐Keller et al. 1998). The phenomenon of bcl‐2 translocation onto the cell surface has to be further studied and confirmed, but we can speculate that it might be connected with the ability of bcl‐2 to prevent ROS‐induced injury of cell membranes (Hockenbery et al. 1993) and/or to form ion‐channels in the membranes (Schendel et al. 1997). In fact, our results showed an association between the intracellular concentration of ROS and the level of surface bcl‐2 during the progress of EL‐4 cell apoptosis (3, 5). At the same time, significant expression of surface bcl‐2 was observed, not only on apoptotic, but also on live cells (Fig. 5e), i.e. in conditions without developing oxidative stress. This may be connected with both high background levels of ROS in EL‐4 cells that induce elevated levels of bcl‐2 production and translocation of this protein to the plasma membrane, and a compensative reaction of in vitro cultivated EL‐4 cells to an imbalance of ionic homeostasis resulting in the formation of additional ion channels in plasma membranes.
Altogether, these data show a strong association between surface HSP70 and bcl‐2 expression and localization, alterations in mitochondrial membrane potential, caspase‐3 and ROS activity during EL‐4 cell apoptosis. The close correlation between these activities and the onset of apoptosis suggests a role for HSP70 in counteracting both the destruction of dying cell membranes and the degradation of cytosolic cytoskeletal elements and proteins. This coordination may be directed both toward protection of the process of cell membrane reorganization during apoptosis and toward prevention of the release of intracellular contents into the intercellular space.
The kinetics of HSP70 expression during EL‐4 cell apoptosis showed decreases of both surface and intracellular pools of this protein at the terminal phase of cell death (Fig. 5a). Taking into account that HSPs are generally long‐lived proteins, we proposed that this decrease is due to shedding of surface HSP70 into the intercellular space. Indeed, our data have demonstrated an increase in the proportion of surface HSP70 relative to the total pool of this protein during apoptosis (Fig. 5b), that in turn indicated an increase in the relative level of cytoplasmic HSP70 translocation to plasma membrane during apoptosis. The simultaneous increase in translocation and reduction of surface HSP70 level at the terminal stage of apoptosis may therefore be connected with shedding of surface HSP70. Immunoblotting, carried out to examine this proposal, showed HSP70 in the supernatant of both viable and apoptotic EL‐4 cell cultures (Fig. 6a). The fraction of this protein was higher in the samples obtained from unattached (apoptotic) EL‐4 cells (Fig. 6b) Analysis of samples from freeze/thawed EL‐4 cells showed intracellular contents characterized by a much wider spectrum of proteins (Fig. 6d) by comparison with normal supernatant (Fig. 6a) and lower levels of HSP70 relative to other proteins, suggesting that necrotic death was not responsible for apoptosis‐associated shedding. These results indicate active HSP70 shedding or exocytosis in EL‐4 cell culture. It could be speculated that the extracellular pool of these protective proteins is adsorbed by live cells and protected these cells from apoptosis. Such an effect of exogenous HSP70 has been described in the literature (Guzhova et al. 1998). According to this hypothesis, HSP70 production by dying cells is directed to protection of the still‐viable component of the cell population against apoptosis. This hypothesis is consistent with the concept of social control of cell survival (Raff 1992), as it is possible that autocrine factors protecting cell populations from apoptosis in normal conditions are supplemented in stressed conditions with dying‐cell production of extracellular protective HSPs. However, this hypothesis remains to be definitively tested.
Acknowledgements
This work was supported by Grant 00‐04‐48898 from the Russian Foundation for Basic Research.
References
- Altmeyer A, Maki RG, Feldweg AM, Heike M, Protopopov VP, Masur SK, Srivastava PK (1996) Tumor‐specific cell surface expression of the KDEL containing, endoplasmic reticular heat shock protein gp96. Int. J. Cancer 69, 340. [DOI] [PubMed] [Google Scholar]
- Asea A, Kraeft SK, Kurt‐Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK (2000) HSP70 stimulates cytokine production through a CD14‐dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6, 435. [DOI] [PubMed] [Google Scholar]
- Banki K, Hutter E, Gonchoroff NJ, Perl A (1999) Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J. Immunol. 162, 1466. [PMC free article] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Suto R., Anderson KM, Srivastava PK (2000) Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF‐κB pathway. Int. Immunol. 12, 1539. [DOI] [PubMed] [Google Scholar]
- Bradford M (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248. [DOI] [PubMed] [Google Scholar]
- Bruce‐Keller AJ, Begley JG, Fu W, Butterfield DA, Bredesen DE, Hutchins JB, Hensley K, Mattson MP (1998) Bcl‐2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid beta‐peptide. J. Neurochem. 70, 31. [DOI] [PubMed] [Google Scholar]
- Chen‐Levy Z, Nourse J, Cleary ML (1989) The bcl‐2 candidate proto‐oncogene product is a 24‐kilodalton integral‐membrane protein highly expressed in limphoid cell lines and lymphomas carrying the t(14; 18) translocation. Mol. Cell. Biol. 9, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig EA, Weissman JS, Horwich AL (1994) Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell 78, 365. [DOI] [PubMed] [Google Scholar]
- Di Cesare S, Poccia F, Mastino A, Colizzi V (1992) Surface expressed heat‐shock proteins by stressed or human immunodeficiency virus (HIV)‐infected lymphoid cells represent the target for antibody‐dependent cellular cytotoxity. Immunology 76, 341. [PMC free article] [PubMed] [Google Scholar]
- Erkeller‐Yeksel FM, Isenberg DA, Dhillon VB, Latchman DS, Lydyard PM (1992) Surface expression of heat shock protein 90 by blood mononuclear cells from patients with systemic lupus erythematosus. J. Autoimmun. 5, 803. [DOI] [PubMed] [Google Scholar]
- Ferrarini M, Heltai S, Zocchi MR, Rugarli C (1992) Unusual expression and localization of heat‐shock proteins in human tumor cells. Int. J. Cancer 51, 613. [DOI] [PubMed] [Google Scholar]
- Gorman AM, Heavey B, Creagh E, Cotter TG, Samali A (1999) Antioxidant‐mediated inhibition of the heat shock response leads to apoptosis. FEBS Lett. 445, 98. [DOI] [PubMed] [Google Scholar]
- Guzhova IV, Arnholdt ACV, Darieva ZA, Kinev AV, Lasunskaia EB, Nilsson K, Bozhkov VM, Voronin AP, Margulis BA (1998) Effects of exogeneous stress protein 70 on the functional properties of human promonocytes through binding to cell surface and internalization. Cell Stress Chap. 3, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzhova IV, Darieva ZA, Rocha Melo A, Margulis BA (1997) Major stress protein 70 kDa and subunits of NF‐κB regulatory complex are associated in human T‐lymphoma cells. Cell Stress Chap. 2, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton MB, Orrenius S (1997) Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett. 414, 552. [DOI] [PubMed] [Google Scholar]
- Hantschel M, Pfister K, Jordan A, Scholz R., Andreesen R., Schmitz G, Schmetzer H, Hiddenman W, Multhoff G (2000) Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chap. 5, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU (1996) Molecular chaperones in cellular protein folding. Nature 381, 571. [DOI] [PubMed] [Google Scholar]
- Hightower LE, Guidon PT (1989) Selective release from cultured mammalian cells of heat‐shock (stress) proteins that resemble glia‐axon transfer proteins. J. Cell. Physiol. 138, 257. [DOI] [PubMed] [Google Scholar]
- Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okasaki T, Yamamoto K, Sasada M (1998) Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas‐induced apoptosis. J. Exp. Med. 187, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmayer SJ (1993) Bcl‐2 functions in an antioxidant pathway to prevent apoptosis. Cell 75, 241. [DOI] [PubMed] [Google Scholar]
- Ishiyama T, Koike M, Akimoto Y, Fukuchi K, Watanabe K, Yoshida M, Wakabayashi Y, Tsuruoka N (1996) Heat shock‐enhanced T cell apoptosis with heat shock protein 70 on T cell surface in multicentric Castleman's disease. Clin. Exp. Immunol. 106, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katon S, Mitsue Y, Kitani K, Suzuki T (1999) Hyperoxia induces the neuronal differentiated phenotype of PC12 cells via a sustained activity of mitigen‐activated protein kinase induced by Bcl‐2. Biochem. J. 338, 465. [PMC free article] [PubMed] [Google Scholar]
- Knight CR, Rees RC, Griffin M (1991) Apoptosis: a potential role for cytosolic transglutaminase and its importance in tumour progression. Biochim. Biophys. Acta 1096, 312. [DOI] [PubMed] [Google Scholar]
- Monney L, Otter I, Oliviert R., Ozer HL, Haas AL, Omura A, Borner C (1998) Defects in the ubiquitin pathway induce caspase‐independent apoptosis blocked by Bcl‐2. J. Biol. Chem. 273, 6121. [DOI] [PubMed] [Google Scholar]
- Morimoto RI, Tissiers A, Georgopoulos C, eds. (1994) The biology of heat shock response and molecular chaperones. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Multhoff G, Botzler C, Jennen L, Schmidt J, Ellwart J, Issels R (1997) Heat shock protein 72 on tumor cells: a recognition structure for natural killer cells. J. Immunol. 158, 4341. [PubMed] [Google Scholar]
- Multhoff G, Hightower LE (1996) Cell surface expression of heat shock proteins and the immune response. Cell Stress Chap. 1, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Lindquist S (1993) The function of heat‐shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu. Rev. Genet. 27, 437. [DOI] [PubMed] [Google Scholar]
- Poccia F, Piselli P, Vendetti S, Bach S, Amendola A, Placido R., Colizzi V (1996) Heat‐shock protein expression on the membrane of T cells undergoing apoptosis. Immunology 88, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB, Toft DO (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18, 306. [DOI] [PubMed] [Google Scholar]
- Raff MC (1992) Social controls on cell survival and cell death. Nature 356, 397. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola‐Hartshorn C, Chen A, Lin M, Chen LB (1995) Mitochondrial membrane potential monitored by JC‐1 dye. Meth Enzymol. 260, 406. [DOI] [PubMed] [Google Scholar]
- Rogias J, Wallen ES, Loening SA, Moseley PL (1998) Heat shock proteins (HSP72) surface expression enhances the lysis of a human renal cell carcinoma by IL‐2 stimulated NK cells. Adv. Exp. Med. Biol. 451, 225. [DOI] [PubMed] [Google Scholar]
- Rothe G, Valet G (1990) Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2′,7′‐dichlorofluorescin. J. Leukoc. Biol. 47, 440. [PubMed] [Google Scholar]
- Sapozhnikov AM, Ponomarev ED, Tarasenko TN, Telford GW (1999) Spontaneous apoptosis and expression of cell surface heat shock proteins in cultured EL‐4 lymphoma cells. Cell Prolif. 32, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC (1997) Channel formation by anti‐apoptotic protein Bcl‐2. Proc. Natl Acad. Sci. USA 94, 5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger MJ (1990) Heat shock proteins. J. Biol. Chem. 265, 12111. [PubMed] [Google Scholar]
- Tõrõk Z, Horváth I, Goloubinoff P, Kovács E, Glatz A, Balogh G, Vígh L (1997) Evidence for a lipochaperonin: association of active protein‐folding GroESL oligomers with lipids can stabilize membranes under heat shock conditions. Proc. Natl Acad. Sci. USA 94, 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]