

Abstract
Objectives
The aim of this study was to investigate whether in vitro stimulation of dental pulp stem cells (DPSCs) by tumour necrosis factor alpha (TNF‐α) would induce secretion of EphB2/ephrin‐B1 signalling.
Materials and methods
Dental pulp stem cells isolated from human dental pulp were treated with TNF‐α (5–100 ng/ml) over 2–48 h. EphB2/ephrin‐B1 mRNA and protein levels were measured by real‐time polymerase chain reaction (RT‐PCR) and western blot analysis respectively. Additionally, DPSCs were pre‐incubated with TNF‐α receptor neutralizing antibodies or infected with nuclear factor‐kappa B (NF‐ĸB) inhibitor, p38 MAPK inhibitor, Jun N‐terminal kinase (JNK) inhibitor and MEK inhibitor before TNF‐α treatment. Results were analysed by one‐way ANOVA.
Results
Tumour necrosis factor alpha increased EphB2 mRNA expression in DPSCs at concentrations up to 20 ng/ml and ephrin‐B1 at concentrations up to 40 ng/ml (P < 0.05). Its mRNA expression reached maximum at 24 h when treated with TNF‐α at 20 ng/ml (P < 0.05). EphB2/ephrin‐B1 protein expression levels were high at 16 and 24 h as shown by western blotting. Neutralizing antibodies for TNFR1/2 receptors down‐regulated EphB2/ephrin‐B1 mRNA expression (P < 0.05) and ephrin‐B1 protein expression, but not EphB2 protein expression. JNK‐inhibitor inhibited EphB2 mRNA expression only (P < 0.05).
Conclusions
EphB2/ephrin‐B1 were invoked in DPSCs with TNF‐α treatment via the JNK‐dependent pathway, but not NF‐ĸB, p38 MAPK or MEK signalling.
Introduction
Dental pulp is a loose connective tissue rich in blood vessels, nerve fibres, undifferentiated mesenchymal cells and fibroblasts, organized within an intricate extracellular matrix framework 1. When teeth suffer traumatic injury or undergo bacterial infiltration along dentinal tubules, enclosed pulp can become perpetually inflamed and infected with pathogens. Subsequent immunity reactions emit strong signals for expression of pro‐inflammatory cytokines such as tumour necrosis factor alpha (TNF‐α) 2, which promotes secretion of various essential adhesion molecules for leukocyte adhesion and diapedesis 3. During pathological events of pulpitis, there are gradients of TNF‐α concentration in dental pulp tissue. Lower concentration of TNF‐α in the inner pulp may promote migration of mesenchymal stem cells (MSCs) into the affected odontoblast site 4. TNF‐α, in a relatively higher concentration, facilitates homing of migrating stem cells and promotes local odontogenesis 5. However, under steady‐state conditions, dental pulp stem cells (DPSCs) are retained within their perivascular niche by bi‐directional signalling of EphB/ephrin‐B molecules that restrict their attachment and migration. Following caries infection or dentin injury, EphB/ephrin‐B signalling is activated, which drives DPSCs in pulp tissue to move from the perivascular region to site of injury and deeper into it 6. Other studies also have suggested that forward EphB2 signalling enhances stem cell migration, and reverses ephrin‐B signalling suppressed stem cell attachment, and spreading 7, 8, 9.
Effects of TNF‐α are mediated by activation of caspases, nuclear factor‐kappa B (NF‐ĸB) pathway and mitogen‐activated protein kinase (MAPK). Binding TNF‐α to TNF receptor‐1 (TNFR1) and/or ‐2 (TNFR2) initiates apoptosis and anti‐inflammatory responses through caspase activation, while anti‐apoptosis and inflammatory responses occur via activation of MAPK and NF‐κB pathways 10. In endothelial cells, it has been shown that ephrin‐A1 expression is up‐regulated by TNF‐α stimulation 11, thus inducing angiogenesis 12. Neutralizing antibodies against ephrin‐A1 suppress TNF‐α‐induced angiogenesis, indicating the role of ephrin‐A1 in TNF‐α‐induced angiogenesis 11. Further study confirmed that ephrinA1 up‐regulation by TNF‐α in endothelial cells was mediated through both p38 MAPK and stress‐activated protein kinases (SAPK)/c‐Jun N‐terminal kinases (JNK), but not p42/44 MAPK or NF‐κB pathways 12. In the light of this information, we speculated that inflammation within the tooth, caused by dental caries or traumatic injury, could invoke secretion of TNF‐α from inflammatory cells and, in turn, promote genesis of EphB2/ephrin‐B1, which eventually awakens endogenous pulp regeneration. However, interplay of TNF‐α and EphB/ephrin‐B signalling in DPSCs in dentin bio‐mineralization and repair had not up to now been investigated. Thus, the aim of the present study was to investigate whether TNFα regulates EphB2/ephrin‐B1 signalling in DPSCs, and how this process operates at a molecular level.
Materials and methods
Isolation of DPSCs
Extracted third molars were collected from 10 healthy donors (18–40 years of age) after obtaining their informed consent (IRB UW09‐340). Tooth surfaces were cleaned and cracked open, using a vice, to reveal the pulp chamber. Pulp tissue was gently separated from crowns and roots, then digested in a solution of 3 mg/ml collagenase type I (GIBCO‐Invitrogen, Carlsbad, CA, USA) and 4 mg/ml dispase (GIBCO‐Invitrogen), for 1 h at 37 °C, to generate a pulp cell mass, free of extracellular matrix. Single‐cell suspensions were obtained by passing the cell mass through a 70 μm strainer (BD Biosciences, Franklin Lakes, NJ, USA) to yield a homogenous population of individual cells. Cultures were established by seeding single‐cell suspensions (102–105) into T‐25 flasks in growth medium, α‐modification of Eagle's medium supplemented with 20% foetal calf serum (SAFC), 100 μm L‐ascorbic acid 2‐phosphate, 2 mm L‐glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (SAFC), then incubated at 37 °C in 5% CO2. Before using DPSCs for experiments, the freshly isolated cells were assessed for their stem cell properties, by using flow cytometry, to show expression of CD73, CD90, CD105, STRO‐1 and CD45. Cells between passages 3–6 were used in all experiments as, at higher passages, extracted stem cells are highly likely to differentiate and lose their stem cell phenotype.
Reverse‐transcription real‐time PCR
Dental pulp stem cells were seeded into six‐well culture plates at 1 × 105 cells/cm2. Cells were cultured to 80% confluence, starved for 16 h in medium deficient of growth factors, pre‐incubated with cycloheximide (CHX) (Sigma, St. Louis, Missouri, USA) at 10 μg/ml for 30 min, then stimulated with recombinant human TNF‐α (R&D Systems, Minneapolis, Minnesota, USA) ranging from 5 to 100 ng/ml, for 2 h or without TNF‐α, or stimulated with TNF‐α at 20 ng/ml over periods ranging from 15 min to 48 h.
Total RNA was extracted from DPSCs using RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and mRNA was converted to complementary DNA (cDNA) by SuperScript®VILO™Mastermix (Invitrogen, Carlsbad, CA, USA). RT‐PCR reactions were performed with SyberGreen (Applied Bio‐Systems, Carlsbad, CA, USA) and ABI Prism 7000 Sequence Detection System (Applied Bio‐Systems). Primers used were as follows: human EphB2 (Forward 5′–3′ATGAACACGATCCGCACGTA; Reverse 3′–5′ TTGGTCCGTAGCCAGTTGTTCT; AF_025304), human ephrin‐B1 (Forward 5′–3′AGCTCCCTCAACCCCAAGTT; Reverse 3′–5′GGCAGATGATGTCCAGCTTGT; NM_004429). All samples were run in triplicate in 96‐well plates, with each well containing 0.8 μl cDNA for a total reaction volume of 20 μl. RT‐PCR was performed as follows: initial denaturation 95 °C for 10 min, cycles with denaturation at 95 °C for 15 s and annealing at 64 °C for 1 min. Amplifications were checked by melting point analysis with subsequent heating at 95 °C for 15 s, then 60 °C for 1 min, and extension period from 95 °C to 60 °C for 1 min. Reactions for each sample were performed in triplicate. All samples were normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) “housekeeping” gene.
Western blot analysis
Dental pulp stem cells were cultured to 80% confluence, starved for 16 h in medium deficient in growth factors, then 20 ng/ml TNF‐α was added to each well. Cells were collected using MPER Reagent (Thermo Scientific, Rockford, IL, USA) after 2, 4, 8, 16, 24 and 48 h. Total protein content was measured using a Pierce® BCA protein assay kit (Thermo Scientific). Further, equal amounts of protein were fractionated by SDS‐polyacrylamide gel electrophoresis (EphB2: 10 μg protein and 7.5% gel; ephrin‐B1: 20 μg protein and 12% gel) and transferred to nitrocellulose membranes, which were blocked for 1 h at room temperature with Tris‐buffered saline containing 0.1% Tween and 5% powdered non‐fat milk. Then they were incubated with anti‐EphB2 polyclonal antibody (1:200; Cell Signaling Technology, Danvers, MA, USA), anti‐ephrin‐B1 polyclonal antibodies (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and β‐actin polyclonal antibodies (1:200; Santa Cruz Biotechnology). Secondary antibodies were anti‐rabbit IgG and anti‐mouse IgG (1:3000; Cell Signaling Technology).
RT‐PCR for TNF‐α receptor, NF‐ĸB, JNK, p38 MAP and MEK activation
Dental pulp stem cells were pre‐incubated with 5 μg/ml neutralizing antibodies (R&D Systems) to human TNFR1 or TNFR2 for 1 h, then stimulated with 20 ng/ml TNF‐α for 48 h. For chemical inhibitors prior to CHX and TNF‐α stimulation, DPSCs were treated with NF‐ĸB inhibitor (oxaprozin; Abcam Ltd., Hong Kong, China) at 30 μm, JNK inhibitors (SP600125; Abcam Ltd., Hong Kong, China) at 30 μm, p38 MAPK inhibitor (SB 203580; Abcam Ltd., Hong Kong, China) at 2.5 μm and mitogen‐activated protein kinase kinase (MEK) inhibitor (PD98059; Cell Signaling Technology) at 50 μm for 1 h. Total RNA was used for RT‐PCR. Reactions for each sample were performed in triplicate. GAPDH was used as a loading control.
Western blotting for TNF‐α receptors and JNK inhibition
Dental pulp stem cells were cultured to 80% confluence and starved for 16 h in medium deficient of growth factors. One hour prior to TNF‐α stimulation, human TNFR1 or TNFR2 antibody (R&D Systems) and JNK inhibitors (SP600125; Abcam Ltd.) were added respectively. Total proteins were extracted from cell cultures using MPER reagent. Western blot analysis was carried out as described above.
Statistical analysis
Data expressed in bar graphs are mean ± SD of triplicate experiments. Statistical significance was assessed using one‐way ANOVA. P < 0.05 was considered to be statistically significant.
Results
Human DPSCs expressed mesenchymal stem cell markers
The freshly isolated DPSCs, as analysed by flow cytometry, expressed mesenchymal stem cell markers CD90 (99.91%), CD73 (99.80%), CD105 (99.87%) and STRO‐1 (21.31%). They were negative for haematopoietic marker, CD45 (0.97%). Multilineage differentiation capacity of the cells was confirmed by using osteo/odontogenic, adipogenic and neurogenic induction media (results not shown).
EphB2/ephrin‐B1 expression stimulated by TNF‐α
Dental pulp stem cells were cultured in the presence of five concentrations of TNF‐α (0, 5, 10, 20, 40 or 100 ng/ml) and EphB2/ephrin‐B1 induction was assessed using RT‐PCR. We found that 5, 10 and 20 ng/ml TNF‐α induced EphB2 mRNA expression, whereas 40 and 100 ng/ml TNF‐α did not affect its levels (Fig. 1a,b). However, all TNF‐α concentrations, except 100 ng/ml, affected ephrin‐B1 mRNA expression.
Figure 1.
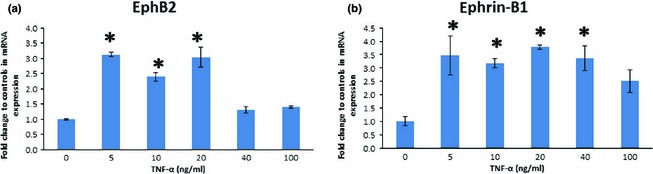
Dose‐dependent tumour necrosis factor alpha (TNF‐α)‐induced expression of EphB2/ephrin‐B1 mRNA in dental pulp stem cells. TNF‐α increased EphB2 mRNA expression at concentrations of 5, 10 and 20 ng/ml, and ephrin‐B1 at 5, 10, 20 and 40 ng/ml. (a) Relative expression of EphB2. (b) Relative expression of ephrin‐B1 (*P ˂ 0.05 versus control).
Cumulative effects of EphB2/ephrin‐B1 secretion over time, following treatment with TNF‐α (20 ng/ml), caused significant induction, seen as early as 4 h (EphB2) and 16 h (ephrin‐B1) after treatment (Fig. 2a,b). Maximum levels of EphB2 and ephrin‐B1 were observed at 24 h, followed with significant reduction by 48 h.
Figure 2.
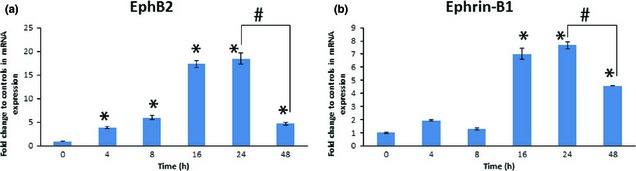
Time course of tumour necrosis factor alpha (TNF‐α)‐induced expression of EphB2/ephrin‐B1 mRNA in dental pulp stem cells. mRNA expression of EphB2/ephrin‐B1 reached its maximum at 24 h when treated with TNF‐α at 20 ng/ml. (a) Relative expression of EphB2. (b) Relative expression of ephrin‐B1 (*P ˂ 0.05 versus control; #P ˂ 0.05, as indicated bracketed).
In the western blot analyses, the EphB2 band was detected at 105 kDa (Fig. 3), and ephrin‐B1 at 45 kDa (Fig. 4). Following a time‐course experiment, comparable levels of EphB2 protein expression were observed until 24 h, which then decreased by 48 h (Fig. 3). In contrast, ephrin‐B1 protein levels were expressed increasing for 24 h, compared to protein level at 0 h (Fig. 4).
Figure 3.

Time course of tumour necrosis factor alpha (TNF‐α)‐induced expression of EphB2 protein in dental pulp stem cells. Western blot analysis of EphB2 protein expression in dental pulp stem cells after treating with 20 ng/ml of TNF‐α for 4, 8, 16, 24 and 48 h.
Figure 4.

Time course of tumour necrosis factor alpha (TNF‐α)‐induced expression of ephrin‐B1 protein in dental pulp stem cells. Western blot analysis of ephrin‐B1 protein expression in dental pulp stem cells after treating with 20 ng/ml of TNF‐α for 4, 8, 16, 24 and 48 h.
EphB2/ephrin‐B1 mRNA expression blocked by TNF‐α receptors and JNK inhibitor, but not NF‐ĸB, p38 MAPK and MEK inhibitors
To determine which receptor was involved in regulation of EphB2/ephrin‐B1 expression, DPSCs were treated with neutralizing monoclonal antibodies specifically against either TNFR1 or TNFR2; as shown in Fig. 5, these inhibited EphB2/ephrin‐B1 mRNA expression. These results indicate that both TNF‐α receptors are involved in regulating EphB2/ephrin‐B1 mRNA expression.
Figure 5.
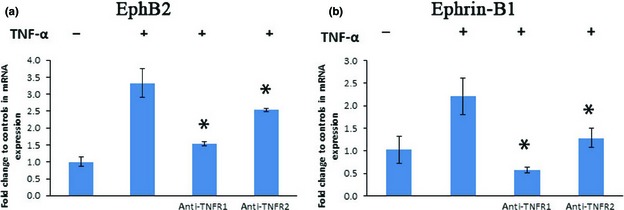
Induction of EphB2/ephrin‐B1 mRNA expression by tumour necrosis factor alpha (TNF‐α) was mediated through both TNFR1 and TNFR2. Neutralizing antibodies for TNFR1 or TNFR2 receptors down‐regulated EphB2/ephrin‐B1 mRNA expression (*P ˂ 0.05, compared to TNF‐α‐positive control group).
To investigate whether MAP kinases mediated TNF‐α‐induced EphB2/ephrin‐B1 expression, a chemical inhibitor (SP600125) was used to inhibit the JNK pathway. As shown in Fig. 6, EphB2 mRNA expression was inhibited, but not ephrin‐B1 mRNA expression. Furthermore, NF‐ĸB inhibitor (oxaprozin) had no impact on secretion of TNF‐α‐induced EphB2/ephrin‐B1. Also, p38 MAP inhibitor (SB 203580) and MEK inhibitor (PD98059) did not down‐regulate EphB2/ephrin‐B1 mRNA expression.
Figure 6.
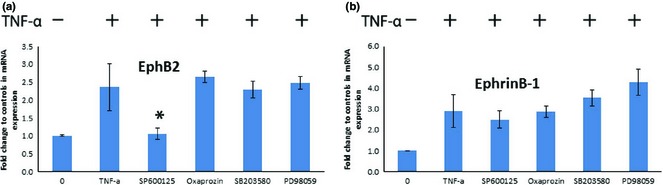
Inhibition of NF‐ĸB, p38 MAP, JNK and MEK activation on EphB2/ephrin‐B1 mRNA expression. JNK inhibitor (SP600125) shown to inhibit EphB2 mRNA expression but not ephrin‐B1 expression (P < 0.05). However, NF‐ĸB inhibitor (oxaprozin), p38 MAPK inhibitor (SB 203580) and MEK inhibitor (PD98059) did not affect EphB2/ephrin‐B1 mRNA expression. (*P ˂ 0.05, compared to TNF‐α positive control group).
After addition of human TNFR1 or TNFR2 antibodies to block the respective receptors, protein levels of ephrin‐B1 decreased accordingly without any significant change in EphB2 protein expression. When JNK inhibitor SP600125 was used, no change in protein expression of either ephrin‐B1 and EphB2 was observed (Figs 7, 8).
Figure 7.
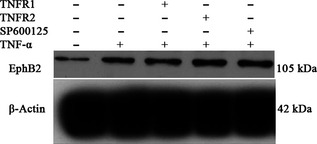
Effects of TNFR1/2 and JNK inhibitors on tumour necrosis factor alpha (TNF‐α)‐mediated EphB2 protein expression in dental pulp stem cells. Western blot analysis of EphB2 protein expression in dental pulp stem cells after stimulation with the human TNF1/2 antibody, JNK inhibitor and TNF‐α.
Figure 8.
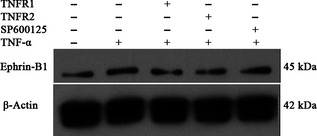
Effects of TNFR1/2 and JNK inhibitors on tumour necrosis factor alpha (TNF‐α)‐mediated ephrin‐B1 protein expression in dental pulp stem cells. Western blot analysis of ephrin‐B1 protein expression in dental pulp stem cells after stimulation with the human TNF1/2 antibody, JNK inhibitor and TNF‐α.
Discussion
Tumour necrosis factor alpha is a multifunctional cytokine that induces a broad spectrum of cellular activities, including mesenchymal stem cell proliferation, survival, apoptosis 13, secretion of other cytokines 14 and angiogenesis 15. In mineralized tissues, TNF‐α signalling protein has been shown to promote bone fracture repair via osteogenic signalling of muscle‐derived stromal cells (MDSCs). TNF‐α was shown first to advance MDSC migration then osteogenic differentiation, at low concentrations. However, excess TNF‐α reduced MDSC migration and pro‐osteogenic effects. Furthermore, administration of high doses of TNF‐α fracture sites accelerated wound healing in vivo 16. Considering the role of TNF‐α on reparative dentin formation, it has been reported that long‐term TNF‐α treatment suppressed expression of osteonectin and bone sialoprotein in dental pulp cells 17, while at early time points, TNF‐α stimulated DPSC over‐expression of odontogenic genes towards odontoblastic differentiation via p38 regulation 18. TNF‐α treatment also activated the NF‐κB pathway, suggesting that DPSCs may be involved in host immune responses when bacterial infection occurs 19. TNF‐α is implicated in directly operating with the Eph/ephrin pathways that signal to cells of calcified tissues 20. However, effects of TNF‐α on EphB2/ephrin‐B1 protein secretion in DPSCs, and underlying mechanism are not known.
In the current study, dose‐dependent and time‐dependent investigations were performed to discover expression patterns of EphB2/ephrin‐B1 after stimulation with TNF‐α. We observed that low concentrations of TNF‐α induced both EphB2 and ephrin‐B1 mRNA expression, whereas 40 and 100 ng/ml TNF‐α did not affect EphB2 levels, and 100 ng/ml did not affect ephrin‐B1 mRNA expression. We further treated DPSCs with TNF‐α (20 ng/ml), and found that significant secretion of EphB2 was observed as early as by 4 h, while ephrin‐B1 expression started at 16 h. Western blot analyses also confirmed concomitant protein expression levels arising from translation of mRNA transcripts. Balance of EphB2 and ephrin‐B1 is one of the critical factors for retention of DPSCs in the perivascular niche 7, 21. Once the balance is disrupted, a EphB2/ephrin‐B1 signalling pathway could be activated. Subsequently, forward EphB2 signalling promoted MSC migration 8, 9, 22. As shown in the present study, TNF‐α treatment broke down the EphB2/ephrin‐B1 balance, with significant enhancement of EphB2 gene and protein expression at earlier time points. It is assumed that over‐expression of forward EphB2 signalling promoted DPSCs migration away from the perivascular niche, although as yet we have no direct evidence of this.
Ephrin‐B1 is strongly expressed by STRO‐1‐positive cells within pulp tissue, while EphB2 is weakly marked but shows a similar pattern in STRO‐1‐positive cells 21. However, in injured pulp tissue, ephrin‐B1 gene expression is suppressed, indicating disruption of EphB/ephrin‐B balance with enhanced DPSC migration to injured sites 23. Calcium hydroxide, as a pulp‐capping agent, promotes dentin bridge formation and stimulates EphB2 gene expression, but inhibits ephrinB1 gene expression in primary pulp cells in proliferation stages 23, 24. It seems that EphB/ephrin‐B controls the balance between active and quiescent stem cells in dental pulp tissue. Tuning EphB/ephrin‐B expression in DPSCs could be a potential way to induce reparative dentin formation.
Inflammatory cytokines released by injured tissue also results in up‐regulation of matrix metalloproteinases (MMPs), enhancing migratory capacity of perivascular cells 25, 26. It has been reported that MMP‐1, ‐2, ‐9 and ‐14 are expressed in healthy dental pulp. MMP‐3 and MMP‐9 mRNA levels were increased in odontoblasts, fibroblasts, inflammatory infiltrate and endothelial cells, in the inflamed pulp 27. Combined with our present results, TNF‐α may stimulate both EphB2/ephrin‐B1 and MMP secretion in DPSCs during the course of inflammation. MMPs may contribute to perivascular cell recruitment at several levels, including extracellular matrix degradation and cleavage of EphB receptors 28. Thus, Eph/ephrin interactions can be abolished by proteolytic cleavage with MMPs 29. However, investigations must still be conducted to determine whether metalloproteinase cleavage of EphB receptors influences signalling events in DPSCs.
Functions of TNF‐α are mediated by two cell surface receptors, TNFR1 or TNFR2. TNF‐α binding to its receptors stimulates receptor aggregation, and succeeds in recruitment of various types of intracellular signal transducer to TNFR complexes. TNF‐α has different affinities to these receptors and has been shown to activate distinct, as well as overlapping, pathways 30. Recent studies have shown that both TNFR1 and TNFR2 are involved in regulating ephrin‐A1 expression in endothelial cells 12. To elucidate the role of TNFR1 or TNFR2 in DPSCs in regulation of EphB/ephrin‐B expression, the cells were treated with neutralizing monoclonal antibodies specifically against either TNFR1 or TNFR2. Neutralizing antibodies against either TNFR1 or TNFR2 inhibited EphB2/ephrin‐B1 mRNA expression.
NF‐κB pathway is one of the main mediators of intracellular functions of TNF‐α in DPSCs, in the host immune response, and up‐regulation of MMP‐1 and MMP‐13 expression 18, 19. As EphB2/ephrinB1 was also up‐regulated by TNF‐α treatment, we interrogated whether the NF‐κB pathway was involved in mediating this cellular function. NF‐κB inhibitor (oxaprozin) was used to block NF‐κB signalling; however, mRNA expression of EphB2/ephrin‐B1 was not affected, indicating that NF‐κB was not the prime mediator in TNF‐α‐induced EphB2/ephrin‐B1 in DPSCs, here.
Tumour necrosis factor alpha is also known to be activated by at least three different subtypes of MAP kinase: p42/p44 MAPK, p38 MAPK and SAPK/JNK in endothelial cells 11. To further study whether MAP kinases mediate TNF‐α function on EphB2/ephrin‐B1 mRNA expression in DPSCs, selective chemical inhibitors, p38 MAPK inhibitor (SB 203580), JNK inhibitor (SP600125) and MEK inhibitor (PD98059) were used to inhibit p38MAPK, JNK and MEK respectively. TNF‐α is a strong and very rapid inducer of EphB2/ephrin‐B1 functional activity in DPSCs. As shown in Fig. 6, JNK inhibitor (SP600125) inhibited EphB2 mRNA expression, but not ephrin‐B1 mRNA expression. In contrast, EphB2/ephrin‐B1 mRNA expression was not affected in the presence of p38 MAPK, MEK kinase and NF‐ĸB inhibitors.
Further, western blot analysis confirmed that blocking TNFR1/2 receptors down‐regulated ephrin‐B1 expression partly in DPSCs, suggesting that the two receptors are involved in TNF‐α stimulation. However, it cannot be completely ruled out of the receptors' role on EphB2 expression, as blocking TNFR1/2 only down‐regulated EphB2 mRNA level but not its protein level. Similarly, SP600125 did not inhibit EphB2/ephrin‐B1 protein expression with stimulation of TNF‐α, despite down‐regulation of EphB2 gene expression.
In conclusion, expression of EphB2/ephrin‐B1 in human DPSCs was up‐regulated by stimulation with TNF‐α, as shown by real‐time PCR and western blot analyses. Both TNFR1 and TNFR2 were involved in regulating cellular mRNA activities, but partly with protein levels. TNF‐α‐induced EphB2/ephrin‐B1 mRNA expression was JNK‐dependent, but was independent of p38 MAPK, MEK and NF‐ĸB signalling.
Conflicts of interest
The authors deny no conflicts of interest that are related to this study.
Acknowledgements
This study was supported by the Seed Funding Programme for Basic Research (Grant number: 201211159043) from the University of Hong Kong.
References
- 1. Orsini G, Mazzoni A, Orciani M, Putignano A, Procaccini M, Falconi M et al (2011) Matrix metalloproteinase‐2 expression induced by two different adhesive systems on human pulp fibroblasts. J. Endod. 37, 1663–1667. [DOI] [PubMed] [Google Scholar]
- 2. Pezelj‐Ribaric S, Anic I, Brekalo I, Miletic I, Hasan M, Simunovic‐Soskic M (2002) Detection of tumor necrosis factor alpha in normal and inflamed human dental pulps. Arch. Med. Res. 33, 482–484. [DOI] [PubMed] [Google Scholar]
- 3. Teo GS, Ankrum JA, Martinelli R, Boetto SE, Simms K, Sciuto TE et al (2012) Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor‐α‐activated endothelial cells via both leukocyte‐like and novel mechanisms. Stem Cells 30, 2472–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cooper PR, Takahashi Y, Graham LW, Simon S, Imazato S, Smith AJ (2010) Inflammation‐regeneration interplay in the dentine‐pulp complex. J. Dent. 38, 687–697. [DOI] [PubMed] [Google Scholar]
- 5. Goldberg M, Farges JC, Lacerda‐Pinheiro S, Six N, Jegat N, Decup F et al (2008) Inflammatory and immunological aspects of dental pulp repair. Pharmacol. Res. 58, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldberg M (2011) Pulp healing and regeneration: more questions than answers. Adv. Dent. Res. 23, 270–274. [DOI] [PubMed] [Google Scholar]
- 7. Arthur A, Koblar S, Shi S, Gronthos S (2009) Eph/ephrinB mediate dental pulp stem cell mobilization and function. J. Dent. Res. 88, 829–834. [DOI] [PubMed] [Google Scholar]
- 8. Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N et al (2006) Ephrin‐B2 controls cell motility and adhesion during blood‐vessel‐wall assembly. Cell 124, 161–173. [DOI] [PubMed] [Google Scholar]
- 9. Chumley MJ, Catchpole T, Silvany RE, Kernie SG, Henkemeyer M (2007) EphB receptors regulate stem/progenitor cell proliferation, migration, and polarity during hippocampal neurogenesis. J. Neurosci. 27, 13481–13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ventura JJ, Kennedy NJ, Lamb JA, Flavell RA, Davis RJ (2003) c‐Jun NH(2)‐terminal kinase is essential for the regulation of AP‐1 by tumor necrosis factor. Mol. Cell. Biol. 23, 2871–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng N, Chen J (2001) Tumor necrosis factor‐alpha induction of endothelial ephrin A1 expression is mediated by a p38 MAPK‐ and SAPK/JNK‐dependent but nuclear factor‐kappa B‐independent mechanism. J. Biol. Chem. 276, 13771–13777. [DOI] [PubMed] [Google Scholar]
- 12. Song Y, Zhao XP, Song K, Shang ZJ (2013) Ephrin‐A1 is up‐regulated by hypoxia in cancer cells and promotes angiogenesis of HUVECs through a coordinated cross‐talk with eNOS. PLoS ONE 8, e74464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang H, Zhao N, Xu X, Xu Y, Li S, Zhang J et al (2011) Dose‐specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif. 44, 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Turner NA, Mughal RS, Warburton P, O'Regan DJ, Ball SG, Porter KE (2007) Mechanism of TNF alpha‐induced IL‐1alpha, IL‐1beta and IL‐6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc. Res. 76, 81–90. [DOI] [PubMed] [Google Scholar]
- 15. Holzman LB, Marks RM, Dixit VM (1990) A novel immediate‐early response gene of endothelium is induced by cytokines and encodes a secreted protein. Mol. Cell. Biol. 10, 5830–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J (2011) TNF‐α promotes fracture repair by augmenting the recruitment and differentiation of muscle‐derived stromal cells. Proc. Natl. Acad. Sci. USA 108, 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Min KS, Kwon YY, Lee HJ, Lee SK, Kang KH, Lee SK et al (2006) Effects of proinflammatory cytokines on the expression of mineralization markers and heme oxygenase‐1 in human pulp cells. J. Endod. 32, 39–43. [DOI] [PubMed] [Google Scholar]
- 18. Paula‐Silva FW, Ghosh A, Silva LA, Kapila YL (2009) TNF‐alpha promotes an odontoblastic phenotype in dental pulp cells. J. Dent. Res. 8, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang J, Zhang C, Tani‐Ishii N, Shi S, Wang CY (2005) NF‐kappaB activation in human dental pulp stem cells by TNF and LPS. J. Dent. Res. 84, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tejero R, Anitua E, Orive G (2014) Toward the biomimetic implant surface: biopolymers on titanium‐based implants for bone regeneration. Prog. Polym. Sci. 39, 1406–1447. [Google Scholar]
- 21. Stokowski A, Shi S, Sun T, Bartold PM, Koblar SA, Gronthos S (2007) EphB/ephrin‐B interaction mediates adult stem cell attachment, spreading, and migration: implications for dental tissue repair. Stem Cells 25, 156–164. [DOI] [PubMed] [Google Scholar]
- 22. Arthur A, Zannettino A, Panagopoulos R, Koblar SA, Sims NA, Stylianou C et al (2011) EphB/ephrin‐B interactions mediate human MSC attachment, migration and osteochondral differentiation. Bone 48, 533–542. [DOI] [PubMed] [Google Scholar]
- 23. Wang X, Jong G, Lin LM, Shimizu E (2013) EphB‐EphrinB interaction controls odontogenic/osteogenic differentiation with calcium hydroxide. J. Endod. 39, 1256–1260. [DOI] [PubMed] [Google Scholar]
- 24. Ahn SJ, Rhim EM, Kim JY, Kim KH, Lee HW, Kim EC et al (2014) Tumor necrosis factor‐alpha induces matrix metalloproteinases‐3, ‐10 and ‐13 in human periodontal ligament cells. J. Periodontol. 85, 490–497. [DOI] [PubMed] [Google Scholar]
- 25. Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P (2007) MMP‐2, MT1‐MMP, and TIMP‐2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood 109, 4055–4063. [DOI] [PubMed] [Google Scholar]
- 26. Takata F, Dohgu S, Matsumoto J, Takahashi H, Machida T, Wakigawa T et al (2011) Brain pericytes among cells constituting the blood‐brain barrier are highly sensitive to tumor necrosis factor‐α, releasing matrix metalloproteinase‐9 and migrating in vitro. J. Neuroinflammation 8, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng L, Amano K, Iohara K, Ito M, Imabayashi K, Into T et al (2009) Matrix metalloproteinase‐3 accelerates wound healing following dental pulp injury. Am. J. Pathol. 175, 1905–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chantrain CF, Henriet P, Jodele S, Emonard H, Feron O, Courtoy PJ et al (2006) Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. Eur. J. Cancer 42, 310–318. [DOI] [PubMed] [Google Scholar]
- 29. Janes PW, Saha N, Barton WA, Kolev MV, Wimmer‐Kleikamp SH, Nievergall E et al (2005) Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 123, 291–304. [DOI] [PubMed] [Google Scholar]
- 30. Dempsey PW, Doyle SE, He JQ, Cheng G (2003) The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 14, 193–209. [DOI] [PubMed] [Google Scholar]