

Abstract
Objectives
Enhancer of zeste homologue 2 (EZH2) is crucially involved in epigenetic silencing by acting as a histone methyltransferase. Although EZH2 is overexpressed in many cancers and is involved in malignant cell proliferation and invasion, the role of EZH2 in senescence induced by DNA damage has up to now remained largely unknown. In this study, we sought to explore the outcome of EZH2 depletion along with exposure of doxorubicin (DOX), and related mechanisms, in gastric cancer cells.
Materials and methods
Here, senescence induced by DNA damage was achieved in gastric cancer cells by DOX treatment. EZH2 was downregulated by transfection with siRNA or treated with (‐)‐epigallocatechin‐3‐gallate, a targeted inhibitor. Senescence‐associated β galactosidase (SA‐β‐gal) and formation of senescence‐associated heterochromatin foci were used to identify cell senescence. To investigate effects of EZH2 depletion on the cell cycle, apoptosis and proliferation, flow cytometry and MTT analysis were employed. Changes in p53–p21 axis activation were detected by Western blotting.
Results
We found that cell proliferative arrest caused by DOX could be promoted by EZH2 depletion. Mechanistically, EZH2 depletion not only worked in coordination with DNA damage during the progression of cell senescence but also promoted apoptosis in p53 mutant cells. However, it had no cooperative relationship with DOX in p53 wild‐type cells.
Conclusions
These data help unravel a crucial role for EZH2 in senescence and apoptosis in gastric cancer cells and that p53 genomic status was associated with different cell responses to EZH2 silencing.
Introduction
Although the incidence of gastric cancer has been substantially declining over the last several decades, it remains the fourth most frequently diagnosed cancer and second leading cause of death from cancer in some parts of the world 1. Chemotherapy is an important treatment for gastric cancer alongside surgical resection. While historically apoptosis has been considered to be the only desirable outcome of any anti‐neoplastic treatment, it has emerged recently that senescence could be a potential alternative outcome for tumour therapy in vivo. As a chemotherapeutic drug, doxorubicin (DOX) is known to induce cell senescence by causing a DNA damage response 2; it causes similar physiological and phenotypic changes with replicative senescence, including growth arrest, enlarged and flattened cell morphology, appearance of senescence‐associated β‐galactosidase (SA‐β‐gal) activity and senescence‐associated heterochromatic foci (SAHF) 3, 4, 5. As DOX induces cell senescence at concentrations significantly lower than those required for induction of apoptosis 6, promoting cancer cell senescence would have the advantage of reducing side effects of DOX treatment and act as an important internal barrier against tumourigenesis, by eliminating premalignant cells.
Enhancer of zeste homologue 2 (EZH2), a component of polycomb repressive complexes 2, is a human homologue of the Drosophila protein ‘enhancer of zest’. In particular, it trimethylates lysine 27 of histone H3 (H3K27me3) via its SET domain, thereby regulating gene expression by an epigenetic regulatory mechanism 7, 8. EZH2 is more highly expressed in malignant than non‐malignant tissues of gastric cancer, and expression of EZH2 closely correlates with tumour size, depth of invasion, vessel invasion, lymph node metastasis and clinical stage 9, 10. Additionally, EZH2 is involved in both in cell senescence and apoptosis. Its depletion inhibits cell proliferation and restores features of the senescent cell phenotype by relieving transcriptional repression of some cell cycle‐related genes, such as p21, p16 and p15 11, 12. In human glioma cells, nasopharyngeal carcinoma cells and some other cancers, inhibition of EZH2 expression can result in apoptosis 13, 14, 15; these reports illustrate the dual character of EZH2 on cancer cells.
Despite accumulating wealth of information concerning influences of EZH2 on progress of cancers, the role of EZH2 in cell response to low DNA damage has not been well characterized. In this study, we have sought to explore the outcome of EZH2 depletion along with exposure to DOX and related mechanisms, in gastric cancer cells.
Materials and methods
Cell culture
The human gastric cancer cell line MKN28 (GeneChem, Shanghai, China) and AGS cell line (Type Culture Collection of the Chinese Academy of Sciences) were maintained in RPMI 1640, supplemented with 10% foetal bovine serum (Hyclone, Logan, UT, USA) in a humidified CO2 (5%) incubator at 37 °C.
Induction of cell senescence
Doxorubicin was purchased from Roche incorporated (Basel, Switzerland). To induce premature senescence, the different cell lines were treated with a variety of concentrations of DOX (MKN28: 25 μm, AGS: 0. 1 μm,) for 2 h, then incubated in drug‐free culture medium for a further 48 h.
Inhibition of endogenous EZH2 by siRNA
Small interfering RNA (siRNA) oligo against the human EZH2 gene (Gene ID: 2146, accession: NM_004456.4) and negative control siRNA were purchased from Cell Signaling Technology (Boston, MA, USA) (6509S, 6201S). Fifty to sixty per cent confluent gastric cancer cells were transfected ith 100 nm of siRNA using Lipofectamine 2000 (Invitrogen, Camarillo, CA, USA) following the manufacturer's instructions. Transfected cells were incubated for 48 h, followed by cell harvesting and analysis.
(−)‐Epigallocatechin‐3‐gallate (EGCG) treatment
The gastric cancer cells were split to 50% confluence 24 h before treatment. Then they were treated with EGCG (Sigma, St. Louis, MO, USA) at 50 μm for 48 h. At the end of treatment, cells were harvested for analysis.
RNA isolation and q‐RT‐PCR
Total RNA was isolated using Trizol (Takara, Dalian, China). cDNA was synthesized using PrimeScript RT reagent kit (Takara) according to the manufacturer's instructions and PCR was conducted with Taq polymerase (Takara). Primers to EZH2 were as follows: 5′‐GACGGCTTCCCAATAACA‐3′ (F) and 5′‐TGAGGCTTCAGCACCACT‐3′ (R). GAPDH was as an internal control using the following primer set: 5′‐AACGGATTTGGTCGTATTG‐3′ (F) and 5′‐GGAAGATGGTGATGGGATT‐3′ (R).
Western blotting
Proteins were lysed in RIPA buffer containing a protease inhibitor cocktail (Sigma); they were resolved using sodium dodecyl sulphate‐poly‐acrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were incubated in the indicated primary antibodies and anti‐rabbit secondary antibodies conjugated to horseradish peroxidase. After development with the ECL system, signals were detected using the GelDoc XR System (Bio‐rad, Hercules, CA, USA). Antibodies used were as follows: rabbit anti‐EZH2 (Cell Signaling Technology), rabbit anti‐p53 (Sigma), rabbit anti‐p21 (RabMAbs, Burlingame, CA, USA) and rabbit anti‐GAPDH (Sigma).
MTT assay
Cell viability was measured using the MTT assay. Cells were plated in 96‐well plates; at the end of treatment, 20 μl MTT (5 mg/ml) was added to each well for an additional 4 h. Blue MTT formazan precipitate was then dissolved in 150 μl of DMSO. Absorbance at 490 nm (A value) was measured on a microplate reader, and cell proliferation inhibition levels were counted and calculated using the following formula:
SA‐β‐gal staining
Cytochemical staining for SA‐β‐gal at pH 6.0 was performed using a senescence‐β‐galactosidase staining kit (Cell Signaling Technology). After staining, cells were imaged using a microscope‐mounted camera. Five fields each were examined with at least 300 cells per field; SA‐β‐gal‐positive cells were counted.
SAHF analysis
Cells were cultured directly on glass coverslips and were fixed in 4% paraformaldehyde. After washing in PBS, DNA was visualized by DAPI (1 μg/ml) (Roche) staining for 5 min, followed by two PBS washes. Samples were then examined using a fluorescence microscope (Olympus, Tokyo, Japan). Five fields were examined with at least 50 cells per field; cells with SAHF were counted along the way.
Cell cycle analysis
Cells were washed in PBS and fixed in ice cold 75% ethanol at 4 °C, overnight. They were then washed twice in PBS and stained with 100 μg/ml propidium iodide (Sigma) containing RNase A (Sigma), for 30 min at 37 °C. Cell cycle distribution in different phases was determined using flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
Analysis of apoptosis
Apoptotic cells were detected using an annexin V‐EGFP apoptosis detection kit (Keygentec, Nanjing, China). Collected cells were washed twice in ice‐cold PBS and re‐suspended in 500 μL binding buffer. FITC annexin V (5 μl) and propidium iodide (5 μl) were added to cell preparations and incubated for 15 min at room temperature in the dark. Samples were then analysed by flow cytometry (Becton Dickinson).
Statistical analysis
All experiments were repeated more than three times. Statistical analysis was carried out using the t‐test for independent samples. P values of <0.05 were considered statistically significant.
Results
DOX induced gastric cancer cells to senesce
Doxorubicin has been shown to suppress cell proliferation through a DNA damage response 2. Our gastric cancer cells were treated with DOX to investigate whether it had the function of inducing senescence in them. After treatment, most cells became flattened and had comparatively enlarged morphology. As shown in (Fig. 1a,b), positive level of senescence‐associated SA‐β‐gal staining was increased from 4.50 ± 2.07 to 34.45 ± 7.05% in MKN28 cells and from 6.54 ± 0.75 to 66.61 ± 2.59% in AGS cells; SAHF formation increased in treated MKN28 cells 30.13 ± 1.8% (Fig. 1a,c). Flow cytometry analysis demonstrated that proportions of cells in G0/G1 phase increased in treated cells. In MKN28 cells, we also noticed a significant increase in proportions of cells in G2/M (Fig. 1d,e). Cell inhibitory levels were 44.18 ± 0.33% in MKN28 cells and 5.81 ± 0.92% in AGS cells. These morphological changes and characteristics of mitosis confirmed that low concentrations of DOX efficiently induced senescence in the gastric cancer cells. Western blotting indicated that DOX induced p53 and protein of its target gene p21 increased slightly in AGS cells, but only p53 increased in MKN28 cells.
Figure 1.
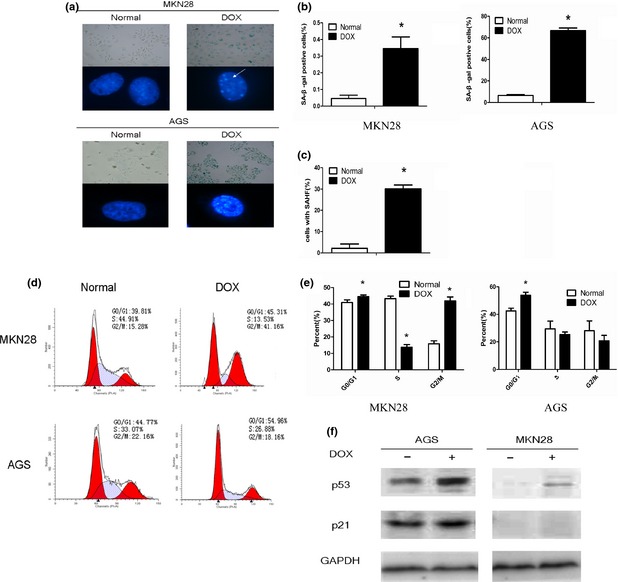
Low concentration of doxorubicin (DOX) induced gastric cancer cells to senescence. Gastric cancer cells were treated with DOX for 2 h (MKN28: 25 μm, AGS: 0.1 μm). After 48 h of culture in drug‐free medium, cells were stained with SA‐β‐gal staining solution (pH 6.0) and DAPI, then cell images were captured. White arrow indicates senescence‐associated heterochromatin foci (a). The positive rate of SA‐β‐gal staining was increased in both cell lines (b). Percentage of senescence‐associated heterochromatin foci‐positive cells scored in normal and DOX group of MKN28 (c). Cell cycle was analysed by flow cytometry and cells arrested in G0/G1 phase were increased. Besides, there was also a significant increase in the proportion of cells in G2/M phase in MKN28 cells (d, e). A representative Western blot shows higher levels of p53 and p21 proteins in AGS, but only p53 increased in MKN28 after treatment of DOX (f). *Indicates a significant (P < 0.05) difference between normal and DOX group.
EZH2 downregulation inhibited proliferation of the gastric cancer cells
MKN28 and AGS cells were transfected with siRNA and scrambled siRNA as negative control (Control siRNA). Transcriptional levels of EZH2 were tested using q‐RT‐PCR and a significant reduction in EZH2 was observed. Lower protein levels were also found by Western blot assay. Then, we used EGCG to inhibit EZH2 pharmacologically. EZH2 expression was downregulated as a result of increase in EGCG concentration. In general, both siRNA and EGCG inhibited EZH2 expression (Fig. 2a,b). To investigate effects of EZH2 depletion on cell proliferation, the MTT viabilty assay was employed. EZH2 depletion by targeted siRNA or EGCG led to reduced proliferation efficiency. Cell inhibitory level is shown in Fig. 2c and 2d.
Figure 2.
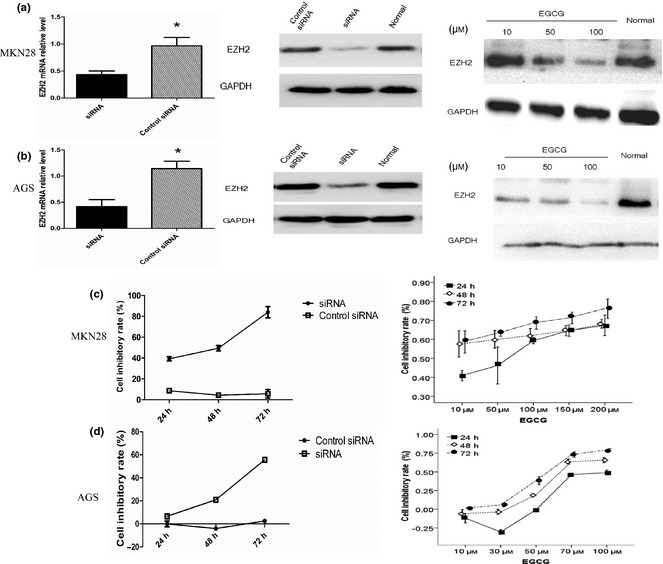
Enhancer of zeste homologue 2 (EZH2) downregulation inhibits the growth of gastric cancer cells. mRNA expression of EZH2 was examined by q‐RT‐PCR in gastric cancer cells transfected with siRNA (100 nm, 2 days). Protein expressions of EZH2 in cells transfected with siRNA or treated with various concentrations of EGCG were measured by Western blot. GAPDH was used as an internal control. *Indicates a significant (P < 0.05) difference between siRNA and control siRNA treatment (a, b). Cellular proliferation inhibited rate increased with time extending in cells transfected siRNA compared with control siRNA. Besides time extending, cell inhibitory rate had a positive correlation with the concentrations of EGCG (c, d).
Inhibition of EZH2 worked in coordination with DOX to arrest cell proliferation
Given the effect of EZH2 on proliferation of our gastric cancer cells, we speculated on possible synergistic effects of EZH2 depletion and DOX. We treated cells with siRNA/EGCG and DOX, independently and in combination. Cell inhibition was most profound in the siRNA + DOX group (60.96 + 2.87% in MKN28 cells, 49.86 + 1.71% in AGS cells) and the EGCG + DOX group (65.22 + 0.25% MKN28 cells, 33.12 + 2.52% in AGSs).
EZH2 depletion promoted DOX ‐induced senescence in cells with mutant p53
To further elucidate reasons why inhibiting EZH2 promoted DOX‐induced cell proliferation inhibition, we counted cells positive for SA‐β‐gal expression, and observed SAHF formation again. In MKN28 cells carrying mutant p53, siRNA and EGCG caused marked increase in positive level of SA‐β‐gal staining and SAHF (Fig. 3a,c,d) and there was increase in percentage of G0/G1 phase cells (Fig. 3e). Remarkably, this effect was greater in cells treated with siRNA/EGCG combined with DOX (Fig. 3b–e). These results suggested that EZH2 depletion promoted DOX‐induced senescence in MKN28 cells. In AGS cells which had wild‐type p53, only EGCG increased positive level of SA‐β‐gal expression and percentage of G0/G1 slightly; small changes were observed when both agents were present (Fig. 3a–c,e).
Figure 3.
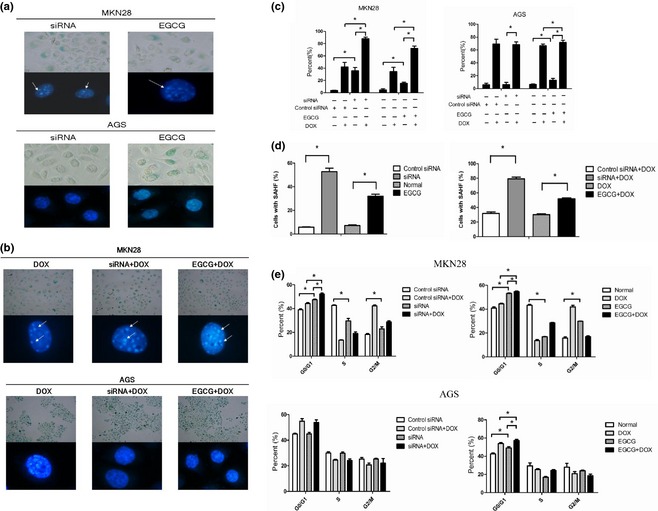
Enhancer of zeste homologue 2 (EZH2) depletion promotes doxorubicin (DOX)‐induced senescence in p53 mutant type cells. After being transfected or treated with EGCG or treatment of siRNA/EGCG combined with DOX, cells stained with SA‐β‐gal staining solution and DAPI were captured. White arrows indicate senescence‐associated heterochromatin foci (a, b). In MKN28 cells, which carry mutant p53, siRNA and EGCG caused a marked increase in positive rate of SA‐β‐gal staining and senescence‐associated heterochromatin foci formation, and this effect was greater in cells treated with siRNA/EGCG combined with DOX. In AGS which had wild‐type p53, only EGCG increased the positive rate of SA‐β‐gal staining slightly, and small change was observed when both agents were present (c, d). Targeted siRNA or EGCG increased percentage of G0/G1 phase cells and promoted DOX‐induced cell cycle arrest in MKN28. However, only EGCG arrested cell cycle in G0/G1 phase, and slight change is observed when both agents were present in AGS (e), *P < 0.05.
Combination of EZH2 inhibition and DOX promoted apoptosis in cells with mutant p53
The above results suggest that senescence was not the mechanism acting to promote growth arrest in AGS cells, so we hypothesized that apoptosis might be the explanation for incremental cell inhibitory level. Flow cytometric analysis showed a significant increase in per cent apoptotic events after siRNA treatment in both types of gastric cancer cell and an additional increase when siRNA and DOX were present in MKN28 cells, but not in AGSs. These results indicate that EZH2 silencing caused apoptosis in p53 mutant and p53 wild‐type cancer cells, and promoted apoptosis more profoundly in wild‐type p53 cells (Fig. 4a,b).
Figure 4.

Combination of Enhancer of zeste homologue 2 (EZH2) inhibition and doxorubicin (DOX) promotes apoptosis in cells with mutant p53. After being transfected targeted siRNA, cells were induced by DOX. Cellular apoptosis was analysed by flow cytometry. Only annexin V–stained cells were considered as apoptotic. siRNA produced a significant accumulation of cells in early apoptosis both in MKN28 and in AGS. Additionally, siRNA and DOX co‐treatment increased the per cent of apoptosis cells compared with single treatment in MKN28, but not in AGS, *P < 0.05. (a, b) Western blot shows that combined EZH2 depletion and DOX alter expression of p53, p21 proteins in AGS, but no change was observed in MKN28. GAPDH was used as an internal control (c).
We then had performed Western blotting to examine expression of p53 in the DOX group and co‐treatment group. As shown in (Fig. 4c), EZH2 depletion increased accumulation of p53 protein and protein of its target gene p21, in AGS cells under co‐treatment. However, expression of the two proteins was not changed in MKN28 cells. These results indicate that EZH2 depletion might have a relationship with activation of the p53 pathway in p53 wild‐type cells.
Discussion
As one of the first protein lysine methylases implicated in human cancers, EZH2 is overexpressed in many kinds of malignancy, playing an important role in tumour cell proliferation, metastasis and senescence 16, 17, thus considered to be a promising potential target in cancer therapy. In this study, we demonstrate that inhibiting EZH2 worked in coordination with DOX to arrest cancer cell proliferation. EZH2 depletion promoted DOX‐induced cell senescence and apoptosis in gastric cancer cells with mutant p53, but had almost no cooperative effect in cells with wild‐type p53. These findings bear important implications for cancer treatment and indicate that the therapeutic inhibition of EZH2 might be an attractive approach to sensitize low‐dose chemotherapy for cancers that overexpress EZH2.
Apoptosis and senescence are both distinct mechanisms for tumour suppression 18, 19 and share one common pathway – p53/p21 20. MKN28 cells have mutant p53, and express mRNAs of p21, CDK2 and G1 cyclins, at high levels. Although MKN28 cells express high levels of p21 mRNA, its protein is not detectable by Western blot analysis 21. AGS cells, as a p53 wild‐type gastric cancer cell line contain a functional p53/p21 pathway and can be activated by EZH2 silencing. Thereby, we consider that p53 genomic status plays an important role in determining response to DNA damage with EZH2 depletion in gastric cancer cells.
p53 is a transcription factor whose ability to suppress tumourigenesis in mammals has been extensively studied 22, and is regarded as a central player in tumour suppression and therapy. As p53 is an integral and central part of a network of proteins that respond to stress stimuli, the decision between proliferative growth arrest, senescence or apoptosis is believed to be determined by the appropriate qualitative status of p53 23. Level of genetic activation of p53 may be decisive for the cellular response: lower levels of p53 may favour growth arrest, whereas higher levels could trigger apoptosis 23. In spite of low concentrations of DOX causing activation of the p53/p21 pathway 24, as p53 mutant type cancer cells, MKN28 cells always keep the p53/p21 pathway inactive Low‐level p53 caused senescence most significantly when both DOX and EZH2 inhibitor were present. On the other hand, the p53/p21 pathway is functional in wild type cells, either DOX or EZH2 inhibition was shown to activate it. Small doses of DOX caused p53 slight activation, and resulted in senescence. EZH2 inhibitors accumulate p53 at high‐expression levels, leading to apoptosis being the most striking feature in AGS cells after treatment. In accordance with our work, Wu et al. 25 reported that EZH2 depletion results in abrogation of both cell cycle G1 and G2/M checkpoints, directing a DNA damage response predominantly toward apoptosis, in both p53‐proficient and p53‐deficient cancer cells, but not in normal cells. Mechanistically, EZH2 regulates the DNA damage response in p53 wild‐type cells mainly through transcriptional repression of FBXO32, which binds to and directs p21 for proteasome‐mediated degradation, whereas it affects p53‐deficient cells by regulating Chk1 activation by a distinct mechanism.
EGCG, a bioactive polyphenolic agent in green tea, and histone lysine methyltransferase that reduces cell survival through reducing Bmi‐1 and EZH2 levels and also a global reduction of H3K27me3 26, 27. In this study, we treated cells with a wide range of EGCG concentrations and found that EGCG depleted EZH2 effectively in the gastric cancer cells. This change induced a diversity of responses at the cellular level. In contrast to siRNA, EGCG increased SA‐β‐gal expression and percentage of G0/G1 slightly; small increases were observed when both agents were present in AGS cells. This suggests that EZH2 is not the only target for EGCG. Further investigation of the molecular mechanisms is likely to provide additional insight into this.
Given the toxic side effect of chemotherapy, any method that can make cancer cells sensitive to even mild DNA damage, and response towards senescence or apoptotic progression, needs serious attention paid to it. Although the responses are different, EZH2 silencing promoted growth arrest by low concentrations of DOX no matter the cells’ p53 status. We consider it as a sensitizer that not only can enhance efficacy of DNA damaging chemotherapeutic agents but also reduce toxic effects. In conclusion, these results suggest that joint application of EZH2 inhibitors and low concentrations of DOX may be a potential novel approach to gastric cancer treatment.
Acknowledgements
This work was supported by Natural Science Foundation of Hubei Province (no. 2010CDB07706).
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J. Clin. 61, 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Demidenko ZN, Blagosklonny MV (2008) Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 7, 3355–3361. [DOI] [PubMed] [Google Scholar]
- 3. Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA et al (2003) Rb‐mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. [DOI] [PubMed] [Google Scholar]
- 4. Campisi J, d'Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. [DOI] [PubMed] [Google Scholar]
- 5. Roninson IB (2003) Tumor cell senescence in cancer treatment. Cancer Res. 63, 2705–2715. [PubMed] [Google Scholar]
- 6. Zhang Y, Gao Y, Zhang G, Huang S, Dong Z, Kong C et al (2011) DNMT3a plays a role in switches between doxorubicin‐induced senescence and apoptosis of colorectal cancer cells. Int. J. Cancer 128, 551–561. [DOI] [PubMed] [Google Scholar]
- 7. Simon JA, Lange CA (2008) Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 647, 21–29. [DOI] [PubMed] [Google Scholar]
- 8. Pal B, Bouras T, Shi W, Vaillant F, Sheridan JM, Fu N et al (2013) Global changes in the mammary epigenome are induced by hormonal cues and coordinated by Ezh2. Cell Rep. 3, 411–426. [DOI] [PubMed] [Google Scholar]
- 9. Choi JH, Song YS, Yoon JS, Song KW, Lee YY (2010) Enhancer of zeste homolog 2 expression is associated with tumor cell proliferation and metastasis in gastric cancer. APMIS 118, 196–202. [DOI] [PubMed] [Google Scholar]
- 10. Matsukawa Y, Semba S, Kato H, Ito A, Yanagihara K, Yokozaki H (2006) Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 97, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC et al (2011) EZH2‐dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol. Cancer Res. 9, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kheradmand Kia S, Solaimani Kartalaei P, Farahbakhshian E, Pourfarzad F, von Lindern M, Verrijzer CP (2009) EZH2‐dependent chromatin looping controls INK4a and INK4b, but not ARF, during human progenitor cell differentiation and cellular senescence. Epigenetics Chromatin 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang R, Wang R, Chang H, Wu F, Liu C, Deng D et al (2012) Downregulation of Ezh2 expression by RNA interference induces cell cycle arrest in the G0/G1 phase and apoptosis in U87 human glioma cells. Oncol. Rep. 28, 2278–2284. [DOI] [PubMed] [Google Scholar]
- 14. Cai K, Wan Y, Sun G, Shi L, Bao X, Wang Z (2012) Let‐7a inhibits proliferation and induces apoptosis by targeting EZH2 in nasopharyngeal carcinoma cells. Oncol. Rep. 28, 2101–2106. [DOI] [PubMed] [Google Scholar]
- 15. Li H, Cai Q, Wu H, Vathipadiekal V, Dobbin ZC, Li T et al (2012) SUZ12 promotes human epithelial ovarian cancer by suppressing apoptosis via silencing HRK. Mol. Cancer Res. 10, 1462–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piunti A, Pasini D (2011) Epigenetic factors in cancer development: polycomb group proteins. Future Oncol 7, 57–75. [DOI] [PubMed] [Google Scholar]
- 17. McCord RP, Nazario‐Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR et al (2013) Correlated alterations in genome organization, histone methylation, and DNA‐lamin A/C interactions in Hutchinson‐Gilford progeria syndrome. Genome Res. 23, 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang L, Pan J, Wang T, Song M, Chen W (2013) Pathological cyclic strain‐induced apoptosis in human periodontal ligament cells through the RhoGDIalpha/caspase‐3/PARP pathway. PLoS One 8, e75973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM et al (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346. [DOI] [PubMed] [Google Scholar]
- 20. Hasty P, Christy BA (2013) p53 as an intervention target for cancer and aging. Pathobiol. Aging Age Relat. Dis. 3, 22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yokozaki H (2000) Molecular characteristics of eight gastric cancer cell lines established in Japan. Pathol. Int. 50, 767–777. [DOI] [PubMed] [Google Scholar]
- 22. Shangary S, Wang S (2008) Targeting the MDM2‐p53 interaction for cancer therapy. Clin. Cancer Res. 14, 5318–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Espinosa JM (2008) Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene 27, 4013–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leontieva OV, Gudkov AV, Blagosklonny MV (2010) Weak p53 permits senescence during cell cycle arrest. Cell Cycle 9, 4323–4327. [DOI] [PubMed] [Google Scholar]
- 25. Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ et al (2011) Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 18, 1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balasubramanian S, Lee K, Adhikary G, Gopalakrishnan R, Rorke EA, Eckert RL (2008) The Bmi‐1 polycomb group gene in skin cancer: regulation of function by (‐)‐epigallocatechin‐3‐gallate. Nutr. Rev. 66(Suppl. 1), S65–S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balasubramanian S, Adhikary G, Eckert RL (2010) The Bmi‐1 polycomb protein antagonizes the (‐)‐epigallocatechin‐3‐gallate‐dependent suppression of skin cancer cell survival. Carcinogenesis 31, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]