

Abstract
Abstract. RAIDD (RIP‐associated ICH‐1 homologous protein with a death domain) is an adaptor molecule that mediates the action of cysteine proteases involved in apoptosis. To study the possibility of a novel system of cell ablation mediated by RAIDD, a preadipocyte cell line (3T3L1) was stably transfected with a plasmid containing the murine Raidd cDNA under the control of the adipocyte specific promoter aP2. Instead of the expected apoptosis, a blockage to differentiation upon hormonal induction was observed as judged by an absence of lipid accumulation, a lack of expression of adipocyte‐specific genes and a fibroblastic appearance. Proliferation rate of Raidd‐transfected clones remained unaffected. Overexpression of Raidd cDNA in 3T3L1 cell therefore inhibited differentiation, suggesting that Raidd plays a role in controlling differentiation of mouse preadipocytes and, perhaps, in other cell types, in addition to its established role in apoptosis.
INTRODUCTION
RAIDD (RIP‐associated ICH‐1 homologous protein with a death domain) is an adaptor molecule that contains an amino‐terminal CARD (Caspase Recruitment Domain) region and a carboxy‐terminal ‘death domain’ (Duan & Dixit 1997). The carboxy‐terminal domain interacts with the homologous death domain region in RIP, a serine/threonine kinase component of the TNFRI signalling complex, and the Caspase‐2 (ICH‐1) molecule associates with the homologous amino‐terminal CARD domain of RAIDD (Cohen 1997). The human death adaptor molecule RAIDD, which shares a very high homology with the mouse Raidd gene (Horvat & Medrano 1998), was tested in MCF‐7 cell lines (a human breast carcinoma cell) to address the function of RAIDD in apoptosis. MCF‐7 cells transiently transfected with a RAIDD construct underwent apoptosis, which was inhibited by the inclusion of several known inhibitors of apoptosis, thus confirming the role of this gene in the apoptotic pathway (Duan & Dixit 1997).
Because RAIDD overexpression led to apoptosis in MCF‐7 cells, we wanted to examine if a similar effect could be seen in mouse preadipocytes (3T3L1 cell line), which would result in cell ablation mediated by RAIDD. A preadipocyte cell line (3T3L1) was stably transfected with a plasmid expressing the murine Raidd cDNA under the control of the adipocyte specific promoter aP2 (Ross et al. 1990). 3T3L1 is a preadipocyte cell line, initially described by Green & Meuth (1974) and later by many others, that differentiates in vitro and can exhibit most of the functions of adipocytes in vivo. Prior to differentiation, the preadipocyte cell line is morphologically similar to fibroblastic preadipose cells in the stroma of the adipose tissue. When appropriately induced with hormonal agents, e.g. glucocorticoids, insulin‐like growth factor‐1, and cyclic AMP, or factors that mimic these agents (Student et al. 1980), committed preadipocytes differentiate into adipocytes in culture.
When preadipocytes stably transfected with Raidd were induced to differentiate in the presence of hormones, most clones failed to differentiate. This effect was judged by the absence of lipid accumulation, a lack of expression of adipocytes‐specific genes and a fibroblastic morphological appearance. In two of six clones, partial differentiation was observed, as judged by the partial accumulation of lipid and the death of differentiated adipocytes that escaped the initial blockage.
MATERIALS AND METHODS
Plasmid construct and transfection experiments
A 0.85‐kb fragment containing the pA signal from SV40 was excised from the pA plasmid (kindly provided by Dr Wei Cui) by double digestion with SmaI/NotI, gel‐purified, and ligated to the aP2 promoter plasmid, kindly provided by Dr R. Graves (Ross et al. 1990) that was also digested with SmaI/NotI. A 0.88‐kb fragment corresponding to the murine Raidd cDNA was excised from the plasmid clone 6–1, EMBL No. AJ224738 (Horvat & Medrano 1998) by double digestion with SmaI/ClaI, end‐filled, and ligated into the unique SmaI site of the newly generated aP2‐pA plasmid.
Stable transfection of 3T3L1 cells was carried out by calcium phosphate precipitation (Sambrook et al. 1989). Cells were transfected with a 5 : 1 molar ratio of chimeric construct/SV2‐neo plasmid DNAs (15 µg per 10‐cm dish). Selection of neomycin resistant foci was carried out with 400 µg/ml of neomycin for approximately 11 days, followed by individual clone expansion in selective medium until analysis.
Cell culture
Culture and differentiation of mouse embryo 3T3L1 preadipocytes (American Type Culture Collection, Rockville, MD, USA) was performed as described previously (Student et al. 1980). Briefly, cells were propagated to confluence in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS). On differentiation day 0, cells were fed with DMEM supplemented with 10% FCS, 0.5 mm methylisobutylxanthine, 10 µg/ml of insulin, and 1 µm dexamethasone. Cells were changed to culture medium lacking methylisobutylxanthine and dexamethasone 2 days later (differentiation day 2) and cultured in this medium for 6 days.
RNA preparation and northern blot analysis
Cell monolayers were washed twice with ice‐cold phosphate buffered saline (PBS), and total RNA extracted with RNAzol B (Biogenesis, Poole, UK) following the manufacturer's instructions. RNA was prepared at confluence (−2), after hormonal induction (+2) and after 6 days of differentiation (+6). Fifteen micrograms of total RNA was fractionated on 1.5% formaldehyde/agarose gels and blotted onto nylon membranes (Boehringer Mannheim, Indianapolis, USA). After UV cross‐linking, the filters were hybridized at 65 °C overnight in 0.5 m sodium phosphate, 7% SDS with 32P‐labelled cDNA probes and washed in 2 × SSC, 0.1% SDS for 10 min, followed by one wash of 15 min in 0.2 × SSC, 0.1% SDS. The membranes were visualized using Phosphorimager (Molecular Dynamics, Sunnyvale, CA, USA) and AGFA X‐ray film. cDNA probes for C/EBPα and C/EBPβ were obtained from the UK HGMP Resource Centre Winston, Cambridge, UK and aP2 cDNA probe was provided by Dr S. Butterwith.
RT‐PCR and QPCR analysis
Total RNA from control and Raidd‐transfected 3T3L1 cells was isolated 8 days after hormonal induction. Five micrograms of total RNA (DNase‐treated) was used to produce cDNA using the SuperScript ™ II RNase H− reverse transcriptase and the conditions given by the provider (Gibco BRL, Rockville, MD, USA). The reverse transcription was performed at 42 °C for 50 min and heated at 70 °C for 15 min to inactivate the reverse transcriptase. Ten per cent of this final reaction was used in a standard polymerase chain reaction (PCR). The cDNA primers for Adipsin were forward (5′‐ATGACGACTCTGTGCAGGTG‐3′) and reverse (5′‐GTATAGACGCCCGGCTTTTT‐3′), whereas the cDNA primers for exogenous Raidd were forward (5′‐AGCACCCTCCTGTGCAG‐3′) that lies in the aP2 promoter, and a reverse primer (5′‐GCGAATGCACGTTGTGGGGA‐3′) corresponding to an internal sequence of Raidd. Relative quantification of exogenous RAIDD mRNA and GAPDH mRNA was carried out using Real‐Time Quantitative PCR (QPCR) (Higuchi et al. 1993).
RNA was subjected to Dnase 1 treatment and cDNA synthesized using the First Strand cDNA synthesis kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The ABI Prism 7700 Sequence Detector was used to monitor the accumulation of the PCR product by measuring the increase in fluorescence resulting from the binding of SYBER Green to double‐stranded DNA. The SYBER Green PCR Core Reagents kit (ABI, Warrington, Cheshire, UK) including the internal reference dye, ROX, was used for all QPCR reactions.
Oil Red O staining
Oil Red O staining was performed according to the method described by Green & Kehinde (1974). Cells were washed twice with PBS and fixed with 10% formalin in PBS for 15 min. After two washes in PBS, cells were stained for approximately 1 h in freshly diluted Oil Red O solution (six parts Oil Red O stock solution in isopropanol). The stain was then removed and the cells washed twice with water and then photographed.
RESULTS
Overexpression of murine Raidd cDNA blocks differentiation of 3T3L1 cells into adipocytes
3T3L1 cells can be induced to differentiate in the presence of agents that promote differentiation such as dexamethasone, insulin and methylisobutylxanthine (Student et al. 1980). Upon differentiation, the cells undergo a dramatic morphological change into adipocytes that results in a massive accumulation of cytosolic triglycerides by day 8 post induction (Fig. 1a). Clones of 3T3L1 cells transfected only with the backbone plasmid containing the antibiotic resistance marker neomycin, differentiated as the untransfected 3T3L1 cells (data not shown). In marked contrast, four out of six 3T3L1 clones (clones 2, 3, 5, 6) stably transfected with the murine Raidd cDNA, failed to differentiate and did not show accumulation of cytosolic triglycerides when treated under identical conditions as the parental cells (two clones shown in Fig. 1b and c). Upon examination by Oil Red O staining, which stains specifically the lipid droplets (Green & Kehinde 1974), parental 3T3L1 cells exhibited strong staining for lipid droplets (Fig. 1d) whereas the Raidd‐transfected clones showed no staining (Fig. 1e and f). Similar results were obtained in a repeated transfection experiment. No staining for lipid droplets was observed in the Raidd‐transfected 3T3L1 cells even 2 weeks post induction or when the induction protocol was initiated 4 or 6 days post‐confluence, instead of 2 days as recommended in the published differentiation protocol (Student et al. 1980).
Figure 1.

Differentiation of 3T3L1 cells into adipocytes upon hormonal induction and lack of differentiation in Raidd‐transfected 3T3L1 cells. (a, b, c) are photographs (100 × magnification) of cells 8 days after hormonal induction to differentiation. (a) shows parental 3T3L1 cells that differentiated normally after hormonal induction. In contrast, cells stably transfected with the murine Raidd cDNA did not undergo adipocyte differentiation in response to hormone treatment [(b) and (c) for clones 2 and 6, respectively]. (d, e, f) show cells stained with Oil Red O, which stains specifically the lipids. Dark staining is observed in the parental 3T3L1 cells (d) that accumulate triglycerides indicating appropriate differentiation into adipocytes. No staining is observed in the Raidd ‐transfected clones [(e, f), for clones 2 and 6, respectively] confirming the blockage of differentiation in these clones.
In two of six Raidd‐transfected clones (clones 1, 4) we observed partial differentiation into adipocytes, in that some morphological change from fibroblast‐like to rounded‐up cells was detected. One of these clones (clone 1) had shown reduced accumulation of cytoplasmic triglycerides, whereas in the other clone (clone 4), partial differentiation into adipocytes occurred, but was followed by pronounced cell death. Overall, four of six clones showed a complete blockage of differentiation with the other two exhibiting only partial differentiation. We conclude that transformation of 3T3L1 cells with the murine Raidd cDNA results in the block of differentiation into adipocytes upon induction of the differentiation programme.
Expression analysis of exogenous Raidd mRNA and adipsin, a marker of terminally differentiated adipocytes
Expression of exogenous Raidd cDNA was assessed by RT‐PCR and by real‐time QPCR. For RT‐PCR, total RNA of the six Raidd‐clones, after 8 days of the induction to differentiation, was used as a template. In order to detect specifically the expression of the exogenous Raidd DNA, a pair of primers was designed to amplify a region containing aP2 promoter (5′‐primer) and a reverse primer corresponding to an internal sequence of Raidd (3′‐primer, see Materials and methods). All transfected clones expressed the Raidd mRNA from the integrated construct (clones 1–6, Fig. 2), whereas, as expected, control parental cells before (Fig. 2, lane B) or after differentiation into adipocytes (lane A) showed no signal for the exogenous Raidd.
Figure 2.
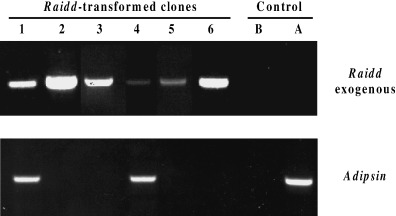
Raidd expression from the introduced construct and adipsin expression, a marker for differentiation. 5 µg of total RNA from cells 8 days after hormonal induction was DNAse treated, reverse transcribed with oligo (dT)12–18 mer (Gibco BRL) and subjected to PCR with primers (see Material and methods) specific for Raidd transcript from the introduced construct and adipsin. All six Raidd ‐transfected clones (1–6) express Raidd transcript from the construct. Adipsin, a marker of terminally differentiated adipocytes, is expressed only in Raidd ‐transfected clones 1 and 4 that partially differentiated, and in control cells 8 days after hormonal induction (A). No adipsin expression is detected in Raidd ‐clones exhibiting a complete blockage to differentiation (clones, 2, 3, 5, 6) and in control cells before hormonal induction (B).
For the real‐time QPCR, the same primers for exogenous Raidd expression as for RT‐PCR were used (see above). Expression of exogenous Raidd cDNA was assessed relative to Gapdh expression in clones 5 and 6 that showed a complete blockage to differentiation and control 3T3L1 cells. Analyses were carried out on cells at both 2 days prior to induction to differentiate (−2) and at 8 days post‐induction (+8). Expression of exogenous Raidd cDNA was evident in clones 5 and 6 both before and after induction, but undetectable in control samples. For individual clones, expression of exogenous Raidd cDNA relative to Gapdh did not change significantly as a result of the induction process, however, expression in clone 6 was approximately 100‐fold greater than that observed in clone 5 (clone 5: −2 = 3 × 10−5, +8 = 3.5 × 10−5; clone 6: −2 = 6 × 10−3, +8 = 1.2 × 10−2). These results confirm the RT‐PCR analysis (Fig. 2) that also showed a higher intensity of the amplified product in clone 6, suggesting a higher expression of exogenous Raidd cDNA in this clone.
Next, we examined if the block of differentiation in Raidd clones is associated with the lack of expression of the adipsin in cells 8 days after hormonal induction. Adipsin is a serine protease whose cDNA was isolated by differential screening of preconfluent and differentiated 3T3L1 cells (Cook et al. 1987). It is found in vivo in the circulation of both animals and humans and is produced and secreted in vitro by differentiated 3T3L1 cells, thus making this gene a marker for terminally differentiated adipocytes. RT‐PCR for adipsin produced a signal only in the two clones that partially differentiated into adipocytes (clones 1 and 4, Fig. 2) and, as expected, in the control parental cells (3T3L1) after differentiation into adipocytes (lane A, Fig. 2). In contrast, no induction of the expression of this gene was observed for the clones 2, 3, 5 and 6, thus confirming a complete block in the differentiation of these cells as observed in the morphological and Oil Red staining analysis (Fig. 1).
Overexpression of Raidd cDNA inhibits expression of various markers during adipogenesis
Adipocyte growth and differentiation follow patterns of sequential B‐ZIP protein expression, including AP‐1 (Distel et al. 1987) and C/EBP family proteins (McKnight et al. 1989). To examine if this pattern of sequential expression is affected in the Raidd‐transfected 3T3L1 clones, we conducted northern analysis of the transcription factors involved in the differentiation of 3T3L1 into adipocytes. RNA from two Raidd‐transfected clones (clones 5, 6, Fig. 3) and parental cells isolated before hormonal induction and 2 and 8 days after induction was analysed.
Figure 3.
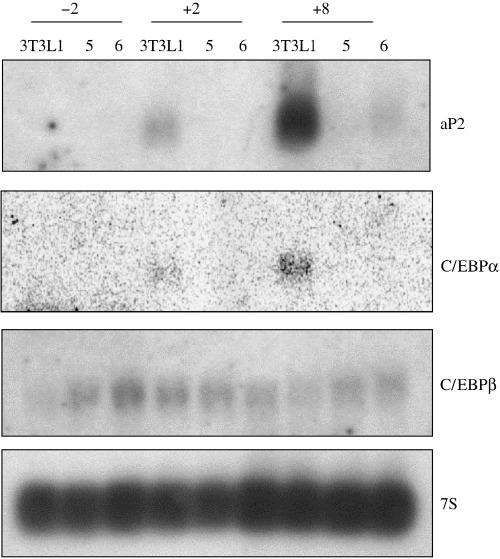
Raidd overexpression inhibits terminal markers of the adipogenic programme. Ten micrograms of total RNA from two Raidd‐ transfected clones and the parental cells 3T3L1 after different days of the differentiation programme were run on a denaturing agarose gel and northern blot hybridized with probes: aP2, C/EBPα, C/EBPβ and 7S as loading control.
In the control cells, expression of the mRNA encoding fatty acid binding‐protein aP2 (Bernlohr et al. 1984) was first detectable at low levels after the induction to differentiate (day 2) and rose to maximal levels by day 8. Control cells differentiated normally and accumulated triglyceride droplets in the cytoplasm of the cells (not shown). In the Raidd‐transfected clones, failure to differentiation was observed again and aP2 expression could not be detected in clone 5, whereas in clone 6 a very low signal after 8 days of hormonal induction was observed. This northern blot was rehybridized with the C/EBPα and C/EBPβ cDNA probes. Consistent with earlier studies done in 3T3L1 cells (Birkenmeier et al. 1989), C/EBPα transcript was not detected prior to the differentiation and was first detected at low levels on day 2 and increased at day 8 in the control cells. However, no signal was detected in the two Raidd‐transfected clones. In contrast, the mRNA encoding for C/EBPβ that is expected to be expressed in undifferentiated 3T3L1 cells was detectable in both control and Raidd‐transfected cells, and remained detectable during the later half of the differentiation programme, consistent with earlier studies (Cao et al. 1991). These findings show that the blockage in the differentiation of the Raidd‐transfected clones correlate with the lack of sequential expression of transcription factors during adipocyte differentiation such as C/EBPα and aP2.
Cell proliferation is not impaired in Raidd‐transfected clones
To assess if cell division was impaired in the Raidd‐transfected cells, the rate of proliferation of the cells was examined at different stages of growth, both before and after induction with hormones. Wild type 3T3L1 cells and Raidd‐transfected cells (clone 6) were plated with the same initial numbers on 6‐well dishes and cells were counted at confluence (−2), after induction with the hormones (+2) and after 6 days of hormonal induction. Figure 4 shows that the number of cells for 3T3L1 cells and Raidd‐transfected clone at confluence (−2) is essentially the same, indicating that overexpression of Raidd cDNA does not affect proliferation rate in the preadipocyte stage. After hormonal induction (Fig. 4, +2, +6) both groups increased the number of cells due to the clonal expansion induced by the combination of mitogenic and adipogenic signals provided in the differentiation medium. However, the number of cells was similar in both groups although parental 3T3L1 cells underwent differentiation into adipocytes, whereas the Raidd‐transfected clone remained at the preadipocyte stage. These results show that cell proliferation is not impaired in the Raidd‐transfected clone, either before or after hormonal induction, suggesting that inhibition of cell division does not cause the blockage to differentiation of Raidd‐transfected clones.
Figure 4.

Cell number quantification at different time points of the differentiation programme. Cell number of the parental cells 3T3L1 (empty bar) and one of the clones transfected with Raidd (solid bar) were counted at different points of the differentiation programme, including at confluence (−2) after hormonal induction (+2) and after 6 days of differentiation (+6). Each bar represents the mean ± SD of duplicates.
DISCUSSION
Transfection of 3T3L1 cells with the murine Raidd cDNA resulted in a complete blockage to differentiation into adipocytes in four of six clones, after exposure to the cocktail of hormones/inducers. In two of six clones, partial differentiation was observed with one clone showing a partial accumulation of triglycerides and the other showing a pronounced cell death of the partially differentiated adipocytes. It is possible that in the two partially differentiated Raidd‐transfected clones either the timing and/or the level of exogenous Raidd cDNA expression was different from the four completely blocked clones, such that it allowed the cells to partially differentiate. Even in these two clones, differentiation was not normal and did not proceed to completion. All of the Raidd‐transfected clones showed either blocked or impaired differentiation. Therefore we demonstrate that stable transformation of 3T3L1 cells with Raidd cDNA has an inhibitory effect on differentiation into adipocytes.
These findings show that overexpression of Raidd cDNA blocks the differentiation of 3T3L1 cells into adipocytes. The Raidd cDNA used was under the control of aP2 promoter (Hunt et al. 1986). The aP2 promoter does not support expression in preadipocytes and its maximum activity is reached after ∼6 days of the differentiation programme. However, Lin & Lane (1992) have observed a low level of aP2 mRNA by RNase protection analysis in confluent 3T3L1 preadipocytes, and we too have observed expression of a fluorescent reporter gene (GFP), driven by the aP2 promoter in preadipocytes (Felmer R. unpublished observations). Therefore, a low level of expression from this promoter can be observed earlier in the adipogenic programme even before transactivation from C/EBPα. Real‐time QPCR analysis also showed that the aP2 promoter in our transgene was already active in early preadipocytes. With QPCR we found that exogenous Raidd was expressed at the same level before and 8 days after induction in two of the Raidd‐transfected clones that failed to differentiate. It is possible that it is this sustained overexpression of exogenous Raidd cDNA, starting from early stage preadipocytes, that inhibits differentiation.
Overexpression of Raidd cDNA could block cell differentiation by perturbing one or several steps that are important for the terminally differentiated state. Raidd cDNA overexpression could be acting by stimulating the expression of anti‐differentiation genes or, alternatively, inhibiting the expression of pro‐differentiation genes. By analysing patterns of gene expression characteristic of each phase of the differentiation programme, it was determined that the Raidd‐mediated block in the differentiation of 3T3L1 cells seems to be manifested during the terminal stages of the differentiation in clones that failed to differentiate. Expression of a very early differentiation marker C/EBPβ was not altered in the Raidd clones, whereas the expression of differentiation marker C/EBPα– expressed during adipogenesis – and terminal stage‐specific markers aP2 and adipsin were absent or decreased.
Inhibition of the differentiation of 3T3L1 was demonstrated in other studies by anti‐sense C/EBPα technique (Lin & Lane 1992) that blocked the expression of C/EBPα. This in turn inhibited expression of aP2, SCD1 and GLUT4 and the accumulation of cytoplasmic triglycerides (Lin & Lane 1992), which confirmed that C/EBPα is required for preadipocyte differentiation. Induction of C/EBPβ and C/EBPδ expression is thought to be necessary for the subsequent induction of C/EBPα and PPARγ, which then act to co‐ordinate the expression of a number of fat specific genes required for lipid metabolism, including aP2, SCD1, and Glut4. In the Raidd clones that did not differentiate, C/EBPβ expression was not altered, but the expression of C/EBPα was undetectable. We can thus speculate that Raidd cDNA overexpression causes down regulation of pro‐differentiation proteins such as C/EBPα and possibly PPARγ (not analysed), preventing transactivation of the downstream targets in the differentiation pathway of adipocytes.
Our study demonstrated that overexpression of Raidd cDNA represses adipogenesis, indicating that Raidd may function, directly or indirectly, in differentiation. It would therefore be of interest to test in future if lack of Raidd function can promote preadipocyte differentiation to confirm our current speculation that Raidd functions as a suppressor of adipocyte differentiation. Raidd contains an amino‐terminal CARD region and a carboxy‐terminal ‘death domain’ (Duan & Dixit 1997) and has been so far only implicated to act as an adapter protein in the apoptotic signalling pathway. Our results suggest that Raidd does not function only in apoptosis but could also be involved in differentiation. One caveat, however, is that Raidd may not play a role in differentiation under physiological condition, with the effects observed in our study only occurring at high levels of exogenous gene expression.
Several studies have shown that the inhibition of cell division of cultured preadipocyte clearly blocks their subsequent differentiation (Timchenko et al. 1996). Nevertheless, we found no evidence of impaired cell division in these clones transfected with Raidd, as analysis of the proliferation rate of Raidd‐transfected clone throughout the different steps of the differentiation programme showed no significant difference with the control cells. Furthermore, blocked cells proliferated normally even after confluent dishes that had been treated with the hormones were plated out at lower densities, suggesting that apoptosis was not triggered in them.
In summary, Raidd cDNA overexpression inhibits 3T3L1 preadipocyte differentiation as demonstrated by cell morphology, lipid accumulation, and expression of adipocyte‐specific markers. The differentiation process in Raidd‐transfected clones is associated with inhibition of expression of differentiation factor C/EBPα and late‐stage differentiation markers aP2 and adipsin. As RAIDD protein expression was not conducted in the present study, further studies are required to examine endogenous and exogenous protein levels before and after hormonal induction, so as to provide additional information regarding the mechanism of action. Such studies should shed light on the mechanism of how the overexpression of Raidd cDNA induces the blockage to preadipocyte differentiation, as well as determining whether the endogenous Raidd gene is involved in the regulation of differentiation in preadipocytes or other cell types.
ACKNOWLEDGEMENTS
We would like to thank Claire Neil for technical assistance and Richard Meehan for helpful discussion. The financial support of INIA, Chile (National Institute of Agriculture Research), and the Biotechnology and Biological Sciences Research Council UK, are gratefully acknowledged.
REFERENCES
- Bernlohr DA, Angus CW, Lane MD, Bolanowski MA, Kelly TJ (1984) Expression of specific mRNAs during adipose differentiation: identification of an mRNA encoding a homologue of myelin P2 protein. Proc. Natl Acad. Sci. USA 81, 5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeier EH, Gwynn B, Howard S, Jerry J, Gordon JI, Landschulz WH, McKnight SL (1989) Tissue‐specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 3, 1146. [DOI] [PubMed] [Google Scholar]
- Cao Z, Umek RM, McKnight SL (1991) Regulated expression of three C/EBP isoforms during adipose conversion of 3T3‐L1 cells, Genes Dev. 5, 1538. [DOI] [PubMed] [Google Scholar]
- Cohen GM (1997) Caspases: the executioners of apoptosis. Biochem. J. 326, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, Spiegelman BM (1987) Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science 237, 402. [DOI] [PubMed] [Google Scholar]
- Distel R., Ro H, Rosen B, Groves D, Spiegelman B (1987) Nucleoprotein complexes that regulate gene expression in adipocyte differentiation: direct participation of c‐fos . Cell 49, 835–844. [DOI] [PubMed] [Google Scholar]
- Duan H, Dixit VM (1997) RAIDD is a new ‘death’ adaptor molecule. Nature 385, 86. [DOI] [PubMed] [Google Scholar]
- Green H, Kehinde O (1974) Sublines of mouse 3T3 cells that accumulate lipid. Cell 1, 113. [Google Scholar]
- Green H, Meuth M (1974) An established pre‐adipose cell line and its differentiation in culture. Cell 2, 127. [DOI] [PubMed] [Google Scholar]
- Higuchi R., Fockler C, Dollinger G, Watson R (1993) Kinetic RCR: real‐time monitoring of DNA amplification reactions. Bio/Technology 11, 1026. [DOI] [PubMed] [Google Scholar]
- Horvat S, Medrano JF (1998) A 500‐kb YAC and BAC contig encompassing the high‐growth deletion in mouse chromosome 10 and identification of the murine Raidd/Cradd gene in the candidate region. Genomics 54, 159. [DOI] [PubMed] [Google Scholar]
- Hunt CR, Ro JH, Dobson DE, Min HY, Spiegelman BM (1986) Adipocyte P2 gene: developmental expression and homology of 5′‐flanking sequences among fat cell‐specific genes, Proc. Natl Acad. Sc.I USA 83, 3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FT, Lane MD (1992) Antisense CCAAT/enhancer‐binding protein RNA suppresses coordinate gene expression and triglyceride accumulation during differentiation of 3T3‐L1 preadipocytes. Genes & Dev. 6, 533. [DOI] [PubMed] [Google Scholar]
- McKnight S, Lane M, Gluecksohn‐Welsch S (1989) Is CCAAT/enhancer‐biding protein a central regulator of energy metabolism? Genes Dev. 3, 2021. [DOI] [PubMed] [Google Scholar]
- Ross SR, Graves RA, Greenstein A, Platt KA, Shyu HL, Mellovitz B, Spiegelman BM (1990) A fat‐specific enhancer is the primary determinant of gene expression for adipocyte P2 i. Proc. Natl Acad. Sci. USA 87, 9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual, 2nd edn New York: Cold Spring. Harbour Laboratory Press. [Google Scholar]
- Student AK, Hsu RY, Lane MD (1980) Induction of fatty acid synthetase synthesis in differentiating 3T3‐L1 preadipocytes. J. Biol. Chem. 255, 4745. [PubMed] [Google Scholar]
- Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ (1996) CCAAT/enhancer‐binding protein alpha (C/EBP alpha) inhibits cell proliferation through the p21 (WAF‐1/CIP‐1/SDI‐1) protein. Genes Dev. 10, 804. [DOI] [PubMed] [Google Scholar]