

Isoprenoid is the largest family of natural compounds, including important bioactive molecules such as vitamins, hormones, and natural medicines. The mevalonate pathway is a target for metabolic engineering because it supplies precursors for isoprenoid biosynthesis. Mevalonate 3-kinase is an enzyme involved in the modified mevalonate pathway specific to limited species of thermophilic archaea. Replacement of a single amino acid residue in the active site of the enzyme changed its substrate preference and allowed the mutant enzymes to catalyze a previously undiscovered reaction. Using the genes encoding the mutant enzymes and other archaeal enzymes, we constructed an artificial mevalonate pathway, which can produce the precursor of isoprenoid through an unexplored route, in bacterial cells.
KEYWORDS: archaea, decarboxylase, isoprenoid, kinase, mevalonate pathway, mutagenesis
ABSTRACT
Mevalonate 3-kinase plays a key role in a recently discovered modified mevalonate pathway specific to thermophilic archaea of the order Thermoplasmatales. The enzyme is homologous to diphosphomevalonate decarboxylase, which is involved in the widely distributed classical mevalonate pathway, and to phosphomevalonate decarboxylase, which is possessed by halophilic archaea and some Chloroflexi bacteria. Mevalonate 3-kinase catalyzes the ATP-dependent 3-phosphorylation of mevalonate but does not catalyze the subsequent decarboxylation as related decarboxylases do. In this study, a substrate-interacting glutamate residue of Thermoplasma acidophilum mevalonate 3-kinase was replaced by smaller amino acids, including its counterparts in diphosphomevalonate decarboxylase and phosphomevalonate decarboxylase, with the aim of altering substrate specificity. These single amino acid mutations resulted in the conversion of mevalonate 3-kinase into 5-phosphomevalonate 3-kinase, which can synthesize 3,5-bisphosphomevalonate from 5-phosphomevalonate. The mutants catalyzing the hitherto undiscovered reaction enabled the construction of an artificial mevalonate pathway in Escherichia coli cells, as was demonstrated by the accumulation of lycopene, a red carotenoid pigment.
IMPORTANCE Isoprenoid is the largest family of natural compounds, including important bioactive molecules such as vitamins, hormones, and natural medicines. The mevalonate pathway is a target for metabolic engineering because it supplies precursors for isoprenoid biosynthesis. Mevalonate 3-kinase is an enzyme involved in the modified mevalonate pathway specific to limited species of thermophilic archaea. Replacement of a single amino acid residue in the active site of the enzyme changed its substrate preference and allowed the mutant enzymes to catalyze a previously undiscovered reaction. Using the genes encoding the mutant enzymes and other archaeal enzymes, we constructed an artificial mevalonate pathway, which can produce the precursor of isoprenoid through an unexplored route, in bacterial cells.
INTRODUCTION
The mevalonate (MVA) pathway is one of the two biosynthetic routes that supply fundamental C5 precursors, isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate, for diverse isoprenoids, which include >70,000 natural compounds (1, 2). The MVA pathway exists in almost all eukaryotes and archaea and in a minor portion of bacteria, whereas the other biosynthetic route, the methylerythritol phosphate pathway, exists in a major portion of bacteria and in the chloroplasts of higher plants. In the MVA pathway, three molecules of acetyl-coenzyme A (CoA) are condensed and reduced to form a central C6 intermediate, MVA. The conversion route from MVA to IPP, however, has shown unexpected variations among biological species (Fig. 1). Almost all eukaryotes, most of the MVA pathway-utilizing bacteria, and the archaea of the order Sulfolobales (3) utilize mevalonate kinase (MVK), phosphomevalonate kinase (PMK), and diphosphomevalonate decarboxylase (DMD) for that purpose. The pathway that includes this set of enzymes is called the “classical” MVA pathway because it was discovered more than half a century ago (4). On the other hand, a portion of the bacteria of the phylum Chloroflexi and all halophilic archaea belonging to the class Halobacteria utilize a modified MVA pathway instead (5, 6). The pathway, which is called modified MVA pathway I, converts MVA into IPP using MVK, phosphomevalonate decarboxylase (PMD), and isopentenyl phosphate kinase (IPK). PMDs from Chloroflexi bacteria and Halobacteria archaea are highly homologous to DMD, while the existence of PMD in the majority of archaea has been questionable due to an absence of DMD homologous genes in their genomes (7, 8). The third option for the conversion of MVA into IPP was recently discovered from archaea in the order Thermoplasmatales (9, 10). In the pathway called modified MVA pathway II, MVA is phosphorylated at the 3-hydroxyl group to yield 3-phosphomevalonate (MVA-3-P) by the action of mevalonate 3-kinase (M3K) rather than at the 5-hydroxyl group as in the reaction of MVK (9–11). Intriguingly, M3K is also homologous to DMD. After the formation of MVA-3-P, another kinase, MVA-3-P 5-kinase (M3P5K), catalyzes its 5-phosphorylation (10), and a subsequent decarboxylation is catalyzed by another DMD homolog, 3,5-bisphosphomevalonate decarboxylase (BMD), to give isopentenyl phosphate (IP) (12). IP is then phosphorylated by IPK to yield IPP (13, 14).
FIG 1.
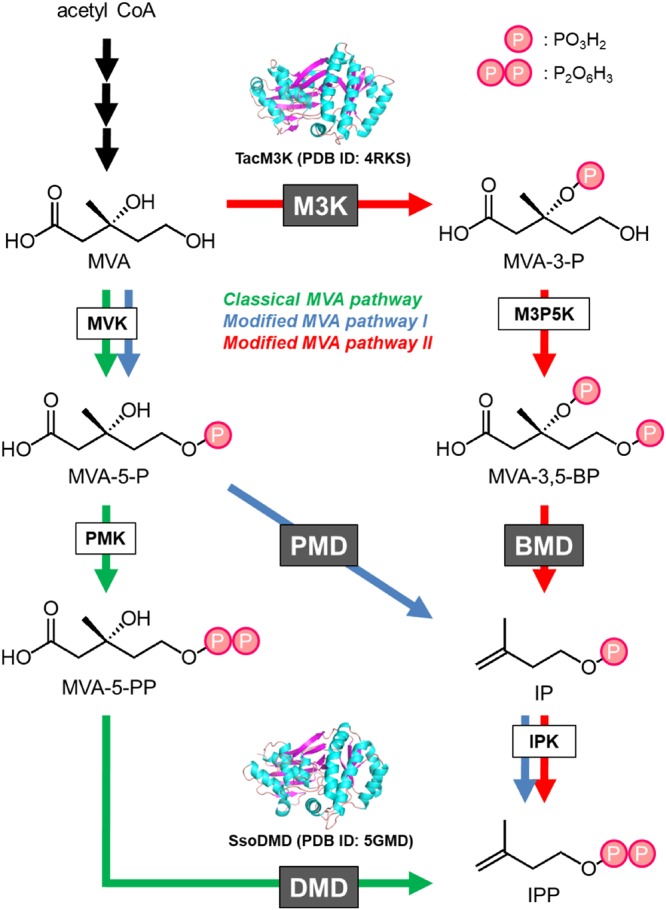
Distribution of DMD homologues in various MVA pathways. The enzyme reactions downstream from MVA in the classical MVA pathway, which is utilized by almost all eukaryotes, a minor part of bacteria, and the archaea of the order Sulfolobales, are indicated in green; those of modified MVA pathway I, which is utilized by halophilic archaea and some Chloroflexi bacteria, are shown in blue; and those of modified MVA pathway II, which is utilized by the archaea of the order Thermoplasmatales, are shown in red. The names of the DMD homologues, DMD, PMD, M3K, and BMD, and the other enzymes are indicated in boxes.
This unexpected variation in the MVA pathway piqued our interest in its molecular evolution. In particular, the evolutionary relationship among the DMD homologues, i.e., DMD, PMD, M3K, and BMD (Fig. 1), is intriguing. These enzymes possess distinct substrate specificities toward 5-diphosphomevalonate (MVA-5-PP), 5-phosphomevalonate (MVA-5-P), MVA, and 3,5-bisphosphomevalonate (MVA-3,5-BP), respectively. Also, DMD and PMD catalyze ATP-dependent decarboxylation reactions unlike either the ATP-dependent kinase M3K or the ATP-independent decarboxylase BMD. Indeed, DMD and PMD engage in the phosphorylation of the 3-hydroxy group of their substrates, but they also catalyze the subsequent decarboxylation in tandem with the elimination of the 3-phosphate group of intermediates. M3K catalyzes only 3-phosphorylation, and BMD catalyzes decarboxylation coupled with 3-phosphate elimination. This means that the reactions of M3K and BMD are similar to the first and second half-reactions of DMD/PMD, i.e., phosphorylation and phosphate elimination/decarboxylation, respectively. Vinokur et al. recently solved the crystal structure of M3K from a thermophilic archaeon, Thermoplasma acidophilum, and compared this structure with the known structures of DMD (15). In their study, the absence of the decarboxylase activity in M3K was in part attributed to the absence of an aspartate residue conserved in DMD and PMD, but they did not test their hypothesis by mutagenesis. In our recent work, the Asp281 residue of DMD from a thermophilic archaeon, Sulfolobus solfataricus, was replaced with, for example, its counterpart in M3K, threonine (16). As a result, the M3K-mimicking mutant still recognized MVA-5-PP as the substrate, but the decarboxylase activity was lost, while the 3-kinase activity that yields 3-phospho-5-diphosphomevalonate (MVA-3-P-5-PP) was retained.
In the present work, we performed a mutational study on T. acidophilum M3K (TacM3K) to elucidate the reason(s) for the difference in the substrate specificities of its homologues. A conserved glutamate residue of M3K, which is involved in substrate recognition, was replaced with other amino acids, including its counterparts in DMD and PMD. The result of the study led to a new insight into the substrate specificity of this family of enzymes. In addition, we succeeded in constructing an artificial mevalonate pathway in Escherichia coli cells using the M3K mutants that catalyze a hitherto undiscovered reaction.
RESULTS
Mutation analysis of TacM3K.
A comparison between the substrate-complex crystal structure of TacM3K (PDB accession number 4RKS) (15) and that of S. solfataricus DMD (SsoDMD) (PDB accession number 5GMD) (16) revealed interesting differences in the structures of the active sites (Fig. 2). The Glu140 residue interacting with the 5-hydroxy group of MVA resides at the active site of TacM3K, while Gly140 is found at the corresponding position of SsoDMD. Furthermore, multiple alignments using the amino acid sequences of DMD homologues showed that Ala138 exists at the corresponding position in Haloferax volcanii PMD (see Fig. S1 in the supplemental material). The steric hindrance introduced by Glu140 seems responsible for excluding larger substrates, such as MVA-5-P and MVA-5-PP, from the active site of TacM3K. This finding suggests that the size of the space regulated by the residues at position 140 in the active sites of TacM3K and SsoDMD contributes to the differences in substrate preferences toward MVA and MVA-5-PP, respectively. To examine this possibility, we constructed three E140 mutants of TacM3K (E140A, E140S, and E140G). The glycine mutant (E140G) and the alanine mutant (E140A) mimic the active sites of SsoDMD and Haloferax volcanii PMD, respectively. The mutants were recombinantly expressed in E. coli, purified by affinity chromatography (Fig. S2), and used in an enzyme assay with radioactive substrates and thin-layer chromatography (TLC) (Fig. 3). M3K activity, which yields MVA-3-P from MVA in the presence of ATP, was observed in the E140S and E140G mutants, whereas the E140A mutant showed no activity, as shown by the normal-phase TLC analysis of their products (Fig. 3 and S3A). In contrast, all of the E140 mutants recognized MVA-5-P and converted it into a highly polar product with an Rf value that was lower than that of MVA-5-PP. The highest conversion activity was observed with the E140G mutant, followed in order by the E140S and E140A mutants (Fig. 3 and S3B). Neither wild-type TacM3K nor any mutants showed reactivity toward MVA-5-PP (Fig. 3).
FIG 2.

Active-site structures of TacM3K and SsoDMD. In the MVA complex structure of TacM3K (PDB accession number 4RKS) (A) and the MVA-5-PP complex structure of SsoDMD (PDB accession number 5GMD) (B), the substrates and the substrate-interacting residues (or their counterparts) are shown as stick models.
FIG 3.

Radio-TLC analysis of the products of the wild-type TacM3K and E140 mutants. Normal-phase TLC plates were developed with n-propanol–28% ammonia water (NH4OH)–H2O (6:3:1). s.f., solvent front; ori, origin.
Analysis of the E140S reaction product from MVA-5-P.
For structural determination of the highly polar products from the E140 mutants, nuclear magnetic resonance (NMR) analysis was carried out using the reaction product of the E140S mutant, which showed relatively weak residual M3K activity but high reactivity toward MVA-5-P (Fig. 3). Prior to the NMR analysis, [U-13C]MVA-5-P was enzymatically synthesized from [U-13C]MVA. In the presence of ATP and Mg2+, purified S. solfataricus MVK was reacted with a 100% 13C-enriched substrate to yield [U-13C]MVA-5-P. After the removal of MVK by filtration, the E140S mutant and additional ATP were added to the reaction mixture to perform the next reaction. Finally, NMR analysis was conducted after the removal of the enzyme (Fig. 4 and Table 1). As a result of the reaction with the E140S mutant, a quartet-of-doublets signal emerged at 78.20 ppm in tandem with a decrease in the C-3 signal of MVA-5-P at 70.90 ppm, which suggested that about half of MVA-5-P had reacted (Fig. 4A and B). The chemical shift of the signal was in good agreement with the reported chemical shift of the C-3 signal of MVA-3,5-BP, the product of M3P5K from T. acidophilum (10), and with that of MVA-3-P-5-PP, which was produced by SsoDMD mutants that lacked decarboxylation activity (16). Moreover, the signal seemed to acquire an additional hyperfine coupling that resulted from the two-bond 13C-31P coupling, which was obvious in the C-3 signals of MVA-3,5-BP and MVA-3-P-5-PP (10, 16). Slight but obvious changes between the spectra of the samples reacted with or without the E140S mutant were also found in the vicinity of the signals of the other carbons of MVA-5-P (Fig. 4C and D). Near the C-1 doublet signal of MVA-5-P at 180.31 ppm, a smaller doublet signal emerged at 179.25 ppm. The peak number of the C-2 doublet-of-doublets signal of MVA-5-P at 48.02 ppm was increased after the reaction with the E140S mutant, probably due to the emergence of a new doublet-of-doublets signal at 48.69 ppm. The apparent C-4 triplet signal of MVA-5-P at 41.26 ppm was shifted to 40.90 ppm, and the C-5 doublet signal of MVA-5-P at 61.15 ppm was slightly shifted to 60.95 ppm after the reaction with the E140S mutant. A new doublet signal came into existence at 25.28 ppm, which approximated the C-6 doublet signal of MVA-5-P at 25.90 ppm. These results suggested that the E140S mutant probably catalyzed the 3-phosphorylation of MVA-5-P to yield MVA-3,5-BP, but we could not exclude the possibility of other modifications such as diphosphorylation or adenylylation because the NMR data were not in complete agreement with that of MVA-3,5-BP reported by Vinokur et al. (10) (Table 1).
FIG 4.

13C-NMR analysis of the E140S reaction product from [U-13C]MVA-5-P. The partial areas of the NMR spectra around the signals of C-3 (A and B) and the other carbons (C and D) of [U-13C]MVA-5-P are shown. For the data shown in panels A and C, the sample from the negative-control reaction with water was used; for the data shown in panels B and D, the sample from the reaction with the E140S mutant was used. Asterisks shown in green and red indicate, respectively, the signals of [U-13C]MVA-5-P and the product.
TABLE 1.
13C NMR spectroscopic data
| Compound | Carbon no. | Chemical shift (ppm) | Coupling patterna |
|---|---|---|---|
| [U-13C]MVA-5-P | 1 | 180.31 | d |
| 2 | 48.02 | dd | |
| 3 | 70.90 | q | |
| 4 | 41.26 | t | |
| 5 | 61.15 | d | |
| 6 | 25.90 | d | |
| MVA-3,5-BPb | 1 | 180.20 | d |
| 2 | 49.74 | m | |
| 3 | 76.73 | qd | |
| 4 | 41.26 | m | |
| 5 | 60.99 | d | |
| 6 | 25.19 | d | |
| E140S reaction product from [U-13C]MVA-5-P | 1 | 179.25 | d |
| 2 | 48.69 | dd | |
| 3 | 78.20 | qd | |
| 4 | 40.90 | t | |
| 5 | 60.95 | d | |
| 6 | 25.28 | d |
Symbols are as follows: d, doublet; dd, doublet of doublets; t, triplet; q, quartet; m, multiplet; qd, quartet of doublets.
From Vinokur et al. (10).
To confirm the formation of MVA-3,5-BP, an electrospray ionization mass spectrometry (ESI-MS) analysis of the product was performed in the negative mode using the direct infusion method. In the MS profile of the sample from the reaction of the E140S mutant with (R,S)-MVA-5-P, an ion peak with an m/z of 307.0 was observed, which was absent from the MS profile of the sample from the reaction of the wild-type TacM3K (Fig. 5A and B). The m/z value corresponded well with the negative ions from both MVA-5-PP and MVA-3,5-BP, but based on the results from the NMR analysis, the ion peak was considered to arise from MVA-3,5-BP. The tandem MS (MS/MS) analysis of the ion gave fragment ions with m/z values of 262.8, 244.8, 208.8, and 164.9, which supposedly derived from [M-CO2]−, [M-CO2-H2O]−, [M-H3PO4]−, and [M-H3PO4-CO2]−, respectively (Fig. 5C). The reaction with the E140A and E140G mutants gave almost the same results as those from a reaction with the E140S mutant (Fig. S4).
FIG 5.

Negative ESI-MS analyses of the E140S reaction product from (R,S)-MVA-5-P. Mass spectra are shown of the samples from the control reaction with the wild-type TacM3K (A) and from the reaction with the E140S mutant (B). (C) MS/MS analysis of the ion of m/z 307.0 from the experiment shown in panel B.
Lycopene production in E. coli harboring the artificial MVA pathway using the TacM3K E140 mutants.
In this work, we obtained the E140A, E140S, and E140G mutants of TacM3K, whose substrate specificities were altered. The mutants catalyze the phosphorylation reaction of the 3-hydroxyl group of MVA-5-P and synthesize MVA-3,5-BP, whereas the wild-type TacM3K catalyzes the 3-kinase reaction of MVA. Interestingly, the MVA-3,5-BP is synthesized through the phosphorylation reaction of the 5-hydroxy group of MVA-3-P that is catalyzed by M3P5K (10) belonging to modified MVA pathway II in the Thermoplasmatales archaea. This means that the same compound can be synthesized via different enzyme reactions or metabolic routes. Therefore, the TacM3K mutants can be used for construction of a previously uncharacterized route to MVA-3,5-BP. In this route, MVA is first converted into MVA-5-P by the action of MVK, which follows a classical MVA pathway. Second, a 3-kinase reaction of MVA-5-P is catalyzed by the TacM3K mutants to synthesize MVA-3,5-BP.
Thus, we tried to create an artificial mevalonate pathway (Fig. 6A, red open arrows) by constructing a plasmid harboring 4 genes: ma0602 encoding Methanosarcina acetivorans MVK, ma0603 encoding M. acetivorans IPK, the gene of the TacM3K mutant, and the codon-optimized pto0478 gene encoding Picrophilus torridus BMD (PtBMD) (Fig. 6B). The ma0602 and ma0603 genes were grouped under an arabinose promoter, and the TacM3K and pto0478 genes were grouped under a tac promoter without an operator sequence for constitutive expression. The TacM3K gene was amplified from the plasmid for the expression of the wild type and the E140A, E140S, and E140G mutants to construct pBAD-MIB-M3K(WT), pBAD-MIB-M3K(E140A), pBAD-MIB-M3K(E140S), and pBAD-MIB-M3K(E140G), respectively (Table 2). The strains, ST-WT, ST-A, ST-S, and ST-G, were constructed by transforming the E. coli cells harboring pBAD-MIB-M3K(WT), pBAD-MIB-M3K(E140A), pBAD-MIB-M3K(E140S), and pBAD-MIB-M3K(E140G), respectively, with the plasmid pACYC-IBEidi expressing the geranylgeranyl diphosphate synthase gene (crtE), the phytoene synthase gene (crtB), the phytoene desaturase gene (crtl), and the IPP isomerase gene (idi), which were sufficient for lycopene production in E. coli (Fig. 6A and B and Table 2). As a control strain, we constructed ST-NC harboring the parental plasmid pBAD18 as well as pACYC-IBEidi. We also constructed the strains expressing known MVA pathways. The pBAD plasmid containing the genes of MVK, PMK, and DMD from Saccharomyces cerevisiae (pBAD-ScMPD) was used for expression of a part of the classical MVA pathway (Fig. 6B). The codon usage of the MVK and PMK genes has been optimized for E. coli. The pBAD-TacM plasmid series contains the genes of M3K, M3P5K, BMD, and IPK for the expression of part of modified MVA pathway II (Fig. 6B). Although the M3K, M3P5K, and IPK genes are derived from T. acidophilum, various BMD genes have been utilized for plasmid construction. Based on the first report on BMD by Vinokur et al. (12), P. torridus BMD, putative full-length T. acidophilum BMD, and truncated T. acidophilum BMD that starts from the Met30 residue were used to construct pBAD-TacM(PtBMD), pBAD-TacM(TaBMDfull), and pBAD-TacM(TaBMDtrunc), respectively. E. coli harboring pACYC-IBEidi was transformed with pBAD-ScMPD, pBAD-TacM(PtBMD), pBAD-TacM(TaBMDfull), and pBAD-TacM(TaBMDtrunc) to construct the strains ScMPD, TacM(PtBMD), TacM(TaBMDfull), and TacM(TaBMDtrunc), respectively (Table 2). These strains were cultivated in 5 ml of LB medium containing 2.5 mg of (R)-mevalonolactone at 37°C for 48 h, and lycopene was extracted from the cells using acetone. As a result of cultivation, the cell pellets of the ST-G strain showed the strongest red among the strains expressing the artificial MVA pathway, followed in order by those of ST-S and ST-A (Fig. 6C), whereas the pellets of ST-NC and ST-WT showed little coloration. As a result of the acetone extraction, the amounts of lycopene extracted from the ST-NC, ST-WT, ST-A, ST-S, and ST-G strains were 0.045 ± 0.003, 0.088 ± 0.018, 0.175 ± 0.032, 0.165 ± 0.081, and 0.347 ± 0.014 μg/mg wet cells, respectively (Fig. 6D), which agreed with the coloration of the cell pellets. These results demonstrated that the ST-G strain efficiently converted the supplied mevalonolactone into lycopene, while the conversion by the ST-A and ST-S strains was more moderate. In contrast, ST-NC and ST-WT produced only slight amounts of lycopene. Given the biological properties of the enzymes, these data suggest that the artificial MVA pathway constructed in E. coli cells functions properly. The amount of lycopene from the ST-G strain was slightly lower than but comparable to that from the ScMPD strain, which expresses the classical MVA pathway (Fig. 6D). The amounts of lycopene extracted from the ScMPD, TacM(PtBMD), TacM(TaBMDfull), and TacM(TaBMDtrunc) strains were 0.491 ± 0.045, 0.059 ± 0.013, 0.007 ± 0.001, and 0.073 ± 0.006 μg/mg wet cells, respectively. Given the facts that the codon-optimized S. cerevisiae MVK and PMK genes are used in pBAD-ScMPD and that pBAD-MIB-M3K(E140G) contains the genes from thermophilic archaea T. acidophilum and P. torridus, it is surprising that lycopene production in E. coli cells via the artificial pathway was comparable to that in the classical MVA pathway, whose ability for the enhancement of isoprenoid production has been established. Optimization of the artificial MVA pathway in the future for expression in, for example, E. coli cells might provide a promising tool to enhance the production of valuable isoprenoids. Lycopene yields from the strains expressing modified MVA pathway II were similar to or lower than those from ST-NC, suggesting that the pathway from thermophilic archaea did not act effectively in E. coli cells.
FIG 6.

Carotenoid production in E. coli strains that possess an artificial MVA pathway. (A) Scheme of lycopene biosynthesis via the artificial or known MVA pathways. The artificial MVA pathway is indicated by red open arrows. The known MVA pathways utilized to construct the strains for comparison are colored as in Fig. 1, and the unused modified MVA pathway I is also indicated. The enzyme catalyzing each reaction is boxed. (B) The plasmids used to construct the lycopene-producing strains. Detailed information about the inserted genes is provided in Table 2. A red asterisk means that the BMD gene from P. torridus or T. acidophilum (in the full-length or truncated form) was used to construct the pBAD-TacM plasmid series. (C) The cell pellets of the strains used, as indicated. (D) Amounts of lycopene extracted from the strains. Cultivation of each strain and extraction of lycopene from the cells were performed in triplicate. DMAPP, dimethylallyl diphosphate.
TABLE 2.
Constructed plasmids and strains in this study
| Name | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | λ– φ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK– mK+) phoA supE44 thi-1 gyrA96 relA1 | Takara Bio, Inc., Japan |
| ST-WT | DH5α/pBAD-MIB-M3K(WT)/pACYC-IBEidi | This study |
| ST-A | DH5α/pBAD-MIB-M3K(E140A)/pACYC-IBEidi | This study |
| ST-S | DH5α/pBAD-MIB-M3K(E140S)/pACYC-IBEidi | This study |
| ST-G | DH5α/pBAD-MIB-M3K(E140G)/pACYC-IBEidi | This study |
| ST-NC | DH5α/pBAD18/pACYC-IBEidi | This study |
| ScMPD | DH5α/pBAD-ScMPD/pACYC-IBEidi | This study |
| TacM(PtBMD) | DH5α/pBAD-TacM(PtBMD)/pACYC-IBEidi | This study |
| TacM(TaBMDfull) | DH5α/pBAD-TacM(TaBMDfull)/pACYC-IBEidi | This study |
| TacM(TaBMDtrunc) | DH5α/pBAD-TacM(TaBMDtrunc)/pACYC-IBEidi | This study |
| Plasmids | ||
| pBAD18 | Cloning and expression vector, arabinose-inducible promoter, pBR322 ori, Ampr | Guzman et al. (26) |
| pACYC184 | p15A ori, Cmr, Tetr | New England BioLabs |
| pET28a(+)-pto0478 | pET28a(+) containing pto0478 encoding P. torridus BMD, fused with N-terminal 6×His tag | Vinokur et al. (12) |
| pBAD-MI | pBAD18 containing ma0602 and ma0603 encoding MVK and IPK from M. acetivorans, respectively | This study |
| pBAD-MI-M3K(WT) | pBAD-MI containing tac promotor sequence excluding operator sequence and TacM3K gene | This study |
| pBAD-MI-M3K(E140A) | pBAD-MI containing tac promotor sequence excluding operator sequence and TacM3K(E140A) gene | This study |
| pBAD-MI-M3K(E140S) | pBAD-MI containing tac promotor sequence excluding operator sequenceand TacM3K(E140S) gene | This study |
| pBAD-MI-M3K(E140G) | pBAD-MI containing tac promotor sequence excluding operator sequence and TacM3K(E140G) gene | This study |
| pBAD-MIB-M3K(WT) | pBAD-MI-M3K(WT) containing pto0478 next to the TacM3K gene | This study |
| pBAD-MIB-M3K(E140A) | pBAD-MI-M3K(E140A) containing pto0478 next to the TacM3K(E140A) gene | This study |
| pBAD-MIB-M3K(E140S) | pBAD-MI-M3K(E140S) containing pto0478 next to the TacM3K(E140S) gene | This study |
| pBAD-MIB-M3K(E140G) | pBAD-MI-M3K(E140G) containing pto0478 next to the TacM3K(E140G) gene | This study |
| pBAD-ScMPD | pBAD18 containing codon-optimized S. cerevisiae MVK and PMK genes and S. cerevisiae DMD gene | This study |
| pBAD-TacM3K5K | pBAD18 containing TacM3K gene and ta0762 | This study |
| pBAD-TacMdelI | pBAD-TacM3K5K containing pto0478 | This study |
| pBAD-TacM(PtBMD) | pBAD-TacMdelI containing ta0103 | This study |
| pBAD-TacM(TaBMDfull) | pBAD-TacM(PtBMD) whose pto0478 gene was replaced with full-length ta0893 | This study |
| pBAD-TacM(TaBMDtrunc) | pBAD-TacM(PtBMD) whose pto0478 gene was replaced with truncated ta0893 | This study |
| pACYC-IBEidi | pACYC184 containing genes for geranylgeranyl diphosphate synthase (crtE), phytoene synthase (crtB), phytoene desaturase (crtl) from Pantoea anantis and IPP isomerase (idi) from E. coli | This study |
DISCUSSION
Our trial to alter the substrate specificity of TacM3K was successful. The Glu140 residue interacts with the 5-hydroxyl group of MVA in the wild-type enzyme, and the replacement of the residue with a smaller amino acid such as Ala, Ser, or Gly seemed to enlarge the volume of the substrate-binding site enough to accommodate larger substrates such as MVA-5-P, as we had expected. The resultant mutants synthesized MVA-3,5-BP via phosphorylation of the 3-hydroxyl group of MVA-5-P. As far as we could ascertain, natural enzymes that catalyze the 3-phosphorylation of MVA-5-P are yet to be identified. Interestingly, however, MVA-3,5-BP is also synthesized via phosphorylation of the 5-hydroxyl group of MVA-3-P by M3P5K in the modified MVA pathway II (Fig. 1, red pathway). On the other hand, contrary to our expectations, the E140G mutant constructed as a mimic of DMD did not recognize MVA-5-PP as a substrate and did not synthesize MVA-3-P-5-PP. Other structural elements seem required for a suitable fit of MVA-5-PP in the substrate-binding site of the E140G mutant. We also tried to change the substrate specificity of SsoDMD by introducing a G140E mutation. The mutant showed only slight reactivity toward MVA and produced MVA-3-P (our unpublished data). The loss of decarboxylase activity of the SsoDMD G140E mutant, which was designed for creating decarboxylase specific to MVA, suggests that the diphosphate group of the substrate is required for decarboxylation or that the fine-tuning of substrate binding is needed in the active site.
Furthermore, by taking advantage of the TacM3K E140 mutants, we succeeded in constructing an artificial MVA pathway, which traces a route that differs from previously identified MVA pathways and also from another modified mevalonate pathway recently discovered from a hyperthermophilic archaeon, Aeropyrum pernix (17). In this artificial pathway, the 5-hydroxyl group of MVA, which is added into a culture medium as mevalonolactone, is phosphorylated by MVK to yield MVA-5-P. Then, MVA-5-P is converted to MVA-3,5-BP by the action of the TacM3K mutant, and thereafter IPP is synthesized via the same route as the modified MVA pathway II of the Thermoplasmatales archaea, which is catalyzed by BMD and IPK. This type of MVA pathway has never been identified from nature, which suggests that it was not selected through the evolution of MVA pathways although they show unexpectedly high levels of diversity. Given the facts that recent metagenomic analyses have discovered new archaeal and bacterial phyla (18) and that the conservation patterns of the MVA pathway-related genes in the genomes of such organisms are highly diverse (19), however, we cannot exclude the possibility that other organisms utilize this MVA pathway.
MATERIALS AND METHODS
Materials.
Precoated normal-phase TLC plates, Silica gel 60, were purchased from Merck Millipore, Germany. [2-14C]MVA-5-P (55 Ci/mol) was purchased from American Radiolabeled Chemicals, USA. [U-13C]MVA was prepared as described elsewhere (20). For the cloning of pto0478 encoding PtBMD, a pET28a(+) plasmid containing the PtBMD gene (12), which was donated by J. M. Vinokur and J. R. Bowie, University of California, Los Angeles, was used as a template DNA. pJBEI-2999 plasmid was purchased from Addgene (Watertown, MA).
General procedures.
Standard techniques of molecular biology, restriction enzyme digestions, transformations, and so on, were performed using the standard method as described by Sambrook et al. (21).
Cloning, recombinant expression, and purification of enzymes.
For the expression of MVK and PMK from S. solfataricus, the pET16b plasmids containing the sso0383 and sso2988 gene, respectively, constructed in our previous work (3), were used to transform E. coli Rosetta2 (DE3). For the expression of TacM3K, E. coli Rosetta2 (DE3) harboring the pET15b plasmid containing the ta1305 gene (pET15b-TacM3K) was used as described elsewhere (9). Partial purification of the enzymes was performed by affinity chromatography using a 1-ml HisTrap FF crude column (GE Healthcare), as described previously (3, 9). Their purity was determined by 12.5% SDS-PAGE.
Site-directed mutagenesis.
To construct the TacM3K E140A, E140S, and E140G mutants, site-directed mutagenesis was carried out with pET15b-TacM3K as a template via the QuikChange method (Stratagene). The oligonucleotide primers used (Table 3) were as follows: for E140A, TacM3K(E140A)-forward and TacM3K(E140A)-reverse; for E140S, TacM3K(E140S)-forward and TacM3K(E140S)-reverse; and for E140G, TacM3K(E140G)-forward and TacM3K(E140G)-reverse. The resulting plasmids were designated pET15b-TacM3K(E140A), pET15b-TacM3K(E140S), and pET15b-TacM3K(E140G), respectively, and the sequences of the mutants were confirmed via DNA sequencing. The processes for the recombinant expression and purification of the mutants were performed using the same methods as for the wild-type enzyme.
TABLE 3.
Primers used to construct plasmids in this study
| Primer function and name | Sequence (5′–3′) |
|---|---|
| Mutation of TacM3K expression plasmid | |
| TacM3K(E140A)-forward | CTCAGGGCGGTCTCCGCGAGCGCAGGCAGGAGC |
| TacM3K(E140A)-reverse | GCTCCTGCCTGCGCTCGCGGAGACCGCCCTGAG |
| TacM3K(E140S)-forward | CTCAGGGCGGTCTCCTCTAGCGCAGGCAGGAGC |
| TacM3K(E140S)-reverse | GCTCCTGCCTGCGCTAGAGGAGACCGCCCTGAG |
| TacM3K(E140G)-forward | CTCAGGGCGGTCTCCGGCAGCGCAGGCAGGAGC |
| TacM3K(E140G)-reverse | GCTCCTGCCTGCGCTGCCGGAGACCGCCCTGAG |
| Construction of artificial MVA pathway | |
| Ma0602-forward | ATCCGAGCTCGGTACAAGGAGTTATATATGATTTCGTGTTCTGCCCCC |
| Ma0602-reverse | TATTTATATCTCCTTTCATTCTAACCTTAAGCCCTGTTC |
| Ma0603-forward | AAGGAGATATAAATAATGAATGCTTCAACCGAACCTG |
| Ma0603-reverse | GGATCCCCGGGTACCTCATACCCTTTTATCCGGACTGATC |
| Ptac-M3K-Fw1 | AAAAGGTACCAATGAGCTGTTG |
| Ptac-M3K-Fw2 | AAGGAGATTATATATGACCTACAGATCAATAGGATC |
| Ptac-M3K-Rv1 | AAAAAAGCTTACTAGTTCACTCTGGTCTTCTATGCCAG |
| Ptac-M3K-Rv2 | ATATAATCTCCTTCCACACATTATACGAGCCGGATGATTAATTGTCAACAGCTCATTGGTACCTTTT |
| Pto0478-Fw | AAAAACTAGTAAGGAGTTATATATGAACGATCTGAACGTATACGG |
| Pto0478-Rv | AAAAAAGCTTTTACAGTTCCTCGATAATAATTTTC |
| Construction of known MVA pathway | |
| ScMVK-forward | GAATTCGAGCTCGGTACAGGAGGAAATAACCATGTCTCTGC |
| ScDMD-reverse | CTAGAGGATCCCCGGTTATTCCTTTGGTAGACCAGTCTTTG |
| Ta1305-forward | CGTTTTTTTGGGCTAGCGATGACCTACAGATCAATAGGATC |
| Ta1305-reverse | GAACATTATCCTGGAGTTCATACTAGTCTCTGGTCTTCTATGCC |
| Ta0762-forward | GCATAGAAGACCAGAGTGAAAGGAGACTAGTATGAACTCCAGGATAATGTTCATC |
| Ta0762-reverse | GGATCCCCGGGTACCGAGCTCGAATTCTCATCTCCTGTCCACAAATTTC |
| Pto0478-MP-forward | CAGGAGATGAGAATTCGAGCTCGGTACAGGAGGAGAATGAACGATCTGAACGTATACG |
| Pto0478-MP-reverse | GTCGACTCTAGAGGATCCCCGGGTACCTTACAGTTCCTCGATAATAATTTTC |
| Ta0103-forward | GGAACTGTAAGGTACCCGGGAAGAGGATGATGATACTGAAGATAGGCG |
| Ta0103-reverse | GCAGGTCGACTCTAGAGGATCCTCATCTTATCACCGTACCTATG |
| Ta0893(full)-forward | AAAAGAGCTCAGGAGGAGAATGGATACAGCCGGAATGGAAC |
| Ta0893(truncated)-forward | AAAAGAGCTCAGGAGGAGAATGGATATTGCAGAGCTTCACAGGG |
| Ta0893-reverse | AAAAGGTACCTCACAGGCGATCCTCTTCGTC |
Preparation of 14C-labeled substrates.
[2-14C]MVA and [2-14C]MVA-5-PP were enzymatically synthesized from [2-14C]MVA-5-P using Antarctic phosphatase (New England Biolabs) and S. solfataricus PMK, respectively, as described in our previous work (3).
Radio-TLC analysis of the reaction products.
To analyze the products from TacM3K mutants, 25 µl of a reaction solution was prepared containing 25 pmol of the 14C-labeled substrate ([2-14C]MVA, [2-14C]MVA-5-P, or [2-14C]MVA-5-PP), 25 pmol (∼1 µg) of the purified recombinant enzyme, 100 nmol of ATP, 1 µmol of sodium phosphate buffer, pH 7.5, and 125 nmol of MgCl2. After the reaction had proceeded for 15 min at 60°C, 10 µl of the solution was spotted onto a Silica gel 60 normal-phase TLC plate, and the plate was developed using a solution of n-propanol–28% ammonia water (NH4OH)–H2O (6:3:1). The distribution of radioactivity on the TLC plates was detected using a Typhoon FLA7000 image analyzer (GE Healthcare).
13C NMR analysis of the product of the E140S mutant.
In a volume of 200 µl, a reaction mixture containing 1 µmol of [U-13C]MVA, 3 nmol of S. solfataricus MVK, 30 µmol of sodium phosphate buffer, pH 7.5, with 5 µmol of ATP, 0.2 µmol of MgCl2, and 10% D2O was prepared for the first reaction and incubated at 60°C for 2 h. After filtration using a 10-kDa-cutoff spin column filter (Vivaspin 500; GE Healthcare), 175 µl of the filtrate was used for the second reaction. Subsequently, 3 nmol of the E140S mutant was added to the filtrate. As a negative control, the same volume of water was added in place of the enzyme. The volume of the solution was adjusted using a 9:1 mixture of H2O-D2O to 600 µl, and the second reaction was conducted at 60°C for 3 h. After filtration, the 13C NMR spectrum of the product from the second reaction was analyzed using an Avance III HD 600 NMR spectrometer equipped with a cryoprobe (Bruker).
MS analysis of the products.
In a 200-µl volume, 0.8 µmol of (R,S)-MVA-5-P was reacted with 2 nmol of either the wild-type TacM3K or the E140A, E140S, or E140G mutant at 60°C for 2 h in a buffer containing 20 µmol of ammonium acetate, pH 7.0, with 4 µmol of ATP and 0.2 µmol of MgCl2. After filtration with a Vivaspin 500 filter (10-kDa-molecular-mass cutoff), a 175-µl volume of the filtrate was diluted with the same volume of acetonitrile and then analyzed by negative ion ESI-MS using an Esquire 3000 mass spectrometer (Bruker) by direct infusion.
Construction of the artificial MVA pathway.
The genes ma0602 and ma0603 were amplified by KOD-plus (Toyobo, Japan) from M. acetivorans genome as a template and using primer pairs shown in Table 3: Ma0602-forward and Ma0602-reverse; Ma0603-forward and Ma0603-reverse, respectively. The PCR product of ma0602 was cloned into KpnI-digested pBAD18 plasmid using an In-Fusion cloning kit (TaKaRa Bio, Japan). After the resulting plasmid was digested with KpnI, the other PCR product of ma0603 was cloned downstream of ma0602 to construct the pBAD-MI plasmid. Then the TacM3K gene was amplified by PCR from pET15b-TacM3K as a template. For the reaction, four primers, Ptac-M3K-Fw1, Ptac-M3K-Fw2, Ptac-M3K-Rv1, and Ptac-M3K-Rv2 (Table 3), were used at the same time to add the tac promoter sequence without an operator sequence upstream of the TacM3K gene. Then the obtained PCR product was digested with KpnI and HindIII and ligated with pBAD-MI digested with the same enzymes using Ligation high, version 2 (Toyobo, Japan). The resulting plasmid was designated pBAD-MI-M3K(WT). The plasmid was digested with SpeI and HindIII and then ligated with a pto0478 fragment, which was amplified from the pET28a(+) plasmid containing pto0478 (12) using the primer pair Pto0478-Fw and Pto0478-Rv. The resulting plasmid was designated pBAD-MIB-M3K(WT). Similarly, pBAD-MIB-M3K(E140A), pBAD-MIB-M3K(E140S), and pBAD-MIB-M3K(E140G) were constructed using pET15b-TacM3K(E140A), pET15b-TacM3K(E140S), and pET15b-TacM3K(E140G), respectively, described above, as a template DNA for amplification of a TacM3K gene.
The pBAD-ScMPD plasmid for the expression of the genes of the classical MVA pathway was constructed as described below. A 3.8-kbp DNA fragment containing the genes of S. cerevisiae MVK, PMK, and DMD was amplified from pJBEI-2999 (Addgene, MA) (22) using ScMVK-forward and ScDMD-reverse primers (Table 3). The amplified fragment was cloned into KpnI-digested pBAD18 plasmid using an In-Fusion cloning kit (TaKaRa Bio, Japan) to construct pBAD-ScMPD. The pBAD-TacM series plasmids for the expression of the genes of modified MVA pathway II were constructed as described below. The wild-type TacM3K gene was amplified from pET15b-TacM3K using the primers Ta1305-forward and Ta1305-reverse. The gene of T. acidophilum M3P5K (ta0762) was amplified from T. acidophilum genomic DNA using Ta0762-forward and Ta0762-reverse primers. Both the amplified TacM3K gene and ta0762 fragments were tandemly cloned into KpnI-digested pBAD18 plasmid to construct plasmid pBAD-TacM3K5K. The gene of P. torridus BMD (pto0478) was amplified from the pET28a(+) plasmid containing pto0478 (12) using primers Pto0478-MP-forward and Pto0478-MP-reverse. The amplified fragment was inserted into KpnI-digested pBAD-TacM3K5K to construct pBAD-TacMdelI. The gene of T. acidophilum IPK (ta0103) was amplified from T. acidophilum genomic DNA using Ta0103-forward and Ta0103-reverse primers (Table 3). The amplified fragment and XbaI-digested pBAD-TacMdelI plasmid were introduced into the competent cells of the E. coli ME9783 strain (National BioResource Project, Japan), which enables in vivo E. coli cloning based on homologous recombination. The plasmid containing the genes of TacM3K, T. acidophilum M3P5K, P. torridus BMD, and T. acidophilum IPK was extracted from the transformant cells and was designated pBAD-TacM(PtBMD). The gene of putative full-length T. acidophilum BMD (ta0893) was amplified from T. acidophilum genomic DNA using Ta0893(full)-forward and Ta0893-reverse primers. Similarly, a shorter ta0893 gene encoding truncated T. acidophilum BMD, which starts from the Met30 residue of the putative full-length protein, was amplified using Ta0893(truncated)-forward and Ta0893-reverse primers. Each of the amplified DNA fragments was cut by KpnI and SacI and then ligated with pBAD-TacM(PtBMD) digested with the same restriction enzymes to construct pBAD-TacM(TaBMDfull) and pBAD-TacM(TaBMDtrunc), which contain the full-length and truncated T. acidophilum BMD genes, respectively, along with the genes of TaM3K, T. acidophilum M3K5P, and T. acidophilum IPK.
For the expression of carotenoid biosynthetic enzymes in E. coli, a plasmid, pACYC-IBEidi, was constructed. A 1.3-kbp EcoRI-HpaI fragment carrying geranylgeranyl diphosphate synthase gene (crtE) was isolated from pCAR25 (23), a plasmid carrying a carotenoid biosynthetic gene cluster from Pantoea ananatis, formerly Erwinia uredovora. The fragment was inserted into the EcoRI-SnaBI site of p3-47-11ΔEcoRI, which harbors the E. coli IPP isomerase gene (idi) (24). The resulting plasmid was cut with EcoRI and PstI to prepare a 2.4-kbp fragment carrying crtE and idi. The fragment was blunt ended and inserted into the ScaI site of pACYC-IB (25) to construct the plasmid pACYC-IBEidi, which contains three carotenoid biosynthesis genes from P. ananatis, i.e., crtE, the phytoene synthase gene (crtB), and the phytoene desaturase gene (crtl), with idi from E. coli.
Analysis of carotenoid production.
Each of the MVA pathway plasmids, pBAD-MIB-M3KWT, pBAD-MIB-M3K(E140A), pBAD-MIB-M3K(E140S), pBAD-MIB-M3K(E140G), pBAD-ScMPD, pBAD-TacM(PtBMD), pBAD-TacM(TaBMDfull), and pBAD-TacM(TaBMDtrunc), and the carotenoid biosynthetic pathway plasmid pACYC-IBEidi were introduced into E. coli DH5α to construct the following strains: ST-WT, ST-A, ST-S, ST-G, ScMPD, TacM(PtBMD), TacM(TaBMDfull), and TacM(TaBMDtrunc), respectively (Table 2). To generate a negative-control strain designated ST-NC, two plasmids, pBAD18 and pACYC-IBEidi, were introduced into E. coli DH5α (Table 2). Each of the strains was precultured in 5 ml of LB medium containing 100 µg/ml of ampicillin and 20 μg/ml of tetracycline. Fifty microliters of the preculture, whose optical density at 600 nm (OD600) was adjusted to 0.4, was inoculated into 5 ml of LB medium containing 2.5 mg of (R)-mevalonolactone (Adeka, Japan), 0.2% (wt/vol) arabinose, 100 μg/ml of ampicillin, and 20 μg/ml of tetracycline and shaken for 48 h at 37°C. After the mixture was centrifuged, the obtained pellet was washed with 5 ml of sterilized water and recentrifuged again. Then the obtained pellet was multiply extracted with ∼3 ml of acetone until almost all lycopene was extracted. The absorbance spectrum of each extract was measured using a UV spectrophotometer (UV-2450; Shimadzu). For the quantification of extracted lycopene, an extinction coefficient ε of 185,000 M−1 cm−1 at 475 nm was used. Cultivation and lycopene measurement of each strain were performed in triplicate.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by JSPS KAKENHI grants 26660060, 16K14882, and 17H05437 and by grants-in-aid from Novozymes Japan and the Institute for fermentation, Osaka, to H.H.
We thank Kazushi Koga, Nagoya University, for help with the NMR analysis and Jeffrey M. Vinokur and James U. Bowie, University of California, Los Angeles, for donation of the pET28a(+) plasmid containing the PtBMD gene.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00256-19.
REFERENCES
- 1.Kuzuyama T, Hemmi H, Takahashi S. 2010. Mevalonate pathway in bacteria and archaea, p 493–516. In Mander L, Liu HW (ed), Comprehensive natural products II chemistry and biology, vol 1 Elsevier, Oxford, UK. doi: 10.1016/B978-008045382-8.00014-9. [DOI] [Google Scholar]
- 2.Vickers CE, Bongers M, Liu Q, Delatte T, Bouwmeester H. 2014. Metabolic engineering of volatile isoprenoids in plants and microbes. Plant Cell Environ 37:1753–1775. doi: 10.1111/pce.12316. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura H, Azami Y, Miyagawa M, Hashimoto C, Yoshimura T, Hemmi H. 2013. Biochemical evidence supporting the presence of the classical mevalonate pathway in the thermoacidophilic archaeon Sulfolobus solfataricus. J Biochem 153:415–420. doi: 10.1093/jb/mvt006. [DOI] [PubMed] [Google Scholar]
- 4.Bloch K. 1965. The biological synthesis of cholesterol. Science 150:19–28. doi: 10.1126/science.150.3692.19. [DOI] [PubMed] [Google Scholar]
- 5.Dellas N, Thomas ST, Manning G, Noel JP. 2013. Discovery of a metabolic alternative to the classical mevalonate pathway. Elife 2:e00672. doi: 10.7554/eLife.00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.VanNice JC, Skaff DA, Keightley A, Addo J, Wyckoff GJ, Miziorko HM. 2014. Identification in Haloferax volcanii of phosphomevalonate decarboxylase and isopentenyl phosphate kinase as catalysts of the terminal enzymatic reactions in an archaeal alternate mevalonate pathway. J Bacteriol 196:1055–1063. doi: 10.1128/JB.01230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lombard J, Moreira D. 2011. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol Biol Evol 28:87–99. doi: 10.1093/molbev/msq177. [DOI] [PubMed] [Google Scholar]
- 8.Matsumi R, Atomi H, Driessen AJ, van der Oost J. 2011. Isoprenoid biosynthesis in Archaea—biochemical and evolutionary implications. Res Microbiol 162:39–52. doi: 10.1016/j.resmic.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Azami Y, Hattori A, Nishimura H, Kawaide H, Yoshimura T, Hemmi H. 2014. (R)-Mevalonate 3-phosphate is an intermediate of the mevalonate pathway in Thermoplasma acidophilum. J Biol Chem 289:15957–15967. doi: 10.1074/jbc.M114.562686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vinokur JM, Korman TP, Cao Z, Bowie JU. 2014. Evidence of a novel mevalonate pathway in archaea. Biochemistry 53:4161–4168. doi: 10.1021/bi500566q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossoni L, Hall SJ, Eastham G, Licence P, Stephens G. 2015. The putative mevalonate diphosphate decarboxylase from Picrophilus torridus is in reality a mevalonate-3-kinase with high potential for bioproduction of isobutene. Appl Environ Microbiol 81:2625–2634. doi: 10.1128/AEM.04033-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vinokur JM, Cummins MC, Korman TP, Bowie JU. 2016. An adaptation to life in acid through a novel mevalonate pathway. Sci Rep 6:39737. doi: 10.1038/srep39737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Poulter CD. 2010. Characterization of thermophilic archaeal isopentenyl phosphate kinases. Biochemistry 49:207–217. doi: 10.1021/bi9017957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mabanglo MF, Schubert HL, Chen M, Hill CP, Poulter CD. 2010. X-ray structures of isopentenyl phosphate kinase. ACS Chem Biol 5:517–527. doi: 10.1021/cb100032g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinokur JM, Korman TP, Sawaya MR, Collazo M, Cascio D, Bowie JU. 2015. Structural analysis of mevalonate-3-kinase provides insight into the mechanisms of isoprenoid pathway decarboxylases. Protein Sci 24:212–220. doi: 10.1002/pro.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motoyama K, Unno H, Hattori A, Takaoka T, Ishikita H, Kawaide H, Yoshimura T, Hemmi H. 2017. A single amino acid mutation converts (R)-5-diphosphomevalonate decarboxylase into a kinase. J Biol Chem 292:2457–2469. doi: 10.1074/jbc.M116.752535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayakawa H, Motoyama K, Sobue F, Ito T, Kawaide H, Yoshimura T, Hemmi H. 2018. Modified mevalonate pathway of the archaeon Aeropyrum pernix proceeds via trans-anhydromevalonate 5-phosphate. Proc Natl Acad Sci U S A 115:10034–10039. doi: 10.1073/pnas.1809154115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castelle CJ, Banfield JF. 2018. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 172:1181–1197. doi: 10.1016/j.cell.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Chen LX, Méndez-García C, Dombrowski N, Servín-Garcidueñas LE, Eloe-Fadrosh EA, Fang BZ, Luo ZH, Tan S, Zhi XY, Hua ZS, Martinez-Romero E, Woyke T, Huang LN, Sánchez J, Peláez AI, Ferrer M, Baker BJ, Shu WS. 2018. Metabolic versatility of small archaea Micrarchaeota and Parvarchaeota. ISME J 12:756. doi: 10.1038/s41396-017-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugai Y, Miyazaki S, Mukai S, Yumoto I, Natsume M, Kawaide H. 2011. Enzymatic total synthesis of gibberellin A4 from acetate. Biosci Biotechnol Biochem 75:128–135. doi: 10.1271/bbb.100733. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 22.Peralta-Yahya PP, Ouellet M, Chan R, Mukhopadhyay A, Keasling JD, Lee TS. 2011. Identification and microbial production of a terpene-based advanced biofuel. Nat Commun 2:483. doi: 10.1038/ncomms1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misawa N, Nakagawa M, Kobayashi K, Yamano S, Izawa Y, Nakamura K, Harashima K. 1990. Elucidation of the Erwinia uredovora carotenoid biosynthetic pathway by functional analysis of gene products expressed in Escherichia coli. J Bacteriol 172:6704–6712. doi: 10.1128/jb.172.12.6704-6712.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemmi H, Ohnuma S, Nagaoka K, Nishino T. 1998. Identification of genes affecting lycopene formation in Escherichia coli transformed with carotenoid biosynthetic genes: candidates for early genes in isoprenoid biosynthesis. J Biochem 123:1088–1096. doi: 10.1093/oxfordjournals.jbchem.a022047. [DOI] [PubMed] [Google Scholar]
- 25.Ohnuma S, Suzuki M, Nishino T. 1994. Archaebacterial ether-linked lipid biosynthetic gene. Expression cloning, sequencing, and characterization of geranylgeranyl-diphosphate synthase. J Biol Chem 269:14792–14797. [PubMed] [Google Scholar]
- 26.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.