

Abstract
Objectives: This study aims to determine the role of macrophage migration inhibitory factor (MIF), a proinflammatory cytokine associated with cell proliferation and tumour growth in vivo.
Materials and methods: Our team used RNA interference technology to knock down MIF expression in human HeLa cervical cancer cells and to establish a stable cell line lacking MIF function.
Results: Our results showed that long‐term loss of MIF had little effect on cell morphology, but significantly inhibited their population growth and proliferation. The HeLa MIF‐knockdown cells retained normal apoptotic signalling pathways in response to TNF‐alpha treatment; however, they exhibited unique DNA profiles following doxorubicin treatment, suggesting that MIF may regulate a cell cycle checkpoint upon DNA damage. Our data also showed that knockdown of MIF expression in HeLa cells led to increased cell adhesion and therefore impaired their migratory capacity. More importantly, cells lacking MIF failed to either proliferate in soft agar or form tumours in vivo, when administered to nude mice.
Conclusion: MIF plays a pivotal role in proliferation and tumourigenesis of human HeLa cervical carcinoma cells, and may represent a promising therapeutic target for cancer intervention.
Introduction
Macrophage migration inhibitory factor, MIF is a small protein hormone of 115 amino acids, encoded by a single functional gene in both human (1) and mouse (2, 3). In spite of T cells being the main source of MIF production, it is also produced and secreted by other cell types (4).
MIF is the first cytokine to be identified that inhibits random migration of macrophages in culture (5, 6). Now it is best known as a proinflammatory cytokine and plays important roles in immune regulation associated with certain inflammatory diseases such as rheumatoid arthritis and atherosclerosis (7). Our earliest observation indicated that MIF was involved in atherogenesis in rabbit (8), and was responsible for destabilization of human atherosclerotic plaques by inducing MMP‐9 expression in macrophages (9). Later, it was found to probably act through the MEK‐ERK MAP kinase pathway (10). After that, we reported that MIF mediated regulation of insulin/Akt signalling by angiotensin converting enzyme 2 (ACE2) (11). More recently, we found that in atrial myocytes MIF was involved in electrical remodelling that accompanies atrial fibrillation (AF), probably by decreasing L‐type Ca2+ channel depression and activation of c‐Src kinase (12). For the same cell type, MIF also seems to be responsible for high glucose‐induced apoptosis (13).
Several lines of evidence have implicated MIF as a regulator of cancer cell expansion. First, it has been found to be highly expressed in many human cancers (14, 15, 16, 17, 18), that is to say, when MIF is overexpressed the cells gain a proliferative advantage. Second, attenuation of cell proliferation and increase in apoptosis have been observed, by genetic mutation/deletion, in MIF‐deficient cells (19, 20), or by effects of anti‐sense MIF plasmid (21, 22) and RNA interference (RNAi) (23, 24). Third, inactivation of MIF by small molecule inhibitors or inhibitory antibodies reduces properties of transformation in some cancer cells (21, 25, 26).
The molecular mechanism of MIF action remains to be elucidated. Within cells, it can specifically interact with Jab1, a co‐activator of the AP‐1 transcriptional complex (27). Outside the cells, secreted MIF protein can bind to CD74, a cell surface receptor, which triggers a number of signal transduction pathways (28). Besides, MIF can physically interact with tumour suppressor p53 and inhibit its transcriptional activity, which overcomes p53‐dependent cell proliferation inhibition or cell death (29, 30, 31, 32).
So far, the nature of cell growth and proliferation regulation by MIF is less well understood. It is therefore important to reconcile how a proinflammatory cytokine like MIF can determine the fate of cancer cells. One possibility is that MIF may promote cell transformation and cancer growth. Alternatively, MIF may prevent cell death by inhibiting p53 function. To address this question, we sought to better characterize the role of MIF in proliferation of human cancer cells. Herein, we focused on HeLa, which is one of the most popularly used human cervical cancer cells, and employed RNAi technology to permanently knockdown its MIF expression. Our findings indicate that although MIF is not absolutely required for growth and proliferation, it is essential for tumourigenesis and probably metastasis of HeLa cells, as well.
Materials and methods
Plasmids
We referenced the input of human MIF cDNA from GenBank (# NM_002415), and designed RNAi target sequences as follows:
-
1
5′‐CTATTACGACATGAACGCG‐3′
-
2
5′‐CAACTCCACCTTCGCCTAA‐3′
The protocol to prepare plasmid constructs coding for shRNA has been described previously (33). In brief, the DNA hairpin was subcloned into a pSUPER/puro vector (Oligoengine, Seattle, WA, USA) and digested with BglII/HindIII to generate pSRP/MIF vector. Similarly, a DNA hairpin with a scrambled sequence was also subcloned into a pSUPER/puro vector and used as negative control (pSRP). All RNAi constructs were confirmed by DNA sequencing.
Cell culture and virus preparation
Human HeLa cervical carcinoma cells (American Type Culture Collection, Manassas, VA, USA; #CCL‐2) were cultured at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum, 2 mm l‐glutamine and antibiotics. All cell culture reagents were purchased from Life Technologies, Inc., Paisley, Scotland. DNA transfections were conducted using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol.
To establish cell lines, pSRP or pSRP/MIF vectors were transfected into Phoenix packaging cells (Orbigen, San Diego, CA, USA) to produce ecotropic retroviral supernatants. Forty‐eight hour post‐transfection, tissue culture medium was filtered through a 0.45 μm filter, and viral supernatants for infection of HeLa cells were used after addition of 4 μg/ml polybrene. Then, cells were selected with 2 μg/ml puromycin until drug‐resistant colonies became visible.
SDS–polyacrylamide gel electrophoresis and immunoblotting
HeLa cells were harvested and lysed in buffer containing 0.5% NP40, 10 mm Tris–HCl (pH 8.0), 50 mm KCl, 1 mm orthovanadate and protease inhibitors. Protein concentrations were quantified by Bio‐Rad assay (Bio‐Rad Laboratories, Hercules, CA, USA) and equal amounts were loaded on 8–15% polyacrylamide gels and subjected to SDS–polyacrylamide gel electrophoresis. Then they were electrotransferred on to PVDF membranes (Millipore Corporation, Bedford, MA, USA) and immunoblotted using: rabbit polyclonal anti‐MIF (Santa Cruz Biotechnology, Santa Cruz, CA, USA; #sc‐20121), anti‐p16 (Santa Cruz Biotechnology; #sc‐468), anti‐p27 (Santa Cruz Biotechnology; #sc‐528), anti‐Src (Abcam, Cambridge, MA, USA; #ab47405) and anti‐FAK (Cell Signaling, Danvers, MA, USA; #3285) antibodies, and mouse monoclonal anti‐cyclin B1 (Santa Cruz Biotechnology; #sc‐245), anti‐cyclin D1 (Santa Cruz Biotechnology; #sc‐8396), anti‐cyclin E (Santa Cruz Biotechnology; #sc‐247), anti‐PCNA (Santa Cruz Biotechnology; #sc‐56) and anti‐c‐Myc (Santa Cruz Biotechnology; #sc‐42) antibodies. All antibodies were used at 1:1000 dilution unless indicated otherwise in the text. Antigen‐antibody complexes were detected using anti‐rabbit or anti‐mouse antibodies conjugated to horseradish peroxidase (HRP) and visualized using SuperSignal chemiluminescent detection kit (Pierce, Rockford, IL, USA). HRP‐conjugated mouse monoclonal anti‐GAPDH (Kangchen Inc., Shanghai, China; #KC‐5G5) antibody was used to ensure equal loading of protein samples.
Cell proliferation assay
HeLa cells were plated in six‐well dishes in duplicate. At various time points, adherent and non‐adherent cells were collected and aliquots were mixed with an equal volume of 0.4% trypan blue (Gibco BRL, Gaithersburg, MD, USA) in phosphate‐buffered saline (PBS), and were counted. Each growth curve is the average of two or three independent experiments.
Analysis of apoptosis
The HeLa cells under normal expansion conditions were harvested, fixed and resuspended in annexin‐binding buffer (10 mm HEPES, 140 mm NaCl, 2.5 mm CaCl2, pH 7.4). Aliquots of 100 μl of cell suspension were treated with 5 μl of anti‐annexin V antibodies conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA), 40 μg of propidium iodide (PI) (Sigma‐Aldrich, St. Louis, MO, USA) and incubated for 15 min at room temperature and cells were analyzed by flow cytometry. Samples were run on a BD Biosciences FACScan (Becton Dickinson Labware, Mountain View, CA, USA) equipped with a 488 nm argon air‐cooled laser and emissions were measured using FL1 channel (530/30 filter). Analysis was performed using ModFit software (BD Biosciences, San Jose, CA, USA).
Cell cycle analysis
In the order of 1 × 106 HeLa cells were detached in 0.25% trypsin and washed in ice‐cold PBS. Following centrifugation at 900 g for 5 min, cells were suspended and fixed in 70% ice‐cold ethanol for 45 min. After several washes in ice‐cold PBS, cells were suspended in 0.5 ml of PBS containing 40 μg of PI (Sigma‐Aldrich) and 100 μg of RNase (Sigma‐Aldrich) per ml. Cells were incubated at 37 °C for 30 min and maintained at 4 °C before flow cytometry analysis on a BD Biosciences FACScan (Becton Dickinson Labware). Analysis was performed using ModFit software (BD Biosciences).
Cell adhesion analysis
One thousand cells were loaded on to vitronectin‐coated 96‐well plates (Cytomatrix cell adhesion strips; Chemicon, Billerica, MA, USA) for 30 min, washed and then subjected to crystal violet staining. Relative number of adhesive cells was measured by optical density under a microplate reader.
Migration assay
Modified Boyden chambers (Millicell‐PCF, 8‐μm pore size; Millipore) were placed in 24‐well plates and coated with 10 μg/ml rat‐tail collagen (Roche Applied Science, Indianapolis, IN, USA) for 16 h at 37 °C. After removal of collagen and washing in PBS, 0.4 ml of migration medium (DMEM with 0.5% bovine serum albumin) was added to the lower chamber. A total of 2 × 105 cells were seeded into the upper compartment in 0.3 ml of migration medium. The plates were then incubated at 37 °C for 16 h, to allow migration to take place. Cells on the upper membrane surface were removed using a cotton tip applicator and washing in PBS, whereas cells on the lower membrane surface were fixed in 4% formaldehyde and stained with 0.1% crystal violet in 20% ethanol.
Wound closure assay
Wounds were created on near to confluent monolayers of cells by scraping a gash using a sterile micropipette tip. Speed of wound closure was monitored and recorded every 24 h. Photographs were taken under 100× magnifications using a Cannon digital camera attached to a phase‐contrast microscope, immediately after wound incision, and later at various time points.
Anchorage‐independent growth assay
5 × 104 cells were re‐suspended in 3 ml of complete DMEM containing 0.3% Noble agar (BD Biosciences; Difco™) and plated on 6‐cm dishes with a solidified bottom layer made of 0.6% agar in complete DMEM. Cells were fed with 1 ml of 0.25% agar in complete DMEM every 4 days. After 20 days, colonies were stained with 0.005% crystal violet for 1 h and were counted by bright field light microscope illumination low power (40×).
Tumour formation in nude mice
Female nude mice 6–8 weeks of age, and weighing 17–21 g, were provided by the National Rodent Laboratory Animal Resources, Shanghai Branch (Shanghai, China) and housed in specific pathogen‐free (SPF) conditions at a temperature of 22–24 °C and humidity of 45–70%. All IVC supplies were also sterilized and autoclaved before entering the cage. A total of 5 × 106 cells were injected subcutaneously into nude mice, twice over a 2‐day interval. During the next 20 days, mice were observed daily to examine tumour growth; eventually animals were euthanized. Digital images were taken post mortem.
Statistical analysis
Results are expressed as mean ± standard error, from three or four independent experiments. Data were subjected to one‐way or two‐way analysis of variance using GraphPad Prism 1.1 program (GraphPad Software, Inc., La Jolla, CA, USA). P‐values <0.05 were considered significant.
Results
Generation of human HeLa cervical cancer cells lacking MIF expression
To study the role of MIF in cell population growth, we decided to knockdown MIF expression in human HeLa cervical cancer cells using RNAi technology. In brief, we selected two different RNAi target sites based on nucleotide sequence of human MIF cDNA to design DNA oligos, which were then synthesized and subcloned into the pSUPER/puro plasmid vector. Retroviruses were produced by transfecting plasmids into Phoenix packaging cells according to manufacturer’s protocol, which were then used to infect HeLa cells and select them in the presence of 2 μg/ml puromycin antibiotics. Cell clones with significant MIF knockdown results were chosen to form a stable cell line, which was used in further experiments and named pSRP/MIF. In parallel, a control cell line named pSRP was established using a scrambled RNAi sequence. Figure 1a shows that viral infection and the following antibiotic selection procedure did not change the steady‐state level of MIF in cells, since parent HeLa cells (non‐infected) and HeLa pSRP control cells (infected) had identical levels of MIF expression (lanes 1 and 2), whereas MIF expression in HeLa pSRP/MIF cells was diminished nearly 10‐fold (compare lanes 3 to 1 and 2).
Figure 1.
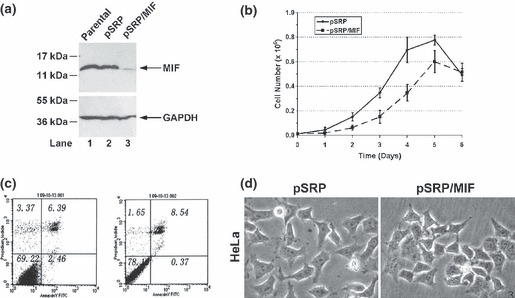
Generation and growth analysis of HeLa MIF‐knockdown cells. (a) MIF expression. HeLa parent, pSRP control and pSRP/MIF knockdown cells were harvested and subjected to western blotting analysis with anti‐MIF antibody. Western blotting for GAPDH was performed to ensure equal loading of protein samples. (b) Growth curve. HeLa pSRP control and pSRP/MIF knockdown cells were seeded in 6‐cm plates and cell number was counted at 0, 1, 2, 3, 4, 5 and 6 days. (c) Analysis of apoptosis. Cells under normal growth conditions were harvested and immuno‐labelled with anti‐annexin V antibody conjugated to Alexa fluor 488. Expression of annexin V on cell surfaces was revealed by flow cytometry as described in the Materials and methods section. (d) Cell morphology. All photographs of the cells are at the same scale.
Analysis of viability and morphology of MIF‐knockdown cells
To examine the effect of MIF knockdown on proliferation, cell numbers were counted. Figure 1b shows that although the number of cells increased in both HeLa pSRP control and pSRP/MIF knockdown cells for at least 5 days, loss of MIF expression significantly altered proliferation of HeLa cells. On day 6, total cell number decreased in both populations, presumably due to the lack of nutrients and accumulation of metabolic waste in the media. Nonetheless, loss of MIF expression did not result in extensive early cell death, as percentages of cells that became permeable to trypan blue was almost the same for the two cell lines (data not shown), and both populations did not exhibit any significant apoptosis, as indicated in Fig. 1c following measurement for phosphatidylserine on cell surfaces. When examined under a bright light microscope, both cell types displayed characteristic epithelial phenotypes except that individual control cells were more dispersed, whereas MIF‐knockdown cells formed somewhat scattered colonies on the plates (Fig. 1d). Thus, it appears that loss of MIF expression caused only minor changes to cell morphology, but significantly affected cell proliferation.
MIF‐knockdown affect on cell cycle and DNA damage response in HeLa cells
Studies by others, on a knockout mouse model, have shown that MIF coordinates the cell cycle with DNA damage checkpoints, which may be beneficial for cancer cells to maintain certain levels of genomic stability and therefore prevent cell death caused by adverse conditions (34). In this regard, we examined the effect of long‐term loss of MIF on cell cycle and DNA damage response in HeLa cells.
Our western blot analysis shown in Fig. 2 indicated that cyclin B1 level was not affected by knockdown of MIF expression in our HeLa cells. However MIF knockdown decreased cyclin D expression but increased cyclin E in the same cells. Next, we examined levels of CDK inhibitors p16 and p27. As shown in Fig. 2, p16 expression was significantly diminished in MIF‐knockdown cells. However, loss of MIF did not seem to affect p27 expression at all. In addition, it also did not alter PCNA expression, suggesting that HeLa cells were able to grow and replicate to some degree in the absence of MIF. More importantly, we found that MIF knockdown strongly affected c‐Myc expression, which has been reported to be involved in cell transformation in some cases (35), suggesting that MIF may contribute tumourigenic potential of HeLa cells.
Figure 2.
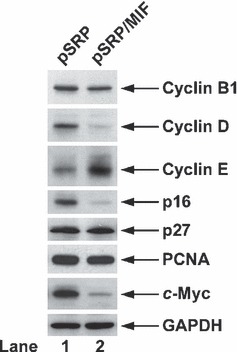
Cell cycle marker analysis. HeLa pSRP control and pSRP/MIF knockdown cells under normal growth conditions were harvested and subjected to western blotting for cyclin B1, cyclin D, cyclin E, p16, p27, PCNA and c‐Myc expression. Western blotting for GAPDH was performed to ensure the equal loading of protein samples.
Next, we examined the response of MIF‐knockdown cells to DNA damage. To this end, we treated cells with doxorubicin at different dosages for 24 h, and subsequently cells were harvested and subjected to cell cycle analysis by fluorescence‐activated cell sorting (FACS). As shown in Fig. 3, under normal conditions, population of G1‐phase MIF‐knockdown cells increased from 51% to 63%, whereas percentage of S‐ and G2/M‐phases cells decreased from 19% and 20% to 15% and 14%, respectively. Consistent with results shown in Fig. 1c, absence of sub‐G1 cells indicated that both HeLa control and MIF‐knockdown cells were devoid of apoptosis. When treated with doxorubicin at low concentrations, control cells ceased to grow and halted in S‐ and G2/M‐phases. When treated with maximal dosage of doxorubicin in this experiment, cells arrested in G1‐ and G2/M‐phases. In contrast, MIF‐knockdown cells exhibited different DNA profiles following DNA damage: when treated with low concentration of doxorubicin, MIF‐knockdown cells mainly arrested in S phase. However higher amounts of doxorubicin led to arrest in all three cell cycle phases or in G1. Interestingly, when treated with 0.5 μm of doxorubicin, MIF‐knockdown cells exhibited a wide spectrum of DNA content ranging from 2N to >4N, which indicated that DNA re‐replication might have occurred in S phase. Taken together, MIF appears to affect cell cycle regulation and DNA damage response in HeLa cells.
Figure 3.
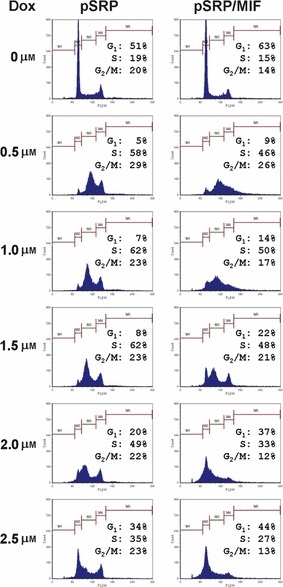
Cell cycle analysis in response to DNA damage. HeLa pSRP control and pSRP/MIF knockdown cells were treated with doxorubicin as indicated in the figure, then harvested and prepared for FACS analysis as described in the Materials and methods section.
MIF‐depletion affected focal adhesion and migration of HeLa cells
Broken cell‐cell and/or cell‐matrix adhesions and increased cell migration are key characteristics of cancer cells (36). To further characterize the HeLa MIF‐knockdown cells, we performed cell adhesion assays to test focal adhesion formation of these cells associating with extracellular matrix (ECM). Figure 4a shows that numbers of HeLa pSRP/MIF knockdown cells that had been attached to bottom of plates were almost three times higher than of HeLa pSRP control cells, suggesting that loss of MIF allowed the cells to form somewhat tighter adhesions to the extracellular matrix.
Figure 4.
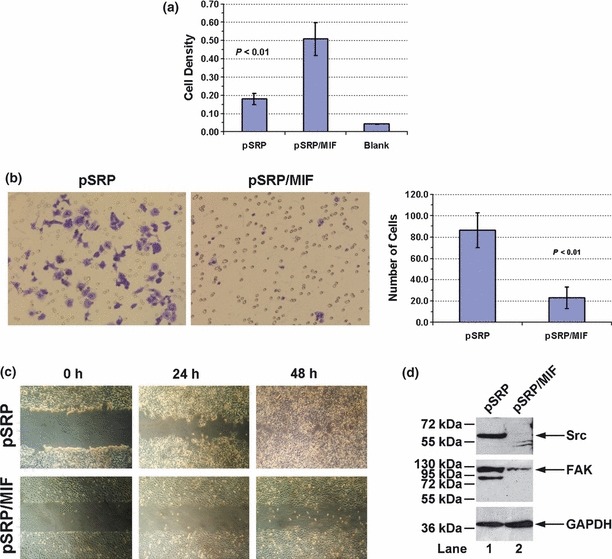
Cell mobility analysis. (a) Cell adhesion assay. HeLa pSRP control and pSRP/MIF knockdown cells were placed in 96‐well plates and stained as described in the Materials and methods section. Cell density was represented by optical value of staining. ‘Blank’ refers to the background level taken by micro‐plate reader. (b) Migration assay. HeLa pSRP control and pSRP/MIF knockdown cells were placed in the upper chamber of a Boyden chamber and allowed to migrate through a microporous membrane. Those that had migrated to the lower side of the membrane were revealed by crystal violet staining, left. On the right, the number of migrating cells per field under bright field microscopy was counted. Error bars indicate standard deviation of the mean from three independent experiments. (c) Wound closure assay. Light microscopy images were taken of HeLa pSRP control and pSRP/MIF knockdown cells at 0, 24 and 48 h after wounding. All photographs of cells are at the same scale. (d) HeLa pSRP control and pSRP/MIF knockdown cells under normal growth conditions were harvested and subjected to western blotting with anti‐Src and anti‐FAK antibodies. Western blotting for GAPDH was performed to ensure the equal loading of protein samples.
Next, we performed a migration assay, which measured ability of cells to migrate through a porous membrane between two modified Boyden chambers. Figure 4b shows that loss of MIF significantly impaired the migratory capacity of the cells. Numbers of migrating control cells was more than 4‐fold compared to those lacking MIF, over a given time period (Fig. 4b, right panel). To confirm this result, we carried out a wound closure assay. As shown in Fig. 4c, HeLa pSRP control cells extensively migrated into the denuded area within 48 h (upper panels), whereas the migratory capacity of HeLa pSRP/MIF knockdown cells was severely compromised (lower panels).
Dynamic assembly and disassembly of focal adhesions play a central role in cell migration, and thus a number of intracellular signalling proteins are involved in forming focal adhesion complexes (37). Among them, Src and focal adhesion kinase (FAK) are two key players and are known to be heavily involved in tumour metastasis (37, 38). Indeed, by western blotting analysis we found that HeLa pSRP/MIF knockdown cells had reduced levels of Src and FAK expression compared to the pSRP control cells (Fig. 4d).
Loss of MIF prevents anchorage‐independent growth and in vivo tumour formation of HeLa cells
To determine transforming capacity of HeLa MIF‐knockdown cells, we carried out an anchorage‐independent growth assay. As shown in Fig. 5a, HeLa pSRP control cells formed sizable colonies in soft agar, whereas HeLa pSRP/MIF knockdown cells failed to do so (left panel). Regardless of their size, numbers of colonies formed by HeLa pSRP/MIF knockdown cells was also significantly less than of HeLa pSRP control cells (right panel).
Figure 5.
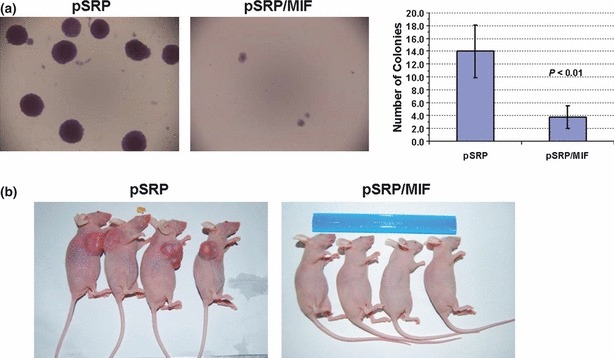
Transformation analysis. (a) Anchorage‐independent growth assay. HeLa pSRP control and pSRP/MIF knockdown cells were plated in soft agar as described in the Materials and methods section. Fifteen days later, colony formation was examined and subjected to microscope photography. All photographs of cells are at the same scale. Numbers of colonies per examined field are shown at the right. (b) In vivo tumour formation assay. HeLa pSRP control and pSRP/MIF knockdown cells were injected into nude mice as described in the Materials and methods section. Twenty days later, animals were sacrificed and photographs were taken during post‐mortem examination.
Next, we examined the ability of HeLa MIF‐knockdown cells to form tumours in vivo. As shown in Fig. 5b, HeLa pSRP control cells readily gave rise to massive tumours in immune‐compromised nude mice, whereas HeLa pSRP/MIF knockdown cells displayed no tumour growth. Taken together, these findings suggest that MIF is essential for tumourigenesis of HeLa cells.
Discussion
Compelling evidence has suggested that malignant processes can be associated with chronic inflammation (39). Thus, a proinflammatory cytokine like MIF has the potential for playing a role in tumour progression (40, 41). In the present study, we utilized an RNAi approach to examine the role of MIF in cancer cell proliferation using human HeLa cervical cancer cells as a model system.
It has been shown by others that cyclin D1 levels are much less in primary fibroblasts isolated from MIF−/− mice than those from MIF+/+ animals (40, 42). Consistent with these findings, we found that cyclin D1 expression was significantly reduced in HeLa MIF‐knockdown cells (Fig. 2), but it is not clear how MIF might have affected cyclin D1 expression in the cells. Some studies have suggested that this could be regulated through an E2F‐dependent pathway (43), or by a mechanism involving Rho GTPase‐mediated activation of MAP kinase and its downstream effector, FAK (42, 44). To test the second possibility, we performed western blotting using antibodies specific to FAK and as a result, we found that loss of MIF indeed affected expression of this protein (Fig. 4d). However, it is premature to conclude that MIF regulates cyclin D1 expression through FAK‐mediated adhesion‐dependent signalling in HeLa cells. Further studies need to be carried out to examine the effect of MIF on other cell cycle factors, as well as to know how these factors might contribute to MIF‐induced cell proliferation and its response to DNA damage.
Cell cyclin analysis by FACS allowed us to examine apoptotic cell death, measured by the proportion of sub‐G1 cells. Previous studies by others have shown that transient suppression of MIF function by RNAi or MIF inhibitors often induced apoptosis in human and mouse cells. In a mouse knockout model, loss of MIF resulted in several folds increase of apoptotic cell death, and such cell death was further increased upon DNA damage (34). In this study we found that, however, long‐term loss of MIF did not show significant amount of apoptotic cell death in the human cancer cells we used, no matter presence/absence of DNA damage. The discrepancy between mouse and human cells was not clear. In our case, we believe that apoptotic cells, if any, might have been eliminated during establishment of stable cell lines. Furthermore, we found that when cells were treated with low doses of doxorubicin, MIF‐deficient HeLa cells underwent DNA re‐replication in S phase (Fig. 3), suggesting the role of MIF in DNA damage response in the cells. The underlying molecular mechanism, however, was not clear.
Our studies demonstrate that although HeLa MIF‐knockdown cells could grow in Petri dishes at a much slower rate compared to controls (Fig. 1a), they did not form any measurable size tumours in vivo after being transplanted into nude mice (Fig. 5b), suggesting that MIF is essential for tumourigenesis of HeLa cells. In addition to modulating c‐Myc expression, we tend to believe that MIF has multiple targets on malignant transformation of human cells. There are reports to show that loss of MIF reduces angiogenesis, which is critical for development and growth of tumours (21, 23, 45).
In conclusion, we have presented evidence that MIF protein is linked to growth and tumourigenesis of HeLa cells, possibly in concert with other intracellular and extracellular growth factors (22, 46, and see review in 47). To confirm this notion, we are currently examining the effect of MIF‐knockdown in other human cancer cell lines using a similar method. In our perspective, cells derived from different types of tissues are of particular interest, since expression of MIF may vary from cell to cell. Nonetheless, the present data suggest that MIF may represent a novel target for therapeutic intervention for human cancer.
Acknowledgements
We thank Mr. Ailin Guo for his assistance in animal experiments and Ms. Zhi Xie for her technical support for histology assessment. This project was supported by National Natural Science Foundation of China (No.81070103) (XYY), Guangdong Natural Science Foundation (No. 06020897) (DZX, BD) and the Medical Science Foundation of Guangdong Province, China (No. A2009030) (DZX, WH).
References
- 1. Paralkar V, Wistow G (1994) Cloning the human gene for macrophage migration inhibitory factor (MIF). Genomics 19, 48–51. [DOI] [PubMed] [Google Scholar]
- 2. Kozak CA, Adamson MC, Buckler CE, Segovia L, Paralkar V, Wistow G (1995) Genomic cloning of mouse MIF (macrophage inhibitory factor) and genetic mapping of the human and mouse expressed gene and nine mouse pseudogenes. Genomics 27, 405–411. [DOI] [PubMed] [Google Scholar]
- 3. Mitchell R, Bacher M, Bernhagen J, Pushkarskaya T, Seldin MF, Bucala R (1995) Cloning and characterization of the gene for mouse macrophage migration inhibitory factor (MIF). J. Immunol. 154, 3863–3870. [PubMed] [Google Scholar]
- 4. Larson DF, Horak K (2006) Macrophage migration inhibitory factor: controller of systemic inflammation. Crit. Care 10, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bloom BR, Bennett B (1966) Mechanism of a reaction in vitro associated with delayed‐type hypersensitivity. Science 153, 80–82. [DOI] [PubMed] [Google Scholar]
- 6. David JR (1966) Delayed hypersensitivity in vitro: its mediation by cell‐free substances formed by lymphoid cell‐antigen interaction. Proc. Natl. Acad. Sci. USA 56, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morand EF, Leech M, Bernhagen J (2006) MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 5, 399–410. [DOI] [PubMed] [Google Scholar]
- 8. Lin SG, Yu XY, Chen YX, Huang XR, Metz C, Bucala R et al. (2000) De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ. Res. 87, 1202–1208. [DOI] [PubMed] [Google Scholar]
- 9. Kong YZ, Yu X, Tang JJ, Ouyang X, Huang XR, Fingerle‐Rowson G et al. (2005) Macrophage migration inhibitory factor induces MMP‐9 expression: implications for destabilization of human atherosclerotic plaques. Atherosclerosis 178, 207–215. [DOI] [PubMed] [Google Scholar]
- 10. Yu X, Lin SG, Huang XR, Bacher M, Leng L, Bucala R et al. (2007) Macrophage migration inhibitory factor induces MMP‐9 expression in macrophages via the MEK‐ERK MAP kinase pathway. J. Interferon Cytokine Res. 27, 103–109. [DOI] [PubMed] [Google Scholar]
- 11. Zhong JC, Yu XY, Lin QX, Li XH, Huang XZ, Xiao DZ et al. (2008) Enhanced angiotensin converting enzyme 2 regulates the insulin/Akt signalling pathway by blockade of macrophage migration inhibitory factor expression. Br. J. Pharmacol. 153, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rao F, Deng CY, Wu SL, Xiao DZ, Yu XY, Kuang SJ et al. (2009) Involvement of Src in L‐type Ca2+ channel depression induced by macrophage migration inhibitory factor in atrial myocytes. J. Mol. Cell. Cardiol. 47, 586–594. [DOI] [PubMed] [Google Scholar]
- 13. Liang JL, Xiao DZ, Liu XY, Lin QX, Shan ZX, Zhu JN et al. (2010) High glucose induces apoptosis in AC16 human cardiomyocytes via macrophage migration inhibitory factor and c‐Jun N‐terminal kinase. Clin. Exp. Pharmacol. Physiol. 37, 969–973. [DOI] [PubMed] [Google Scholar]
- 14. Shimizu T, Abe R, Nakamura H, Ohkawara A, Suzuki M, Nishihira J (1999) High expression of macrophage migration inhibitory factor in human melanoma cells and its role in tumor cell growth and angiogenesis. Biochem. Biophys. Res. Commun. 264, 751–758. [DOI] [PubMed] [Google Scholar]
- 15. Bando H, Matsumoto G, Bando M, Muta M, Ogawa T, Funata N et al. (2002) Expression of macrophage migration inhibitory factor in human breast cancer: association with nodal spread. Jpn. J. Cancer Res. 93, 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer‐Siegler KL, Bellino MA, Tannenbaum M (2002) Macrophage migration inhibitory factor evaluation compared with prostate specific antigen as a biomarker in patients with prostate carcinoma. Cancer 94, 1449–1456. [DOI] [PubMed] [Google Scholar]
- 17. Markert JM, Fuller CM, Gillespie GY, Bubien JK, McLean LA, Hong RL et al. (2001) Differential gene expression profiling in human brain tumors. Physiol. Genomics 5, 21–33. [DOI] [PubMed] [Google Scholar]
- 18. Han I, Lee MR, Nam KW, Oh JH, Moon KC, Kim HS (2008) Expression of macrophage migration inhibitory factor relates to survival in high‐grade osteosarcoma. Clin. Orthop. Relat. Res. 466, 2107–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor JA III, Kuchel GA, Hegde P, Voznesensky OS, Claffey K, Tsimikas J et al. (2007) Null mutation for macrophage migration inhibitory factor (MIF) is associated with less aggressive bladder cancer in mice. BMC Cancer 7, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fingerle‐Rowson G, Petrenko O (2007) MIF coordinates the cell cycle with DNA damage checkpoints. Lessons from knockout mouse models. Cell Div. 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogawa H, Nishihira J, Sato Y, Kondo M, Takahashi N, Oshima T et al. (2000) An antibody for macrophage migration inhibitory factor suppresses tumour growth and inhibits tumour‐associated angiogenesis. Cytokine 12, 309–314. [DOI] [PubMed] [Google Scholar]
- 22. Takahashi N, Nishihira J, Sato Y, Kondo M, Ogawa H, Ohshima T et al. (1998) Involvement of macrophage migration inhibitory factor (MIF) in the mechanism of tumor cell growth. Mol. Med. 4, 707–714. [PMC free article] [PubMed] [Google Scholar]
- 23. Hagemann T, Robinson SC, Thompson RG, Charles K, Kulbe H, Balkwill FR (2007) Ovarian cancer cell‐derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol. Cancer Ther. 6, 1993–2002. [DOI] [PubMed] [Google Scholar]
- 24. Rendon BE, Roger T, Teneng I, Zhao M, Al‐Abed Y, Calandra T et al. (2007) Regulation of human lung adenocarcinoma cell migration and invasion by macrophage migration inhibitory factor. J. Biol. Chem. 282, 29910–29918. [DOI] [PubMed] [Google Scholar]
- 25. Chesney J, Metz C, Bacher M, Peng T, Meinhardt A, Bucala R (1999) An essential role for macrophage migration inhibitory factor (MIF) in angiogenesis and the growth of a murine lymphoma. Mol. Med. 5, 181–191. [PMC free article] [PubMed] [Google Scholar]
- 26. Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R et al. (2008) A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 68, 7253–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kleemann R, Hausser A, Geiger G, Mischke R, Burger‐Kentischer A, Flieger O et al. (2000) Intracellular action of the cytokine MIF to modulate AP‐1 activity and the cell cycle through Jab1. Nature 408, 211–216. [DOI] [PubMed] [Google Scholar]
- 28. Meyer‐Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL (2006) Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU‐145 prostate cancer cells. J. Immunol. 177, 8730–8739. [DOI] [PubMed] [Google Scholar]
- 29. Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH (1999) A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 190, 1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Talos F, Mena P, Fingerle‐Rowson G, Moll U, Petrenko O (2005) MIF loss impairs Myc‐induced lymphomagenesis. Cell Death Differ. 12, 1319–1328. [DOI] [PubMed] [Google Scholar]
- 31. Nemajerova A, Moll UM, Petrenko O, Fingerle‐Rowson G (2007) Macrophage migration inhibitory factor coordinates DNA damage response with the proteasomal control of the cell cycle. Cell Cycle 6, 1030–1034. [DOI] [PubMed] [Google Scholar]
- 32. Jung H, Seong HA, Ha H (2008) Critical role of cysteine residue 81 of macrophage migration inhibitory factor (MIF) in MIF‐induced inhibition of p53 activity. J. Biol. Chem. 283, 20383–20396. [DOI] [PubMed] [Google Scholar]
- 33. Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553. [DOI] [PubMed] [Google Scholar]
- 34. Nemajerova A, Mena P, Fingerle‐Rowson G, Moll UM, Petrenko O (2007) Impaired DNA damage checkpoint response in MIF‐deficient mice. EMBO J. 26, 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gustafson WC, Weiss WA (2010) Myc proteins as therapeutic targets. Oncogene 29, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weber GF (2008) Molecular mechanisms of metastasis. Cancer Lett. 270, 181–190. [DOI] [PubMed] [Google Scholar]
- 37. Lock JG, Wehrle‐Haller B, Stromblad S (2008) Cell‐matrix adhesion complexes: master control machinery of cell migration. Semin. Cancer Biol. 18, 65–76. [DOI] [PubMed] [Google Scholar]
- 38. Brunton VG, Frame MC (2008) Src and focal adhesion kinase as therapeutic targets in cancer. Curr. Opin. Pharmacol. 8, 427–432. [DOI] [PubMed] [Google Scholar]
- 39. Hussain SP, Harris CC (2007) Inflammation and cancer: an ancient link with novel potentials. Int. J. Cancer 121, 2373–2380. [DOI] [PubMed] [Google Scholar]
- 40. Mitchell RA (2004) Mechanisms and effectors of MIF‐dependent promotion of tumourigenesis. Cell. Signal. 16, 13–19. [DOI] [PubMed] [Google Scholar]
- 41. Bucala R, Donnelly SC (2007) Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity 26, 281–285. [DOI] [PubMed] [Google Scholar]
- 42. Liao H, Bucala R, Mitchell RA (2003) Adhesion‐dependent signaling by macrophage migration inhibitory factor (MIF). J. Biol. Chem. 278, 76–81. [DOI] [PubMed] [Google Scholar]
- 43. Petrenko O, Moll UM (2005) Macrophage migration inhibitory factor MIF interferes with the Rb‐E2F pathway. Mol. Cell 17, 225–236. [DOI] [PubMed] [Google Scholar]
- 44. Swant JD, Rendon BE, Symons M, Mitchell RA (2005) Rho GTPase‐dependent signaling is required for macrophage migration inhibitory factor‐mediated expression of cyclin D1. J. Biol. Chem. 280, 23066–23072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishihira J, Ishibashi T, Fukushima T, Sun B, Sato Y, Todo S (2003) Macrophage migration inhibitory factor (MIF): its potential role in tumor growth and tumor‐associated angiogenesis. Ann. N Y Acad. Sci. 995, 171–182. [DOI] [PubMed] [Google Scholar]
- 46. Mitchell RA, Metz CN, Peng T, Bucala R (1999) Sustained mitogen‐activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J. Biol. Chem. 274, 18100–18106. [DOI] [PubMed] [Google Scholar]
- 47. Bach JP, Rinn B, Meyer B, Dodel R, Bacher M (2008) Role of MIF in inflammation and tumorigenesis. Oncology 75, 127–133. [DOI] [PubMed] [Google Scholar]