

Abstract
Abstract. Objectives: The role of stem cells in regenerative medicine is evolving rapidly. Here, we describe the application, for kidney regeneration, of a novel non‐genetically modified stem cell, derived from human amniotic fluid. We show that these pluripotent cells can develop and differentiate into de novo kidney structures during organogenesis in vitro. Materials and methods: Human amniotic fluid‐derived stem cells (hAFSCs) were isolated from human male amniotic fluid obtained between 12 and 18 weeks gestation. Green fluorescent protein and Lac‐Z‐transfected hAFSCs were microinjected into murine embryonic kidneys (12.5–18 days gestation) and were maintained in a special co‐culture system in vitro for 10 days. Techniques of live microscopy, histology, chromogenic in situ hybridization and reverse transcriptase polymerase chain reaction were used to characterize the hAFSCs during their integration and differentiation in concert with the growing organ. Results: Green fluorescent protein and Lac‐Z‐transfected hAFSCs demonstrated long‐term viability in organ culture. Histological analysis of injected kidneys revealed that hAFSCs were capable of contributing to the development of primordial kidney structures including renal vesicle, C‐ and S‐shaped bodies. Reverse transcriptase polymerase chain reaction confirmed expression of early kidney markers for: zona occludens‐1, glial‐derived neurotrophic factor and claudin. Conclusions: Human amniotic fluid‐derived stem cells may represent a potentially limitless source of ethically neutral, unmodified pluripotential cells for kidney regeneration.
INTRODUCTION
Recent advances in understanding developmental pathways of the kidney and the establishment of novel in vitro model systems will have significant implications for kidney tissue engineering in the future. This is an important objective to pursue as end stage renal disease has reached epidemic proportions in the USA and, currently, dialysis or allogenic transplantation remain the only viable treatments despite there being significant complications with both (National Kidney and Urologic Diseases Information Clearinghouse 2004). This, along with an increasing shortage of donor organs, has heightened interest in developing novel methods of therapy for kidney tissue replacement. However, because of its complex structure and function, the kidney still remains a challenging organ to recreate in vitro. Many new concepts, including delineation of the role of growth factors during kidney organogenesis, importance of the extracellular matrix and the potential role of stem cells, have suggested novel organ‐based and cell‐based approaches to kidney regeneration, to which we have applied ourselves in the current study.
Over the last decade, stem cells and their possible role in regenerative medicine for reconstruction of bio‐artificial tissues and organs have emerged (Stocum 2001). For example, embryonic stem cells (ESCs), derived from blastocyst‐stage embryos, propagate readily and are capable of forming aggregates (embryoid bodies) that generate a variety of different cell types, including neural cells, cardiomyocytes and islet‐like clusters (Wobus et al. 1991; Thomson et al. 1998; McDonald et al. 1999; Reubinoff et al. 2000; Soria et al. 2001). However, extraction of these cells is ethically controversial. Progenitor cells also exist in many adult tissues where they contribute to tissue regeneration after normal cellular senescence or injury, and possess more plasticity than was originally thought (Orlic et al. 2003; Zhao et al. 2003). These adult stem cells reside in various organs of the body, but it is still unknown whether they can reliably generate mature cell types of other tissues. For these reasons, investigators continue to search out new cell types that can offer better alternatives for bioengineering applications.
In a collaborative effort over the last 7 years with investigators at Wake Forest University, our group has derived a novel stem cell population, recently described as originating from human amniotic fluid, that exhibits both embryonic and mesenchymal stem cell characteristics (Delo et al. 2006; De Coppi et al. 2007a,b). These human amniotic fluid‐derived stem cells (hAFSCs) can easily be propagated in vitro and maintain ESC‐like properties. In addition, hAFSCs have the capacity to differentiate into many different cell types derived from all the three germ layers. Another important advantage of these cells is that they are easily retrieved during amniocentesis with no injury to the embryo. hAFSCs may, therefore, represent a new pluripotential cell source for tissue regeneration and a viable alternative for whole organ engineering in the future. To test this hypothesis, we applied these cells in an in vitro system aimed at regenerating kidney cells and structures in the laboratory.
In order to establish a model for in vitro kidney regeneration and investigate the pluripotential capacity of hAFSCs, we combined technologies of tissue engineering with those of developmental and stem cell biology, based on the principle that stem cells will develop more appropriately in an embryonic tissue environment. An in vitro system of renal organogenesis was established to demonstrate this concept and to assist in differentiating hAFSCs down a kidney lineage: hAFSCs were injected into mouse embryonic kidneys at E12.5–E18‐day gestation, to determine the ability of these stem cells to survive, replicate and contribute to the formation of primordial renal structures during organ development.
MATERIALS AND METHODS
Isolation and labelling of hAFSCs, pre‐injection
Samples of human amniotic fluid from male foetuses (12–18‐week gestation) were provided to our laboratory by Genzyme Genetics Corporation (Monrovia, CA, USA). The stem cell population was separated from the general cellular milieu using standard magnetic‐activated cell sorting techniques (Miltenyi Biotech, Auburn, CA, USA) against cell surface marker c‐kit as described (De Coppi et al. 2007a). Pluripotential characteristics of the clonal and subclonal groups were tested according to protocols also outlined (De Coppi et al. 2007a). Clones were then cultured in petri dishes in medium containing α‐MEM supplemented with 20% Chang B and 2% Chang C solutions, 20% foetal bovine serum (FBS), 1%l‐glutamine and 1% antibiotics (Pen‐Strep) (Gibco/BRL, Rockville, MD, USA). Labelling of cloned hAFSCs with green fluorescent protein (GFP) and Lac‐Z was achieved through infection with a retrovirus using standard protocols. Expression of GFP and Lac‐Z was confirmed by microscopy under fluorescent light in the infected cells, 20 h post‐infection. Clonal hAFSC populations were labelled with a cell surface marker CM‐DiI (Molecular Probe, Eugene, OR, USA) following the manufacturer's instructions, in order to track the cells during and after injection. Briefly, the cells were incubated with a working solution of CM‐DiI of 1 mg/mL for 5 min at 37 °C followed by incubation for 15 min at 4 °C followed by three washes with phosphate‐buffered saline (PBS).
Embryonic kidney culture system
Wild‐type C57BL/C6 mice metanephric kidneys were dissected under sterile conditions from timed‐pregnant females, at embryonic days 12.5–18 (E12.5 to E18) under a dissecting microscope. Embryonic staging was verified using the criteria of Theiler (1989). These developing mouse kidneys were placed on a 0.4‐µm pore size Transwell membrane (Corning Incorporated, Acton, MA, USA) and were cultivated in the medium‐gas interphase in a 37 °C incubator, fully humidified and 5% CO2. The base media used was Leibovitz's L‐15 (Gibco/Invitrogen, Carlsbad, CA, USA) supplemented with 1% penicillin/streptomycin and 2% FBS (Gibco/Invitrogen). Media were not changed during the culture but were added if needed. Perfusion channels were created under direct microscopy with a 10‐µm diameter glass needle, fashioned in the laboratory, without conveying subsequent damage to the kidney but permitting homogeneous perfusion and bathing throughout the whole organ. In addition, a lower concentration of FBS (2%) significantly decreased the necrosis process over the time span of the culture, which allowed for its long‐term maintenance for an average of 10 days.
Microinjection of hAFSC and co‐culture
Immediately before injection, kidneys were placed on a polyethylene‐terephthalate track‐etched membrane (Sterlitech Corporation, Kent, WA, USA). hAFSCs were trypsinized, counted and loaded into a 15‐µm diameter transfer tip (Eppendorf AG, Hamburg, Germany), guided by a micromanipulator (Eppendorf TransferMan NK2) and a CellTram Oil injector (Eppendorf AG), at a total cell count of 1000 with no serum. Each kidney received a single injection (1–2 µL) and immediately was placed on the top of a Transwell filter in the incubator for 2–10 days.
Live imaging
A Leica® microscope incubator‐chamber (Leica, Cambridge, UK) was used to monitor position, migration, and possible replication of hAFSC within the embryonic kidney environment. Pictures were taken every 4 h for 4 days using Leica Deblur software.
Histology
Kidneys were fixed in 4% buffered paraformaldehyde (Sigma‐Aldrich, St Louis, MO, USA) for 1 h at 4 °C, routinely processed, embedded in paraffin and were sectioned at 5 µm. Briefly, the embryonic kidneys were washed in 70% alcohol for 1 h, followed by two washes in absolute alcohol for 1 h and were placed in xylene twice, for 15 min, then 1 h in a solution of xylene/paraffin and paraffin overnight. The following morning kidneys were entirely embedded in paraffin wax and prepared for sectioning. Sections were stained with haematoxylin and eosin (Sigma‐Aldrich) and toluidine blue (Sigma‐Aldrich) using standard histological protocols. For total organ X‐gal staining, kidneys were fixed in 4% paraformaldehyde (Sigma‐Aldrich) in PBS at 4 °C for 30 min while rocking, then were washed twice for 10 min in PBS at 4 °C, transferred into freshly prepared X‐gal (Research Products International Corporation, Mt. Prospect, IL, USA) solution, and were stained at 37 °C until a clear precipitate formed. For vibratome (Leica) sections, samples were embedded in an albumin (300 mg/mL)–gelatin (5 mg/mL) mix (Sigma‐Aldrich), cross‐linked with glutaraldehyde (0.6%) (Sigma‐Aldrich), and were sectioned at 30–40 µm before viewing by light microscopy.
Chromogenic in situ hybridization
Chromogenic in situ hybridization (CISH) was used to detect the Y‐chromosome in the nuclei of male hAFSC after injection into the embryonic kidney. Formalin‐fixed paraffin‐embedded tissue sections were used for this procedure according to the manufacturer's instructions (Zymed SPOT‐Light CISH, Zymed Laboratories Inc., San Francisco, CA, USA) centromere kit detected Zymed SPOT‐Light Human Chromosome Y Probes (Zymed Laboratories Inc.). Formalin‐fixed paraffin‐embedded sections were incubated at 55 °C overnight, and then slides were deparaffinized in xylene and graded ethanols (Sigma‐Aldrich). Heat pre‐treatment was carried out in buffer at 94–95 °C for 5 min and after application of Zymed SPOT‐Light Chromosome Y Probe the slides were cover‐slipped and edges were sealed with rubber cement. The slides were heated at 95 °C for 5 min followed by overnight incubation at 37 °C using a moisturized chamber. Post‐hybridization washing was performed the next day and was followed by immunodetection using the CISH polymer detection kit (Zymed Laboratories Inc.). CISH signals were detected under light microscopy using a 40× objective lens.
Reverse transcriptase‐polymerase chain reaction
Total mRNA was extracted from the embryonic kidney, using Qiagen mini kit (Qiagen, Hilden, Germany), and was reverse transcribed using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and random hexamers as primers for first strand cDNA synthesis. Amplification of the resulting cDNA was carried out using specific human primers thus not codifying mouse sequences. A polymerase chain reaction thermal cycler (Eppendorf) was employed after an initial denaturation step at 95 °C for 10 min. We used a denaturation step at 95 °C for 30 s, an annealing step at the temperature specific for each primer (ranging from 54 °C to 60 °C) for 45 s, and an extension step at 72 °C for 45 s for a total of 35 cycles. To rule out the possibility of amplifying genomic DNA, RNA samples were treated with a DNA‐free kit (Ambion Inc., Austin, TX, USA). Detection of polymerase chain reaction amplification products was performed by size fractionation on 1% agarose gel electrophoresis. As a housekeeping gene, amplification of fragments of human β‐actin RNA was performed. Primer sequences and predicted sizes of amplicons were as follows in Table 1.
Table 1.
Primers sequences, products size and annealing temperature used for reverse transcription‐polymerase chain reaction
| Gene | Primer sequence | Product size (bp) | Annealing temperature |
|---|---|---|---|
| Claudin | 5′‐ATGATGTCACTCCTCTTGCTGGTG‐3′ | 480 | 60 |
| 5′‐CGCTTCCGTAGGTGGATGTAG‐3′ | |||
| ZO‐1 | 5′‐AGGAGAGGTGTTCCGTGTTG‐3′ | 760 | 59.9 |
| 5′‐GCTGGTTTTGCTGTTGTTGA‐3′ | |||
| GDNF | 5′‐TATGGGATGTCGTGGCTGT‐3′ | 630 | 58.2 |
| 5′‐ACACCTTTTAGCGGAATGCTT‐3′ | |||
| AQP‐1 | 5′‐CACCTCCTCCCTGACTGG‐3′ | 290 | 58.8 |
| 5′‐GGTTGCTGAAGTTGTGTGTGA‐3′ | |||
| AQP‐2 | 5′‐GATCACGCCAGCAGACATC‐3′ | 241 | 59.7 |
| 5′‐GGGCAGGATTCATAGAGCAG‐3′ | |||
| THP | 5′‐TAGACGAGGACTGCAAATCG‐3′ | 223 | 58.8 |
| 5′‐GTCCCGGTTGTCTCTGTCAT‐3′ | |||
| Ang | 5′‐ACTTGTCCACGGACCCAAATC‐3′ | 340 | 58 |
| 5′‐TGGTGTTGTCCACCCAGAACTC‐3′ | |||
| DBAgg | 5′‐CAGCAAGTTACGACTCACAAAAG‐3′ | 380 | 58 |
| 5′‐GAAGCAAAAGACCAAGAGAGGA‐3′ | |||
| PAgg | 5′‐GGTGGGATCCATGCTAGCAGCAAGTAGT‐3′ | 280 | 58.5 |
| 5′‐AACTCGAATTCCTTTCGGGCTTA‐3′ | |||
| Nep | 5′‐ACACGGAGCACACATACCAC‐3′ | 568 | 59.8 |
| 5′‐GGATTGGAGAGGAGCAGAAG‐3′ | |||
| β‐actin | 5′‐AGAAAATCTGGCACCACACC‐3′ | 394 | 55.4 |
| 5′‐CTCCTTAATGTCACGCACGA‐3′ |
Ang, angiotensin; AQP, aquaporin; DBAgg, dolichus biflorus; GDNF, glial‐derived neurotrophic factor; Nep, nephrin; PAgg, peanut agglutinin; THP, Tamm‐Horsfall protein; ZO‐1, zona occludens‐1.
RESULTS
Whole organ embryonic kidney culture
Embryonic kidney rudiments from E12.5 to E18 day embryos of wild‐type C57BL/C6 mice (purchased from Charles River) were successfully microdissected and set into culture (Fig. 1a,b). However, when using previously published culture protocols (Rogers et al. 1991; Hardman et al. 1993; Steer et al. 2002; Gupta et al. 2003), kidney rudiments were only maintained for 4 days before necrosis was observed. In contrast, embryonic kidneys cultured in our protocol (see above) were capable of continued growth for an average of 10 days, without evidence of necrosis, and demonstrated normal glomerular and tubular structures when using haemotoxylin and eosin staining for light microscopy (Fig. 1c,d).
Figure 1.
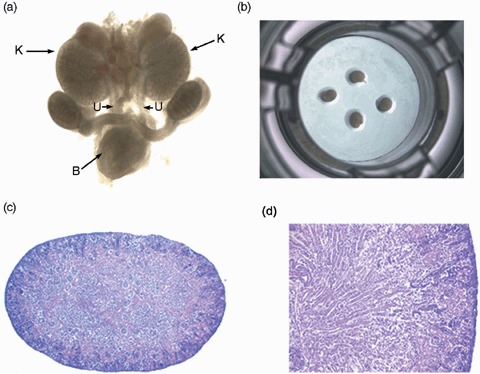
Whole organ embryonic kidney culture, steps in the culture of embryonic kidneys. (a) Entire genitourinary tract en bloc from an E13.5‐day mouse embryo (×2). (b) Embryonic kidneys laid on to a 24‐well plate demonstrating the culture system. (c) Embryonic kidneys with normal morphology when cultured in 24‐well polystyrene plates with PTFE membrane (×4), and new protocols demonstrating no evidence of necrosis. (d) Higher power view demonstrating kidney specimens (×25) after culture using new protocols demonstrating no evidence of necrosis plus maintenance of tubular and glomerular structures highlighted by arrows. H&E, haematoxylin and eosin; PTFE, polytetrafluoroethylene; K, kidney; U, ureters; B, bladder.
Migration pattern of hAFSC in developing embryonic kidney hAFSCs had been selected to be of clonal origin thus in each case guaranteeing a homogeneous population. Prior to injection, expression of GFP and β‐galatosidase Lac‐Z were confirmed in the cells in order to be able to track them in the in vitro culture (Fig. 2a,b). Moreover, hAFSCs also expressed the surface marker CM‐Dil staining positively for it (Fig. 2c) for easy identification under stereo microscopy during the injection phase (Fig. 2d). After 4 days of fluoroscopic live imaging, GFP+ hAFSCs within the injected kidneys exhibited a branching pattern in which cells migrated from the centre to the periphery of the organ (Fig. 2e,f), as shown in the supplementary video. After 3 days of culture, vibratome sections examined by light microscopy also revealed that Lac‐Z+ hAFSCs had migrated from the centre to the periphery of the metanephric kidney in the same centrifugal fashion that normally occurs during embryonic kidney development (Welling & Linshaw 1988; El‐Dahr 2004) (Fig. 3).
Figure 2.
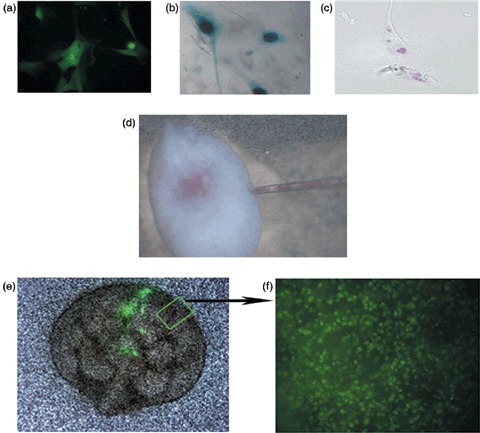
Microinjection of labelled amniotic fluid stem cells, pre‐injected stem cells, the injection in process and post‐injection. (a) hAFSCs transfected with retrovirus carrying the sequence for GFP and β‐galactosidase under fluorescent microscopy (×40). (b) Lac‐Z nuclear staining of hAFSCs (×40). (c) hAFSCs labelled pink for light microscopy with cell surface marker CM‐Dil (×40). (d) Microinjection of hAFSCs labelled with CM‐Dil direct vision (×4) into the centre of the embryonic kidney. (e) GFP labelled cells shown in the embryonic kidney by fluoroscopy at day 0 of injection (×4). (f) Live imaging, at 4 h intervals, of the embryonic kidney after 4 days of culture demonstrating GFP labelled hAFSCs multiplying and spreading throughout the entire organ from the centre to the periphery (×40, see the supplementary video). hAFSCs, human amniotic fluid stem cells; GFP, green fluorescent protein.
Figure 3.

Migration pattern of hAFSCs in developing embryonic kidney, histology of sectioned kidneys post‐injection of stem cells. (a) Lac‐Z staining confirming the presence of hAFSCs after 3 days in culture (×6). (b) Lac‐Z+ hAFSCs migrated to the periphery of the embryonic kidney after injection into the middle of organ (×8). hAFSCs, human amniotic fluid stem cells.
Structural differentiation of hAFSCs within developing embryonic kidneys
Light microscopy with haemotoxylin and eosin counter staining was used to detect Lac‐Z+ cells within important renal precursor elements to both the glomerular and tubular components of the nephron. After 5 days, cells injected into E13 kidneys integrated into C‐ and S‐shaped structures (Fig. 4a), stroma (Fig. 4b) and renal vesicles (Fig. 4c) of the murine embryonic kidney. No Lac‐Z+ cells were detected in the controls (Fig. 4d,e). After 6 days of culture, CM‐Dil labelled hAFSCs had also integrated into embryonic tubular and glomerular structures (Fig. 4f,g). Presence and integration of the hAFSCs in the injected kidneys were further confirmed using chromogenic in situ hybridization (Zymed Laboratories Inc.). The populations of hAFSCs used in all experiments were derived from human male foetuses and, therefore, bore X and Y chromosomes. Specimens were incubated with biotinylated probes against the Y chromosome and preparations were counterstained with DAB(3,3‐diaminobenzidine) in the standard peroxidase reaction. Positively stained cells were visualized using light microscopy, lining walls of the embryonic kidney tubules (Fig. 5a,b). This comprised both medullary and cortical portions of the kidney. No signal was identified in any of the controls (Fig. 5c,d).
Figure 4.
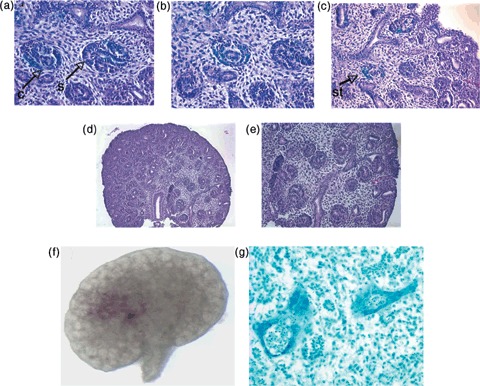
Structural differentiation of amniotic fluid stem cells within developing embryonic kidneys demonstrating integration of stem cells. (a) H&E staining after Lac‐Z staining revealing presence of hAFSCs in C‐ and S‐shaped bodies of the embryonic kidney (×40) (arrows). (b) Lac‐Z+ hAFSCs within the stroma of the developing kidney after H&E staining (×40). (c) Lac‐Z+ hAFSCs within renal vesicles of the developing kidney after H&E staining (×30) (arrow). (d) Controls with no detected Lac‐Z staining (×10). (e) Controls with no detected Lac‐Z stain (×16). (f) hAFSCs labelled with CM‐Dil 1 day after injection (×10). (g) hAFSCs labelled with CM‐Dil, that appears blue with light microscopy after the toluidine treatment, detected in renal primordia (C‐ and S‐shaped bodies) and in the stroma after 5 days of culture within developing tubular nephrons of the embryonic kidney (×40). H&E, haematoxylin and eosin; hAFSCs, human amniotic fluid stem cells; C, C‐shaped body; S, S‐shaped body; St, stroma.
Figure 5.
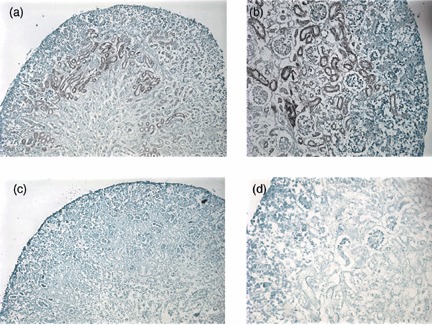
Chromogenic in situ hybridization of injected amniotic fluid stem cells, integration of stem cells into the cultured developing kidneys. (a) CISH for Y chromosome of hAFSCs also confirmed integration into embryonic kidney structures (×20). (b) Primordial tubular nephrons stained positively with CISH confirming integration of hAFSCs into these structures. (c) Controls demonstrated no positive reaction with CISH, ×20 magnification. (d) CISH controls with no positive stain (×30). CISH, chromogenic in situ hybridization; hAFS, human amniotic fluid stem cells.
Molecular evidence of primordial kidney differentiation by the hAFSCs
After 9 days of culture, reverse transcriptase–polymerase chain reaction was performed and expression of a number of specific human kidney genes by the hAFSCs in the injected embryonic kidneys was identified. Zona occludens‐1 (ZO‐1), claudin and glial‐derived neurotrophic factor (GDNF), early markers for kidney differentiation, were detected when compared to controls, which were non‐injected hAFSCs (Fig. 6).
Figure 6.
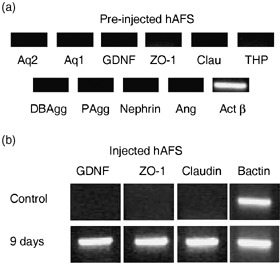
Molecular evidence of primordial kidney differentiation from amniotic fluid stem cells, RT‐PCR markers demonstrating expression of renal specific markers pre‐ and post‐injection and culture. RT‐PCR of hAFSCs prior to injection are negative for most kidney markers (a). hAFSCs, 9 days after injection into embryonic kidneys, demonstrate early kidney gene expression markers, zona occludens‐1 (ZO‐1, 760 bp), glial‐derived neurotrophic factor (GDNF, 630 bp) and claudin (480 bp), compared to no expression of kidney markers in controls (hAFSCs prior to injection) (b). RT‐PCR, reverse transcription‐polymerase chain reaction; hAFSCs, human amniotic fluid stem cells.
DISCUSSION
Bioengineering of the kidney with all of its multifaceted structure and functions, both in vitro and in vivo, still remains a difficult challenge; however, some progress has been made. Both autologous and pluripotential cells have been used to propagate or regenerate, kidney cell lines in culture (MacKay et al. 1998; Lanza et al. 2002). These cells may then be used to create a wide range of filtration devices, some with potential for life‐sustaining properties, or others with more limited functionality (Aebischer et al. 1987; Ip et al. 1988; MacKay et al. 1998; Humes et al. 1999; Amiel et al. 2000). Extracorporeal perfusion circuits constructed from cell‐seeded haemofiltration devices have also been described, but these are only capable of limited perfusion (Amiel & Atala 1999; Amiel et al. 2000; Saito 2004). One of the first applications of an artificial renal device implanted in vivo was recently reported, but only demonstrated limited excretory function (Yoo & Atala 2000; Lanza et al. 2002). Perhaps developing immunocompatible tissues or therapies from stem cells can offer advantages over current methods of allogenic transplantation or dialysis and provide a better method for regenerating organs such as the kidney in the laboratory. Our team combines tissue engineering with the fields of developmental and stem cell biology in a novel approach to re‐engineering kidney tissue prosthetics. A combination of stem cells for tissue regeneration with an understanding of developmental biology and organogenesis is crucial if one chooses to reproduce in vitro a normal embryonic niche in which cells can interact with their environment.
Few studies to date have addressed the application of stem cells for kidney differentiation. Jiang et al. (2002) reported the isolation of a multipotent adult progenitor cell from murine bone marrow and have shown that the cells can differentiate into virtually all somatic tissues of a developing embryo after injection. This study included several typical cell types in the kidney, such as glomerular endothelial and tubular endothelial cells. More recently, exogenously derived genetically modified human mesenchymal stem cells (hMSCs), in combination with whole embryo culture, have been shown to differentiate and contribute to functional complex structures of the kidney during in vitro embryogenesis studies (Yokoo et al. 2005). In some cases, entire nephrons were hMSC‐derived. Thus, hMSCs can be reprogrammed for specific lineage and organ structures, depending on the embryonic environment into which they are placed. ESCs have also been used in cases of direct injection into a mouse embryonic kidney (Steenhard et al. 2005). This established a new model system in which patterns of epithelialization within the developing kidney could be deciphered. Thus, direct injection of cells provides a useful tool for evaluation of steps required for epithelial differentiation and tubulogenesis.
In our laboratory, we have examined this novel stem cell population for kidney differentiation isolated from human amniotic fluid (Siddiqui & Atala 2004). hAFSCs make up a novel population of cells that has recently shown definite promise for regenerative medicine applications (Delo et al. 2006; De Coppi et al. 2007a,b). They possess many of the same properties as traditional ESC lines, for example, hAFSCs are pluripotential and give rise to many different cell types both in vitro and in vivo. hAFSCs express embryonic stem cell markers, Oct‐4 and SSEA‐3 and SSEA‐4. However, hAFSCs possess many important advantages over ESCs: they can be easily isolated from the amnion with no harm to the foetus and can be cultured without a feeder layer, using very simple culture conditions. hAFSCs can also be expanded by multiple passages, and can be frozen and thawed to provide a potentially inexhaustible source of stem cells, when required.
Embryologically, the kidney is derived from the ureteric bud (UB), which forms the collecting system of the kidney, and the metanephric mesenchyme (MM), which gives rise to the glomerular and tubular nephron, or filtration, portion of the kidney. In this study, hAFSCs were physically placed into a developing embryonic kidney in vitro, this then providing the stem cell niche with necessary growth factors, and an appropriate extracellular environment in which renal differentiation could proceed. Many standard protocols are described in the literature outlining commonly applied techniques for whole embryonic kidney culture in vitro, but many of these may not be sufficient for appropriate cellular differentiation to occur (Rogers et al. 1991; Hardman et al. 1993; Steer et al. 2002; Gupta et al. 2003). Embryo organs are fragile by nature and the majority of these culture systems report an average of only 5 days of successful culture before the onset of necrosis in the organ begins. Thus, the majority of these culture systems fails to allow for survival of the organ long enough to enable definitive stem cell differentiation to occur. To provide hAFSC sufficient time for renal differentiation, we established a new, reproducible protocol for long‐term culture of embryonic kidneys in the laboratory. In this context, we could inject hAFSCs with reasonable confidence they would survive and replicate in a normal and well‐preserved environment. Furthermore, it was possible to follow the proliferation and differentiation of these cells during the entire culture period of 10 days.
It is well established that the process of induction, morphogenesis and differentiation of the metanephros occurs in a centrifugal pattern, proceeding from the centre to the periphery of the embryonic organ. This was also observed with our GFP and Lac‐Z‐stained hAFSCs where movement of the injected cells from the site of injection, the centre of the embryonic kidney, to the periphery, was documented by both live imaging and by histological techniques (Potter 1943; Evan et al. 1983; Evan et al. 1991). Thus, hAFSCs, placed into the differentiating MM, seem to follow some very important steps in the development of nephron units. Further evidence of this was conveyed by our histological data that demonstrated that hAFSCs integrated into the renal vesicle, and into C‐ and S‐shaped bodies that are the primordial structures of the kidney that eventually develop into mature glomerular and tubular structures of the nephron. In addition, there was also molecular evidence of renal differentiation exhibited by expression of human GDNF, ZO‐1 and claudin by the hAFSCs within the developing kidney.
In analysing our data, GDNF expression is particularly compelling evidence for successful hAFSCs’ induction into kidney tissue. The majority of GDNF expression is usually seen in only very early embryonic stages during branching of the UB into the MM (Basson et al. 2006; Costantini & Shakya 2006). When the UB branches into the MM, GDNF expression is down‐regulated and other genes induce the final development phase of the kidney. All kidney rudiments in this study (E12.5 to E18) had undergone multiple UB branching already. We therefore conclude that the strong human GDNF expression observed must arise from hAFSCs that had been induced by the surrounding mouse embryonic environment, to undergo renal differentiation. In contrast, expression of late kidney gene markers, aquaporin 1 and aquaporin 2, were not seen, indicating that hAFSCs did not complete a late differentiation phase. We speculate that these later phases of differentiation can not be achieved in the culture system because only early phase development is maintained for the limited lifespan of the culture. Nevertheless, our data suggest that hAFSCs complete at least an initial step essential for commitment towards a renal fate following the natural growth of the organ and that during organ culture, and they further undergo the critical mesenchymal‐to‐epithelial transition that is normally observed in renal development.
Another very important characteristic of hAFSCs demonstrated in this study is their ability to undergo differentiation without prior modification. Recently, published reports have shown that it was possible to obtain kidney cell differentiation from bone marrow‐derived mesenchymal stem cells only when the cells were induced (genetically modified) to produce GDNF (Yokoo et al. 2005). In the latter report, bone marrow stem cells were driven towards a kidney fate before injection. In our study, hAFSCs were not induced to express any kidney genes and were not genetically modified prior to their injection into the organ. This underscores an important principle that hAFSCs do not appear to require any prior manipulation before installation into this particular culture system. It would then appear that hAFSCs posses a greater pluripotentiality than mesenchymal stem cells from bone marrow, because signals from the embryonic renal environment are sufficient to induce hAFSCs towards the differentiation process into kidney cells and structures.
Here, we have demonstrated the ability of hAFSC to survive, proliferate and integrate into the embryonic kidney while it undergoes organ development, in an in vitro culture system. We observed the presence of hAFSC within kidney primordia, including tubules and developing nephrons. Thus, hAFSCs seem to have the capacity to undergo the expected mesenchymal‐to‐epithelial transition that occurs in normal renal development and are induced to express important early kidney markers such as GDNF, ZO‐1 and claudin. Moreover, hAFSCs do not appear to require prior genetic modification or exogenous production of kidney proteins for their differentiation to occur. This is a very important advantage that hAFSCs have for potential future regenerative or bioengineering application. hAFSCs could therefore represent a potentially limitless source of ethically neutral stem cells with many appealing qualities for future studies aimed to achieving viable alternatives in the fields of tissue engineering and cell therapies for the kidney.
Supporting information
Video S1. Live imaging of migrating injected amniotic fluid stem cells. Video created from live images of injected cells under fluorescence taken every 4 h for the first 2 days. Live images of injected hAFSCs taken under fluorescence every 4 h for the first 2 days. Injected cells distribute themselves following a branching pattern natural to the embryonic kidney. hAFSCs, human amniotic fluid stem cells.
This material is available as part of the online article from:
http://www.blackwell‐synergy.com/doi/abs/10.1111/j.1365‐2184.2007.00478.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
ACKNOWLEDGEMENTS
We thank Hiroyuki Shimada, MD/PhD, for his extremely helpful suggestions and Stijn Delanghe, PhD, for his invaluable assistance. In addition, we thank to Francesco Boldrin, PhD, for his assistance in designing our human primers.
Laura Perin and Stefano Giuliani contributed equally to this work.
REFERENCES
- Aebischer P, Ip TK, Panol G, Galletti PM (1987) The bioartificial kidney: progress towards an ultrafiltration device with renal epithelial cells processing. Life Support Syst. 5, 159–168. [PubMed] [Google Scholar]
- Amiel GE, Atala A (1999) Current and future modalities for functional renal replacement. Urol. Clin. North Am. 26, 235–246, xi. [DOI] [PubMed] [Google Scholar]
- Amiel GE, Yoo JJ, Atala A (2000) Renal therapy using tissue‐engineered constructs and gene delivery. World J. Urol. 18, 71–79. [DOI] [PubMed] [Google Scholar]
- Basson MA, Watson‐Johnson J, Shakya R, Akbulut S, Hyink D, Costantini FD, Wilson PD, Mason IJ, Licht JD (2006) Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Dev. Biol. 299, 466–477. [DOI] [PubMed] [Google Scholar]
- Costantini F, Shakya R (2006) GDNF/Ret signaling and the development of the kidney. Bioessays 28, 117–127. [DOI] [PubMed] [Google Scholar]
- De Coppi P, Bartsch G Jr, Siddiqui MM, Xu T, Santos CC, Perin L, Mostoslavsky G, Serrem A, Snyder EY, Yoo JJ, Furth ME, Soker S, Atala A (2007a) Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 25, 100–106. [DOI] [PubMed] [Google Scholar]
- De Coppi P, Callegari A, Chiavegato A, Gasparotto L, Piccoli M, Taiani J, Pozzobon M, Boldrin L, Okabe M, Cozzi E, Atala A, Gamba P, Sartore S (2007b) Amniotic fluid and bone marrow derived mesenchymal stem cells can be converted to smooth muscle cells in the cryo‐injured rat bladder and prevent compensatory hypertrophy of surviving smooth muscle cells. J. Urol. 177, 369–376. [DOI] [PubMed] [Google Scholar]
- Delo DM, De Coppi P, Bartsch G Jr, Atala A (2006) Amniotic fluid and placental stem cells. Meth. Enzymol. 419, 426–438. [DOI] [PubMed] [Google Scholar]
- El‐Dahr SS (2004) Spatial expression of the kallikrein‐kinin system during nephrogenesis. Histol. Histopathol. 19, 1301–1310. [DOI] [PubMed] [Google Scholar]
- Evan, AP , Gattone VH 2nd, Schwartz GJ (1983) Development of solute transport in rabbit proximal tubule. II. Morphologic segmentation. Am. J. Physiol. 245, F391–F407. [DOI] [PubMed] [Google Scholar]
- Evan, AP , Satlin LM, Gattone VH 2nd, Connors B, Schwartz GJ (1991) Postnatal maturation of rabbit renal collecting duct. II. Morphological observations. Am. J. Physiol. 261, F91–F107. [DOI] [PubMed] [Google Scholar]
- Gupta IR, Lapointe M, Yu OH (2003) Morphogenesis during mouse embryonic kidney explant culture. Kidney Int. 63, 365–376. [DOI] [PubMed] [Google Scholar]
- Hardman P, Klement BJ, Spooner BS (1993) Growth and morphogenesis of embryonic mouse organs on non‐coated and extracellular matrix‐coated Biopore membrane. Dev. Growth Differ. 35, 683–690. [DOI] [PubMed] [Google Scholar]
- Humes HD, Buffington DA, MacKay SM, Funke AJ, Weitzel WF (1999) Replacement of renal function in uremic animals with a tissue‐engineered kidney. Nat. Biotechnol. 17, 451–455. [DOI] [PubMed] [Google Scholar]
- Ip TK, Aebischer P, Galletti PM (1988) Cellular control of membrane permeability. Implications for a bioartificial renal tubule. ASAIO Trans. 34, 351–355. [PubMed] [Google Scholar]
- Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz‐Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418, 41–49. [DOI] [PubMed] [Google Scholar]
- Lanza RP, Chung HY, Yoo JJ, Wettstein PJ, Blackwell C, Borson N, Hofmeister E, Schuch G, Soker S, Moraes CT, West MD, Atala A (2002) Generation of histocompatible tissues using nuclear transplantation. Nat. Biotechnol. 20, 689–696. [DOI] [PubMed] [Google Scholar]
- MacKay SM, Funke AJ, Buffington DA, Humes HD (1998) Tissue engineering of a bioartificial renal tubule. ASAIO J. 44, 179–183. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D, Gottlieb DI, Choi DW (1999) Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat. Med. 5, 1410–1412. [DOI] [PubMed] [Google Scholar]
- National Kidney and Urologic Diseases Information Clearinghouse (2004) Kidney and Urologic Diseases Statistics for the United States. National Institute of Health, National Institute of Diabetes and Digestive and Kidney Diseases, website: http://kidney.niddk.nih.gov/kudiseases/pubs/kustats/index.htm#kp
- Orlic D, Kajstura J, Chimenti S, Bodine DM, Leri A, Anversa P (2003) Bone marrow stem cells regenerate infarcted myocardium. Pediatr. Transplant. 7 (Suppl. 3), 86–88. [DOI] [PubMed] [Google Scholar]
- Potter EL (1943) Glomerular development in the kidney as an index of fetal maturity. J. Pediatr. 22, 695. [Google Scholar]
- Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A (2000) Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro . Nat. Biotechnol. 18, 399–404. [DOI] [PubMed] [Google Scholar]
- Rogers SA, Ryan G, Hammerman MR (1991) Insulin‐like growth factors I and II are produced in the metanephros and are required for growth and development in vitro . J. Cell Biol. 113, 1447–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A (2004) Research into the development of a wearable bioartificial kidney with a continuous hemofilter and a bioartificial tubule device using tubular epithelial cells. Artif. Organs 28, 58–63. [DOI] [PubMed] [Google Scholar]
- Siddiqui MM, Atala A (2004) Amniotic fluid derived pluripotential cells In: Lanza R, Gearhart J, Hogan B, eds. Handbook of Stem Cells. Philadelphia, PA: Academic Press, pp. 175–179. [Google Scholar]
- Soria B, Skoudy A, Martin F (2001) From stem cells to beta cells: new strategies in cell therapy of diabetes mellitus. Diabetologia 44, 407–415. [DOI] [PubMed] [Google Scholar]
- Steenhard BM, Isom KS, Cazcarro P, Dunmore JH, Godwin AR, St John PL, Abrahamson DR (2005) Integration of embryonic stem cells in metanephric kidney organ culture. J. Am. Soc. Nephrol. 16, 1623–1631. [DOI] [PubMed] [Google Scholar]
- Steer DL, Bush KT, Meyer TN, Schwesinger C, Nigam SK (2002) A strategy for in vitro propagation of rat nephrons. Kidney Int. 62, 1958–1965. [DOI] [PubMed] [Google Scholar]
- Stocum DL (2001) Stem cells in regenerative biology and medicine. Wound Repair Regen. 9, 429–442. [DOI] [PubMed] [Google Scholar]
- Theiler K (1989) The House Mouse: Atlas of Embryonic Development. New York: Springer‐Verlag, pp. 23–29. [Google Scholar]
- Thomson JA, Itskovitz‐Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Welling LW, Linshaw MA (1988) Structural and functional development of outer versus inner cortical proximal tubules. Pediatr. Nephrol. 2, 108–114. [DOI] [PubMed] [Google Scholar]
- Wobus AM, Wallukat G, Hescheler J (1991) Pluripotent mouse embryonic stem cells are able to differentiate into cardiomyocytes expressing chronotropic responses to adrenergic and cholinergic agents and Ca2+ channel blockers. Differentiation 48, 173–182. [DOI] [PubMed] [Google Scholar]
- Yokoo T, Ohashi T, Shen JS, Sakurai K, Miyazaki Y, Utsunomiya Y, Takahashi M, Terada Y, Eto Y, Kawamura T, Osumi N, Hosoya T (2005) Human mesenchymal stem cells in rodent whole‐embryo culture are reprogrammed to contribute to kidney tissues. Proc. Natl. Acad. Sci. USA 102, 3296–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JJ, Atala A (2000) Tissue engineering applications in the genitourinary tract system. Yonsei Med. J. 41, 789–802. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Glesne D, Huberman E (2003) A human peripheral blood monocyte‐derived subset acts as pluripotent stem cells. Proc. Natl. Acad. Sci. USA 100, 2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Live imaging of migrating injected amniotic fluid stem cells. Video created from live images of injected cells under fluorescence taken every 4 h for the first 2 days. Live images of injected hAFSCs taken under fluorescence every 4 h for the first 2 days. Injected cells distribute themselves following a branching pattern natural to the embryonic kidney. hAFSCs, human amniotic fluid stem cells.
This material is available as part of the online article from:
http://www.blackwell‐synergy.com/doi/abs/10.1111/j.1365‐2184.2007.00478.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item