

Summary
Although the neurovascular unit was originally developed as a conceptual framework for stroke, it is now recognized that these cell–cell interactions play critical roles in many other CNS disorders as well. In brain trauma, perturbations within the neurovascular unit may be especially important. Changes in neurovascular coupling may disrupt blood flow and metabolic regulation. Disruption of transmitter release‐reuptake kinetics in neurons and astrocytes may augment excitotoxicity. Alterations in gliovascular signaling may underlie blood–brain barrier disruptions and traumatic edema. Perturbations in cell–cell signaling between all neuronal, glial, and vascular compartments may increase susceptibility to cell death. Finally, repairing the brain after trauma requires the integrated restoration of all neural, glial, and vascular connectivity for effective functional recovery. Just as in stroke, saving neurons alone may also be insufficient for treating brain trauma. In this minireview, we attempt to briefly highlight some of these pathways to underscore the importance of rescuing the entire neurovascular unit in brain trauma.
Keywords: Endothelial, Glia, Neuron, Neuroprotection, Traumatic brain injury
Introduction
Over the past two decades, various molecular mechanisms of neuronal death after traumatic brain injury (TBI) have been dissected by many groups. These mechanisms often involve complex combinations of necrosis, apoptosis, necroptosis, and autophagy 1, 2. Defining these pathways offer the hope that effective therapeutic targets can be identified. However, it is now increasingly recognized that in addition to neuronal death, responses in glial and vascular compartments should also play a key role in the progression of secondary injury after TBI. Indeed, when TBI patients are in the intensive care unit, major clinical challenges are often centered on cerebrovascular instability perhaps related to glial swelling, blood–brain barrier leakage, and disruptions in flow‐metabolism regulation. Taken together with emerging data in a wide range of experimental models, it is likely that developing therapies for brain trauma should require a careful rescue of the entire neurovascular unit, comprising interactions between all cell types in the damaged brain.
Cerebral Blood Flow and Metabolism
After TBI, the regulation of cerebral blood flow and metabolism can become disordered in dispersed brain regions, with the suppression of glucose metabolism along with reductions in cerebral blood flow (CBF). However, it is unclear whether metabolism is reduced because of insufficient flow or flow is reduced because of decreased demand in injured neurons. Evidence for actual “ischemia” post‐TBI remains rare and controversial 3. But it is likely that simultaneous disruption in neuronal metabolism and vascular flow occurs.
In terms of organ injury, CBF that is too low or too high can both be deleterious. Hyperperfusion may lead to increased capillary leakage and/or bleeding. Hypoperfusion may lead to dangerous metastable states of oligemia or, if CBF levels drop far enough, may even trigger outright ischemia in already vulnerable and traumatized brain tissue. It has also been proposed that CBF response after TBI is nonlinear, and acute hypoperfusion can be followed by secondary hyperperfusion 4. Finally, traumatic vasospasm may also occur, leading to delayed hypoperfusion again at even later timepoints 4. If true, then targeting these multiphasic CBF profiles after TBI may be extremely challenging.
From a functional viewpoint, these perturbations in metabolism and CBF may be due in part to traumatic effects on autoregulation. The brain is a high‐energy organ. So, autoregulation provides a critical mechanism for maintaining balanced levels of energy supply and demand. Once the system is damaged by trauma, autoregulation may be impaired, although autoregulatory failure may not always be correlated with injury severity 5. In rodent models of TBI, CBF may no longer be able to respond to changes in blood pressure, hyperventilation, or hypercapnia 6, 7, 8. The underlying mechanisms will surely be complex and multifactorial. For example, TBI can trigger widespread microthrombosis in a disseminated manner, either due to activation of coagulation cascades in blood or upregulation of adhesion molecules in affected endothelium 9. Recent studies further suggest that endothelin signaling may be involved. Endothelin receptor antagonists may be able to prevent upregulation of vasoconstrictive smooth muscle actin in microvessel pericytes post‐TBI 10. Regardless of the exact molecular mechanisms involved, data from many experimental models now strongly suggest that dispersed and widespread alterations in microvascular perfusion occur after trauma.
Although the spatiotemporal profiles and underlying mechanisms of CBF response after TBI are complex and remain to be fully elucidated, many studies have suggested that in human patients, CBF‐metabolic coupling can sometimes predict long‐term outcomes 11, 12. Hence, finding ways to ameliorate CBF and metabolic dysregulation should be an important therapeutic direction for TBI. Because CBF metabolism function is fundamentally regulated by the entire neurovascular unit, this simply underscores the importance of targeting all neural, glial, and vascular elements to treat traumatized brain.
Blood–Brain Barrier
Perturbations in BBB function have been extensively documented after TBI in both experimental models and human patients 13. From a clinical perspective, BBB leakage is most often associated with edema and brain swelling, which may lead to detrimental elevations in intracranial pressure (ICP) and corresponding reductions in cerebral perfusion pressure (CPP) 14. From a molecular perspective, BBB permeability can be affected by TBI in multifactorial ways 13, 15. Many BBB‐modifying factors can be upregulated after injury, spanning the range from damaging free radicals and inflammatory cytokines to BBB gene modifiers and neurovascular matrix proteases. Additionally, after TBI, mechanical impact may induce immediate BBB damage; microbleeding, vascular inflammation, and secondary ischemia/hypoxia may induce prolonged BBB dysfunction 13, 15, 16.
TBI is known to enhance the generation of reactive oxygen species (ROS) that can damage cerebral endothelium 17. Downregulation of tight junction proteins has also been described in various models of TBI 16. Under some conditions, TBI may upregulate matrix metalloproteinases (MMPs) that degrade both tight junction proteins and associated neurovascular matrix substrates 18. Correspondingly, a wide spectrum of therapeutic approaches may protect the BBB post‐trauma, at least in experimental model systems. For example, free radical scavengers reduced Evans blue leakage in rat‐controlled cortical impact 19, blocking the effects of inflammatory cytokines with neuregulin appears to decrease endothelial tight junction permeability 20, and deleting the gene for MMP9 improved outcomes in transgenic mice 21.
Ultimately, the BBB phenotype is dependent on cell–cell signaling between neuronal, glial, and vascular cell types. Hence, BBB disruptions after TBI may be best interpreted as an integrative response within the entire neurovascular unit. Emerging findings now demonstrate that astrocytes and pericytes are essential for promoting BBB maturation in cerebral endothelium 22, 23, 24. Cell–cell signaling mechanisms in this regard are complex, but broadly involve TGF‐beta cascades 25. More recently, it has also been proposed that oligodendrocyte precursor cells (OPCs) may also contribute to BBB function 26. Hence, any disruption of neurovascular unit signaling may end up interrupting vital cross talk between cerebral endothelium and associated astrocytes, pericytes, and OPCs, thus affecting BBB homeostasis after TBI.
Cell–Cell Signaling Within the Neurovascular Unit
As a conceptual construct, the neurovascular unit emphasizes the importance of cell–cell signaling between all cells in neural, glial, and vascular compartments 27. Historically, the cell type most recognized for its neurosupportive function is the brain astrocyte. Without astrocytes, release‐reuptake kinetics for various neurotransmitters cannot be refined for effective neurotransmission. Any disruption of release‐reuptake profiles will surely affect neurotransmitter networks and overall brain function after trauma. Furthermore, because dysregulated glutamate handling may be a key event in TBI, rescuing astrocyte function may represent a logical target for ameliorating secondary excitotoxicity in traumatized brain tissue. Proof‐of‐concept studies using microdialysis demonstrated that extracellular glutamate levels are rapidly elevated following traumatic injury 28. Simple cell culture studies demonstrate that the presence of astrocytes can defend neurons against glutamate overload 29. Hence, upregulating astrocytic glutamate transporters may represent a potential therapeutic target for neuroprotection 30. In addition to buffering glutamate imbalance, astrocytes may also be neuroprotective via antioxidative mechanisms as these cells can express relatively high levels of antioxidant genes such as superoxide dismutase and glutathione 31. Because excitotoxic and oxidative injury are key components of TBI pathophysiology, targeting astrocytes may be an important direction for pursuing future therapeutic approaches.
An essential part of cell–cell communication in the neurovascular unit comprises gliovascular signaling. Without proper signaling between the astrocytic endfeet and microvessel endothelium, BBB function cannot be maintained. After trauma, astrocyte dysfunction is a major event that underlies BBB leakage and vasogenic edema 32. Some reports suggested that aquaporin channels may underlie some of these pathophysiological responses in TBI 33. Furthermore, outright astrocyte swelling may also contribute to cytotoxic forms of brain edema 34, and targeting different types of astrocyte channels may prevent cytotoxic edema 35. More recently, pericytes have also been recognized as being important contributors to CNS function and dysfunction. In developing brain, pericytes assist with BBB maturation 36. In adult brains, pericytes may assist with vasoregulation for CBF control 37. Hence, changes in pericyte signaling after TBI may further disturb BBB and CBF regulation. Altogether, disruption of signaling between astrocytes, pericytes, and endothelium may underlie gliovascular pathophysiology that further exacerbates secondary injury.
Another important glial reaction to injury comprises microglial activation. Activation of these resident CNS inflammatory cells is well documented in TBI. In animal models, microglia can release a wide spectrum of neurotoxic cytokines that further contribute to neuroinflammation and cell death 38. In humans, rapid microglia activation occurs within hrs and can even persist for up to months or years after injury 39, 40. Because microglia are known to play a role in pruning and maintaining synaptic homeostasis, it is attractive to hypothesize that dysfunctional microglia can contribute to prolonged neuronal dysfunction in TBI. Although blocking microglia may offer therapeutic opportunities for TBI, it is now appreciated that not all of these cells are damaging, and biphasic properties may emerge in injured brain tissue as part of an endogenous inflammatory/wound‐healing response. Experimental findings now suggest that deleterious M1‐like phenotypes can be contrasted against prorecovery M2‐like phenotypes in CNS disorders 41. Defining the pathways that regulate the microglial switch may be an important target for TBI.
Although glial interactions are well dissected in gray matter, it is increasingly recognized now that analogous cell–cell signaling pathways may be essential for understanding function and dysfunction in white matter. In TBI, white matter damage is especially important. In this regard, oligodendrocyte precursor cells (OPCs) may be significant targets for TBI. OPCs are thought to comprise a general stem/progenitor pool for maintaining mature oligodendrocyte populations in white matter 42. Therefore, TBI‐mediated damage to these cells may compromise the ability of damaged white matter to heal. Furthermore, OPCs may also support BBB function 26 , so any disruption of oligovascular cross talk may lead to edema in traumatized white matter.
After TBI, severely injured neurons in the core die rapidly. Then, over the course of hrs to days or even weeks, secondary pathways of injury often occur. The standard hypothesis and hope has been that blocking intraneuronal mechanisms of cell death should be the logical therapeutic target. Increasingly, however, both clinical and experimental data now strongly suggest that much of the pathophysiology of TBI is not only due to neurons dying, but also mediated by perturbations in cell–cell signaling in the entire neurovascular unit. Hence, restoring the homeostasis of these signals should offer important therapeutic directions in TBI.
Remodeling the Neurovascular Unit
The concept of the neurovascular unit has been mostly applied in the context of acute brain injury and neurodegeneration. However, it is increasingly recognized that analogous cell–cell cross talk between neural, glial, and vascular elements is absolutely required for repair and remodeling as well 43, 44. In the mature mammalian brain, neurogenesis is mostly inactive, except for pockets of ongoing neuroblast activity in the subventricular and subgranular zones. After TBI, neurogenesis may be increased in these areas as the damaged brain attempts to heal itself. In experimental models of cortical impact or fluid percussion injury, neuroblasts can be observed to migrate from these subventricular and subgranular regions toward traumatized areas 45. Recently, emerging data now support the notion that after acute brain injury from either stroke or trauma, neurogenesis and angiogenesis appear to be tightly co‐regulated, especially during the recovery phase. Migrating neuroblasts move along perivascular pathways 46. Promoting neurogenesis seems to augment angiogenesis and vice versa 47. Indeed, many mediators that assist with neurogenesis and neural patterning may also assist with angiogenesis and vessel guidance as well, including semaphorins, netrins, VEGF, and BDNF 48.
A prominent example of multicellular cross talk in the neurovascular unit involves the interactions between the brain microvessel and surrounding glia (astrocytes, pericytes, and OPCs) that support BBB maturation. During recovery, BBB function also needs to be restored. But this may require careful attention to the biphasic effects of some neurovascular and gliovascular signals. For example, while VEGF promotes potentially beneficial angiogenesis, incorrect timing of VEGF signals may increase BBB leakage and worsen edema 49. As a general principle, many mediators that are activated after TBI may follow this biphasic rule, that is, deleterious in the acute phase while beneficial in the delayed phase. In this regard, the neurovascular unit may provide a conceptual framework for defining the shifting profiles of pathophysiology over time.
Beyond CBF and BBB function, cerebral microvessels may also subserve many other CNS functions. Recently, the concept of the vasculome has been proposed, which states that the cerebral endothelium may not just comprise “empty pipes” for blood flow 50. Instead, the entire microvessel network may represent an intricate paracrine organ that supports neuronal function and homeostasis 51, 52. Hence, protecting the vasculome in TBI may ultimately defend neurons against cell death as well as augment neurorecovery as the entire neurovascular unit attempts to rebuild itself. Furthermore, the cerebral microvascular system may act as sensors and integrators of brain injury and dysfunction, releasing measurable signals into the circulation 53. Therefore, mining the vasculome may also provide a database for TBI biomarkers 50.
Plastic cross talk can also be found between neurons and astrocytes. Regulation of neurotransmitter release–reuptake kinetics cannot take place without coordinated interactions between neurons and astrocytes. During the remodeling phase post‐TBI, astrocytic cross talk may be important as neuronal circuits are reconstructed. Traditionally, reactive astrocytes are thought to be deleterious because they underlie the inhibitory glial scar 31, 32. However, emerging data now suggest that their roles may be more complex. Under some conditions, astrocytes can be helpful for brain repair 31, 32. Transgenic mice that suppress astrocyte GFAP activation surprisingly show improved outcomes after cortical impact injury 54. The underlying mechanisms may be complex. For example, astrocytes release thrombospondin‐1 which promotes synaptic function and maturation 55, and tissue plasminogen activator which contributes to dendritic plasticity in recovering neurons 56. Indeed, several studies have suggested that the therapeutic benefits of stem cell therapies may depend in part on the ability of astrocytes to amplify the effects on neuronal remodeling 57. Ultimately, the roles of activated glia will surely be complex comprising both damaging and beneficial effects over time as the entire neurovascular unit attempts to heal itself.
For the large human brain, what happens in white matter may be even more important. Emerging data now suggest that besides a neurovascular niche, an analogous oligovascular niche may also exist. Astrocytes and cerebral endothelial cells secrete many trophic factors that support oligodendrocyte precursor cells and vice versa 58. And after injury, vascular endothelial growth factor (VEGF)‐mediated endothelial recovery is linked with the proliferation and migration of oligodendrocyte precursor cells 59. Oligodendrogenesis and angiogenesis are intricately linked. Without proper remodeling in white matter, the recovering brain cannot reconnect.
Finally, accumulating data is beginning to suggest that non‐cell‐autonomous mechanisms may extend beyond the narrow confines of the CNS itself. The neurovascular unit does not stand in isolation. It is in constant communication with the entire systemic physiology as well. For example, peripheral tissue injury and inflammation can alter BBB function 60. After focal brain injury, reactive astrocytes can also release HMGB1 as a signal to promote endothelial progenitors for neurovascular remodeling 61. After brain injury, detectable responses should be present in circulating blood cells and components. Beyond the obvious implications for biomarkers, these systemic blood responses may also have therapeutic implications. Modifying circulating blood responses may potentially influence neuroinflammation and secondary brain pathophysiology. Cross talk between multiple cell types in CNS and non‐CNS compartments will be exceedingly complex (Figure 1). In the end, a systems biology approach may be required to truly dissect the mechanisms involved.
Figure 1.
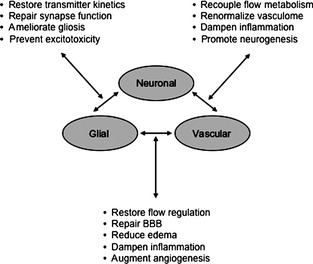
Cross talk between multiple cell types in CNS and non‐CNS compartments after TBI. After TBI, perturbations in cell–cell signaling between all neuronal, glial, and vascular compartments may increase susceptibility to cell death. Changes in neurovascular coupling may disrupt blood flow and metabolic regulation. Disruption of transmitter release‐reuptake kinetics in neurons and astrocytes may augment excitotoxicity. Alterations in gliovascular signaling may underlie blood–brain barrier disruptions and traumatic edema. Finally, repairing the brain after trauma requires the integrated restoration of all neural, glial, and vascular connectivity for effective functional recovery.
Conclusions and Opportunities
Neuronal death provides a cell biology correlate for neurological dysfunction after TBI. However, emerging data in both experimental models and patients now clearly demonstrate that a singular focus on neurons alone may not be enough. Pure neuroprotection (even if it is effective) may not translate into improved outcomes. What may be required is a rigorous consideration of the entire neurovascular unit. In this minireview, we attempted to briefly survey and highlight examples of cell–cell signaling phenomenon relevant for neuronal, glial, and vascular responses after TBI. Targeting these multicellular interactions may provide opportunities for developing therapeutics for ameliorating acute injury as well as promoting delayed repair.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The study is based in part on ideas and sections drawn from Lok et al., Neurochem Res 2006; Xing et al., Neurol Res 2012; Lok et al., Vasc Mech CNS Trauma 2014; and Wang et al., Vasc Mech CNS Trauma 2014 and supported in part by grants from NIH, AHA and the Rappaport Foundation.
References
- 1. Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol 2004;14:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 2010;31:596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: A combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab 2005; 25:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin NA, Patwardhan RV, Alexander MJ, et al. Characterization of cerebral hemodynamic phases following severe head trauma: Hypoperfusion, hyperemia, and vasospasm. J Neurosurg 1997;87:9–19. [DOI] [PubMed] [Google Scholar]
- 5. Golding EM, Robertson CS, Bryan RM Jr. The consequences of traumatic brain injury on cerebral blood flow and autoregulation: A review. Clin Exp Hypertens 1999;21:299–332. [DOI] [PubMed] [Google Scholar]
- 6. Prat R, Markiv V, Dujovny M, Misra M. Failure of cerebral autoregulation in an experimental diffuse brain injury model. Acta Neurochir Suppl 1998;71:123–126. [DOI] [PubMed] [Google Scholar]
- 7. Forbes ML, Hendrich KS, Schiding JK, et al. Perfusion MRI assessment of cerebral blood flow and CO2 reactivity after controlled cortical impact in rats. Adv Exp Med Biol 1997;411:7–12. [DOI] [PubMed] [Google Scholar]
- 8. Engelborghs K, Haseldonckx M, Van Reempts J, et al. Impaired autoregulation of cerebral blood flow in an experimental model of traumatic brain injury. J Neurotrauma 2000;17:667–677. [DOI] [PubMed] [Google Scholar]
- 9. Halpern CH, Reilly PM, Turtz AR, Stein SC. Traumatic coagulopathy: The effect of brain injury. J Neurotrauma 2008;25:997–1001. [DOI] [PubMed] [Google Scholar]
- 10. Dore‐Duffy P, Wang S, Mehedi A, et al. Pericyte‐mediated vasoconstriction underlies TBI‐induced hypoperfusion. Neurol Res 2011;33:176–186. [DOI] [PubMed] [Google Scholar]
- 11. Kelly DF, Martin NA, Kordestani R, et al. Cerebral blood flow as a predictor of outcome following traumatic brain injury. J Neurosurg 1997;86:633–641. [DOI] [PubMed] [Google Scholar]
- 12. Glenn TC, Kelly DF, Boscardin WJ, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: Indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab 2003;23:1239–1250. [DOI] [PubMed] [Google Scholar]
- 13. Chodobski A, Zink BJ, Szmydynger‐Chodobska J. Blood‐brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res 2011;2:492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience 2004;129:1021–1029. [DOI] [PubMed] [Google Scholar]
- 15. Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr Opin Neurol 2010;23:293–299. [DOI] [PubMed] [Google Scholar]
- 16. Alves JL. Blood‐brain barrier and traumatic brain injury. J Neurosci Res 2014;92:141–147. [DOI] [PubMed] [Google Scholar]
- 17. Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta 2012;1822:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma 1995;12:833–842. [DOI] [PubMed] [Google Scholar]
- 19. Smith SL, Andrus PK, Zhang JR, Hall ED. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood‐brain barrier disruption following unilateral cortical impact head injury in the rat. J Neurotrauma 1994;11:393–404. [DOI] [PubMed] [Google Scholar]
- 20. Lok J, Zhao S, Leung W, et al. Neuregulin‐1 effects on endothelial and blood‐brain‐barrier permeability after experimental injury. Transl Stroke Res 2012;3(Suppl 1):S119–S124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Jung J, Asahi M, et al. Effects of matrix metalloproteinase‐9 gene knock‐out on morphological and motor outcomes after traumatic brain injury. J Neurosci 2000; 20:7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia 2013;61:1939–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abbott NJ. Astrocyte‐endothelial interactions and blood‐brain barrier permeability. J Anat 2002;200:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci 2011;14:1398–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beck K, Schachtrup C. Vascular damage in the central nervous system: A multifaceted role for vascular‐derived TGF‐beta. Cell Tissue Res 2012;347:187–201. [DOI] [PubMed] [Google Scholar]
- 26. Seo JH, Maki T, Maeda M, et al. Oligodendrocyte precursor cells support blood‐brain barrier integrity via TGF‐beta signaling. PLoS ONE 2014;9:e103174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lok J, Gupta P, Guo S, et al. Cell‐cell signaling in the neurovascular unit. Neurochem Res 2007; 32:2032–2045. [DOI] [PubMed] [Google Scholar]
- 28. Vespa P, Prins M, Ronne‐Engstrom E, et al. Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: A microdialysis study. J Neurosurg 1998;89:971–982. [DOI] [PubMed] [Google Scholar]
- 29. Rosenberg PA, Aizenman E. Hundred‐fold increase in neuronal vulnerability to glutamate toxicity in astrocyte‐poor cultures of rat cerebral cortex. Neurosci Lett 1989;103:162–168. [DOI] [PubMed] [Google Scholar]
- 30. Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 2006;48:394–403. [DOI] [PubMed] [Google Scholar]
- 31. Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 2010;7:494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shields J, Kimbler DE, Radwan W, Yanasak N, Sukumari‐Ramesh S, Dhandapani KM. Therapeutic targeting of astrocytes after traumatic brain injury.Transl Stroke Res 2011;2:633–642. [DOI] [PubMed] [Google Scholar]
- 33. Ren Z, Iliff JJ, Yang L, et al. ‘Hit & Run’ model of closed‐skull traumatic brain injury (TBI) reveals complex patterns of post‐traumatic AQP4 dysregulation. J Cereb Blood Flow Metab 2013;33:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus 2007;22:E1. [DOI] [PubMed] [Google Scholar]
- 35. Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004;129:877–896. [DOI] [PubMed] [Google Scholar]
- 36. Zlokovic BV. The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron 2008;57:178–201. [DOI] [PubMed] [Google Scholar]
- 37. Itoh Y, Suzuki N. Control of brain capillary blood flow. J Cereb Blood Flow Metab 2012;32:1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: A prime target for Alzheimer's disease prevention. J Neuroinflammation 2012;9:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Engel S, Schluesener H, Mittelbronn M, et al. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage‐related proteins MRP8 and MRP14. Acta Neuropathol 2000;100:313–322. [DOI] [PubMed] [Google Scholar]
- 40. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol 2011;70:374–383. [DOI] [PubMed] [Google Scholar]
- 41. Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: The good, the bad, and the resting. J Neuroimmune Pharmacol 2009;4:389–398. [DOI] [PubMed] [Google Scholar]
- 42. Boulanger JJ, Messier C. From precursors to myelinating oligodendrocytes: Contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 2014;269:343–366. [DOI] [PubMed] [Google Scholar]
- 43. Xing C, Hayakawa K, Lok J, Arai K, Lo EH. Injury and repair in the neurovascular unit. Neurol Res 2012;34:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disord Drug Targets 2013;12:302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu D, Mahmood A, Qu C, Goussev A, Schallert T, Chopp M. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma 2005;22:1011–1017. [DOI] [PubMed] [Google Scholar]
- 46. Thored P, Wood J, Arvidsson A, Cammenga J, Kokaia Z, Lindvall O. Long‐term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke 2007;38:3032–3039. [DOI] [PubMed] [Google Scholar]
- 47. Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- 48. Liu J, Wang Y, Akamatsu Y, et al. Vascular remodeling after ischemic stroke: Mechanisms and therapeutic potentials.Prog Neurobiol 2014;115:138–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nag S. The blood‐brain barrier and cerebral angiogenesis: Lessons from the cold‐injury model. Trends Mol Med 2002;8:38–44. [DOI] [PubMed] [Google Scholar]
- 50. Guo S, Zhou Y, Xing C, et al. The vasculome of the mouse brain. PLoS ONE 2012;7:e52665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guo S, Som AT, Waeber C, Lo EH. Vascular neuroprotection via TrkB‐ and Akt‐dependent cell survival signaling. J Neurochem 2012;123(Suppl 2):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guo S, Kim WJ, Lok J, et al. Neuroprotection via matrix‐trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A 2008;105:7582–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ning M, Sarracino DA, Kho AT, et al. Proteomic temporal profile of human brain endothelium after oxidative stress. Stroke 2011;42:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006;129(Pt 10):2761–2772. [DOI] [PubMed] [Google Scholar]
- 55. Christopherson KS, Ullian EM, Stokes CC, et al. Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell 2005;120:421–433. [DOI] [PubMed] [Google Scholar]
- 56. Xin H, Li Y, Shen LH, et al. Increasing tPA activity in astrocytes induced by multipotent mesenchymal stromal cells facilitate neurite outgrowth after stroke in the mouse. PLoS ONE 2010;5:e9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li Y, Liu Z, Xin H, Chopp M. The role of astrocytes in mediating exogenous cell‐based restorative therapy for stroke. Glia 2014;62:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arai K, Lo EH. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci 2009;29:4351–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hayakawa K, Pham LD, Som AT, et al. Vascular endothelial growth factor regulates the migration of oligodendrocyte precursor cells. J Neurosci 2011;31:10666–10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCaffrey G, Staatz WD, Sanchez‐Covarrubias L, et al. P‐glycoprotein trafficking at the blood‐brain barrier altered by peripheral inflammatory hyperalgesia. J Neurochem 2012;122:962–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high‐mobility group box 1 promotes endothelial progenitor cell‐mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A 2012;109:7505–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]