

Abstract.
Background: Epithelial cells are critically dependent upon cell‐matrix and cell‐cell adhesion for growth and survival. Anoikis is programmed cell death caused by disruption of cell‐substrate adhesion in normal epithelial cells.
Methods: We studied the induction of anoikis in vitro in two cell lines; HaCaT and SW742. PI3K, JAK2 and PKC are key elements in signalling pathways regulating cell survival, and using specific inhibitors we also examined their potential role in the induction of anoikis.
Results: When prevented from adhesion by culture on polyHEMA, HaCaT cells underwent apoptosis selectively from the proliferating population; surviving cells underwent cell cycle arrest. In SW742 cells anoikis also occurred, but was balanced by increased cycling. The effects of specific kinase inhibitors indicated that both Janus kinase 2 and protein kinase C partially protect HaCaT cells from anoikis through inducing cell cycle arrest of surviving nonadherent cells; inhibition of Phosphatidylinositol 3‐kinase did not induce cycling in HaCaTs prevented from adhesion but did stimulate anoikis. SW742 cells showed markedly different responses: Janus kinase 2 inhibition activated apoptosis directly, Phosphatidylinositol 3‐kinase inhibition stimulated both cell cycling and apoptosis, while protein kinase C inhibition stimulated cycling but inhibited apoptosis.
Conclusions: Susceptibility to cell death in adhesion‐prevented epithelial cells may thus be regulated by signalling pathways involving Phosphatidylinositol 3‐kinase, Janus kinase 2 and protein kinase C. The ability of epithelial tumour cells to invade and metastasize may therefore result from disruption of these pathways.
Introduction
Epithelial cells are critically dependent upon cell–matrix and cell–cell adhesion for growth and survival. Disruption of substrate adhesion in normal epithelial cells rapidly induces programmed cell death; a response that has been termed anoikis (Frisch & Francis 1994). In vivo, anoikis would result from detachment of viable epithelial cells from the basement membrane and could thus play an important protective role in preventing actively proliferating cells from re‐attachment and growth in inappropriate environments. In vitro, immortalized epithelial cells adapt to growth on tissue culture plastic, upon which they may deposit matrix proteins to which they remain adherent. Disruption of their adhesion to this growth substratum has been shown to induce anoikis implying (Bates et al. 1994; Frisch & Francis 1994) that the intracellular signalling mechanisms regulating survival and death are responsive to the interaction of transmembrane adhesion molecules with tissue culture substrate in these cells.
The extracellular signals, cell surface receptors and signalling pathways involved in this regulation of epithelial cell survival have yet to be fully elucidated. Frisch and Francis showed that Madin‐Darby canine kidney (MDCK) epithelial cells circumvented anoikis by acquiring expression of cell motility factor, suggesting that abrogation of anoikis may occur as a result, or in concurrence with, a gain in anchorage independence or cell motility (Frisch & Francis 1994). This has also been demonstrated by Behrens et al. (1989) who showed that by specific inhibition of intercellular adhesion, MDCK cells could be made capable of invading both collagen gels and embryonic heart tissue. There is considerable evidence to implicate integrins in the transmission of adhesion‐related survival signals (Frisch et al. 1996a; Frisch & Ruoslahti 1997). Focal adhesion kinase (FAK) is believed to relay these signals, as activation of FAK will transform MDCK cells by increasing their resistance to anoikis (Frisch et al. 1996b).
Many cell membrane receptors transmit effects induced on ligand binding by activating G‐proteins or by direct phosphorylation of target proteins. Large proportions of receptors contain tyrosine kinase domains, which are activated by autophosphorylation on receiving an external signal mediated by receptor–ligand interactions (Stryer 1993). The tyrosine kinases can activate target proteins either directly, by transferring a terminal phosphate group from ATP to the hydroxyl group on tyrosine residues of selected proteins, or through interactions with G‐proteins. The signal transduction cascades that are induced by tyrosine phosphorylation regulate cell proliferation, differentiation and migration (Streuli 1996). The extent of phosphorylation of individual components of each cascade is determined by the balance of the activities of specific protein tyrosine kinase and protein tyrosine phosphatases.
For anoikis to occur, epithelial cells activate jun‐N‐terminal kinases (JNKs) after detachment from the matrix, and activation of both the JNK pathway and caspase 7 is required (Frisch et al. 1996a). Inhibitors of caspases and over expression of bcl‐2 inhibit both JNK activation and induction of anoikis, suggesting that the JNK pathway activates anoikis via classical apoptotic regulatory mechanisms. Transformation with v‐Ha‐ras or v‐src also protects normal epithelial cells from anoikis, and this protection in ras‐transformed cells has been attributed to activation of phosphatidylinositol 3‐kinase (PI3K) (Khwaja et al. 1997). Significantly, recent work by Davies et al. (1998) has shown that anoikis is induced in glioma cells by reintroduction of the phosphatidylinositol 3‐phosphatase tumour suppresser gene PTEN. One of the downstream effectors of the PI3K pathway, PKB/Akt phosphorylates the pro‐apoptotic protein Bad, and the mechanism by which PTEN stimulates anoikis is thought to be through antagonism of the PI3K‐stimulated activation of PKB and thus the release of Bad from its inactivated state (Franke et al. 1995; Franke et al. 1997; Stokoe et al. 1997).
The model for adhesive interactions that may regulate anchorage dependence proposed in a review by Ruoslahti & Reed (1994) indicates that integrin signalling is mediated by several enzymes including protein kinase C (PKC) and, possibly, other kinases.
Studies on anchorage dependence have been performed in vitro by culturing cells on polyHEMA coated Petri dishes. The nonionic nature of polyHEMA prevents both matrix deposition and subsequent cell attachment. A polyHEMA concentration of 10 mg/ml (1 mg/cm2) has been shown to be sufficient to prevent cell attachment to the tissue culture plastic (Folkman & Moscona 1978; Frisch & Francis 1994). This study aimed to investigate the induction of anoikis in the colon cancer cell line SW742 and the nontransformed immortalized human keratinocyte cell line HaCaT, as HaCaT cells have been shown to retain the anoikis response to culture under nonadherent conditions on polyHEMA. We also investigated the effects of specific inhibition of three key kinases, phosphatidylinositol 3‐kinase (PI3K), Janus kinase 2 (JAK2) and protein kinase C (PKC), on cell susceptibility to anoikis.
MATERIALS AND METHODS
Cell culture
HaCaT cells were obtained from Prof. N. Fusenig at the Deutsch Krebsforschungszentrum, Heidelberg, Germany. The cells were grown in Dulbecco’s Minimal Essential Medium supplemented with 10% foetal calf serum (Life Technologies, Paisley, UK). The colon carcinoma cell line SW742 was maintained in RPMI1640 medium containing 10% foetal calf serum. Both cell lines were passaged on reaching approximately 80% confluence at a density ratio of 1 : 6 using trypsin/EDTA (Life Technologies) for SW742 and at 1 : 4 using EDTA followed by trypsin/EDTA for HaCaT. All cells were used within a passage range of 10 with fresh stocks used after each 10th passage.
Anoikis assay
Six‐well tissue culture plates were coated with polyHEMA (poly(2‐hydroxyethylmethacrylate); Sigma‐Aldrich, Poole, UK) as follows: 1 ml of 10 mg/ml of polyHEMA in 95% ethanol was applied to each well and allowed to dry. The procedure was then repeated using a further 1 ml of polyHEMA solution. After drying, the wells were washed in PBS and stored dry until used.
Cells were grown to 80% confluence in 75 cm2 tissue culture flasks, detached using trypsin/EDTA, and plated onto polyHEMA‐coated or un‐coated control wells at 1 × 105 cells/cm2 for HaCaT and 2 × 105 cells/cm2 for SW742. At the indicated times, cells were harvested from polyHEMA‐coated wells by pipetting and centrifugation, followed by treatment with trypsin/EDTA to yield a single cell suspension. Total cells in the uncoated control wells were harvested using trypsin/EDTA, again to yield single cell suspensions. The numbers of viable cells were determined by trypan blue exclusion and counting on an improved Neubauer haemocytometer.
Flow cytometry
Cells were incubated in control or polyHEMA‐coated wells for the times indicated before harvesting as described above. Cell suspensions were pelleted by centrifugation at 400 g for 5 min and the cells washed twice in PBS. Cells were stained using a propidium iodide DNA‐staining kit (Sigma Aldrich) and analysed using an Orthocyte benchtop flow cytometer. Cell cycle analysis was carried out with the Multicycle software package (Phoenix Flow, San Diego, CA) using data files of 5000–10 000 cells. The percentage of cells in each phase of the cell cycle (G1, S and G2/M) was obtained. Following the convention on nomenclature for DNA flow cytometry coefficients of variation (CV) exceeding eight were rejected and retested.
Incorporation of bromodeoxyuridine
Cells were incubated in control or polyHEMA‐coated wells in medium containing 10 µm Bromodeoxyuridine (BrdU, Boehringer Mannheim UK Ltd, Lewes, UK) for the times indicated, harvested as described above, fixed in 1% paraformaldehyde for 10 min followed by 70% ethanol and cytospun onto microscope slides coated with 3‐aminopropyltriethoxysilane (Sigma‐Aldrich). The cells were incubated in 2 m HCl for 15 min and then 0.2 mg/ml pepsin in 10 mm HCl for 45 min. BrdU was detected using an anti‐BrdU mouse monoclonal antibody (Dako Ltd, Ely, UK) and FITC‐conjugated goat antimouse IgG (Caltag‐Med Systems Ltd, Silverstone, UK). Cells were counter‐stained with 0.25 µg/ml 4′6‐Diamidino‐2‐phenylindole (DAPI; Sigma Aldrich UK Ltd) in tris‐buffered saline and analysed by fluorescence microscopy. The number of BrdU stained cells counted was expressed as a percentage of the total cells present.
Apoptosis assay
Detection of apoptosis was carried out by fluorescence microscopy of DAPI counter‐stained cells (following BrdU detection). Cells showing chromatin condensation, nuclear blebbing, or the presence of apoptotic bodies were easily identifiable by this method. Confirmation of the apoptotic status of cells identified by DAPI staining had previously been carried out by double‐labelling HaCaT cells using fluorescent TUNEL staining for DNA fragmentation in combination with DAPI (Sharrard, unpublished results). The amount of apoptosis observed was determined by expressing the number of apoptotic nuclei counted as a percentage of the total cells visualized.
Kinase inhibitors
The protein kinase inhibitors wortmannin (which inhibits PI3K), Ro 31–8220 (an inhibitor of PKC) and AG490 (tyrophostin, which inhibits JAK2) were generously donated by Dr P.R.M. Dobson, Institute for Cancer Studies, University of Sheffield Medical School. The inhibitors were dissolved in dimethylsulphoxide (DMSO) and diluted in culture medium before addition to cells at the following concentrations: AG490–5 µm, 25 µm; wortmannin, 50 nm, 500 nm; Ro 31–8220, 1 µm, 5 µm.
RESULTS
PolyHEMA coated and uncoated tissue culture plates were used in in vitro studies with HaCaT and SW742 cells to investigate the following: (1) survival and proliferation (growth); (2) apoptosis (DAPI); (3) cell cycle (flow cytometry and BrdU incorporation).
Cell survival under adherent and nonadherent culture conditions
HaCaT and SW742 cells were plated onto polyHEMA‐coated or control tissue culture wells and cultured for 24 h, after which cells were counted. HaCaT cell numbers fell markedly during the 24 h on polyHEMA in relation to the controls (Fig. 1a). Conversely the tumour cell line SW742 showed no decrease in cell numbers over 24 h (Fig. 1b). This supports the proposition that anoikis is a phenomenon exhibited by normal epithelial cells, and is characteristically lost during epithelial tumour cell progression.
Figure 1.
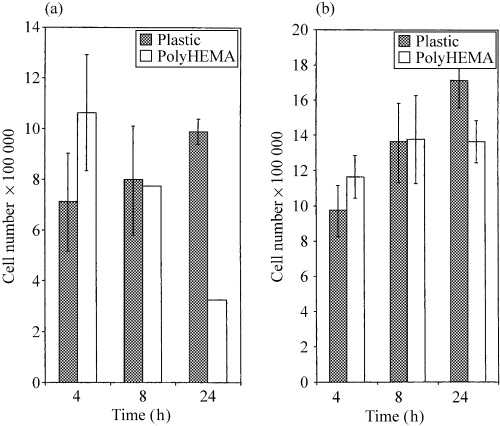
Graphical representation of cell numbers after 24 h for (a) HaCaT and (b) SW742 on plastic and polyHEMA. Bars represent the mean and whiskers standard deviation.
Apoptosis
Cells grown on plastic and polyHEMA were examined for apoptotic nuclear morphology by fluorescence microscopy and apoptotic cells thus detected were expressed as a percentage of the total cells visualized. The levels of apoptosis in HaCaT cells on polyHEMA at 8 h were higher than in the adherent controls and fell at 24 h (Fig. 2a), while SW742 on polyHEMA showed high levels of apoptosis at both 8 and 24 h in comparison to the adherent control cells (Fig. 2b). Typical apoptotic nuclei are shown in Fig. 2c.
Figure 2.
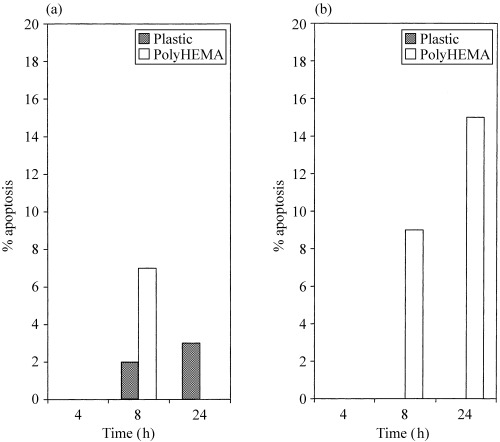
Graphical representation of the percentage apoptosis detected in (a) HaCaT and (b) SW742 cells. Results from a representative experiment are shown. Where no bar is present (for example, SW742 on plastic, HaCaT at 4 h) the value is zero.
Cell cycle analysis
The flow cytometry cell cycle profiles for HaCaT cells at 4 and 8 h after plating were similar for cells on plastic (Fig. 3a) or on polyHEMA (Fig. 3b). The main difference was observed at 24 h, when the cells cultured on polyHEMA remained constant in all phases, in contrast to the adherent controls which had progressed into S‐phase (typically 15% increases to 35% at 24 h).
Figure 3.
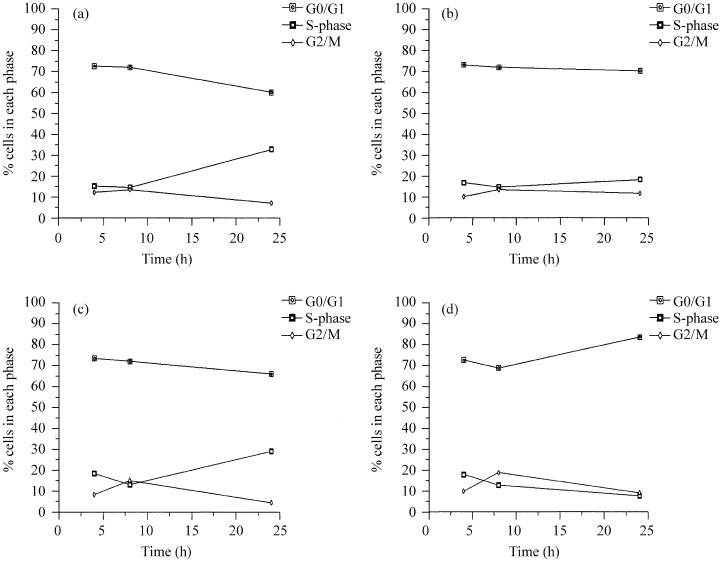
Cells were harvested after 4, 8 and 24 h and cell cycle analysis carried out by flow cytometry. The graphs show HaCaT on (a) plastic and (b) polyHEMA and SW742 on (c) plastic and (d) polyHEMA.
It should be noted here that the high apoptotic rate in nonadherent HaCaT cells reported by Frisch & Francis (1994), detected as a sub‐G1 peak in flow cytometric analysis, was not observed in the present study unless the cells were allowed to become highly confluent prior to plating on polyHEMA‐coated wells. However, we consistently found that HaCaT cells could undergo rapid decreases in cell number with little detectable increase in the number of apoptotic cells present. We conclude that the apoptotic process in these cells must be extremely rapid or, more likely, that cells at an early stage of apoptosis are ingested by their healthy neighbours (Sharrard & Bretland, unpublished observations).
At 4 and 8 h the cell cycle profiles of SW742 cells on plastic and polyHEMA by flow cytometry were very similar. By 24 h there was an increase in the number of cells on plastic that had entered S‐phase (Fig. 3c). After 24 h on polyHEMA the percentage of SW742 cells in S‐phase had decreased with a concomitant accumulation in G0/G1 (typically 10% up to 30% at 24 h; Fig. 3d).
BrdU incorporation
For the nonapoptotic cells it was necessary to establish whether the cells cultured on the polyHEMA‐coated wells were undergoing normal cell cycle progression or whether they had arrested in a specific phase of the cell cycle. This was addressed by DNA flow cytometry (see above). However, cell cycle analysis alone is unable to assess the rate of cell cycle; this can be achieved by measuring BrdU incorporation. BrdU was included in the culture medium at the time at which the cells were plated into the wells. Cells were harvested at 4, 8 and 24 h after plating and stained for BrdU incorporation to obtain a cumulative total of cells passing through S phase. Positive staining was detected by fluorescence microscopy and expressed as a percentage of the total cells visualized.
On polyHEMA‐coated wells the percentage of cells that had incorporated BrdU decreased (Fig. 4a) along with the total number of HaCaT cells as determined by direct cell counting (Fig. 1a). This indicates that the deletion of cells occurred from the cell population that had recently progressed through S‐phase. The cell loss suggests apoptosis but by 24 h the proportion of apoptotic cells had fallen implying that the majority of apoptosis occurred early in the culture period. Flow cytometry data showed an accumulation of cells predominantly in G0/G1 and to a lesser extent in G2/M in comparison to the adherent controls. It would thus appear that surviving HaCaT cells are not undergoing cell cycle as a result of prevention of adhesion.
Figure 4.
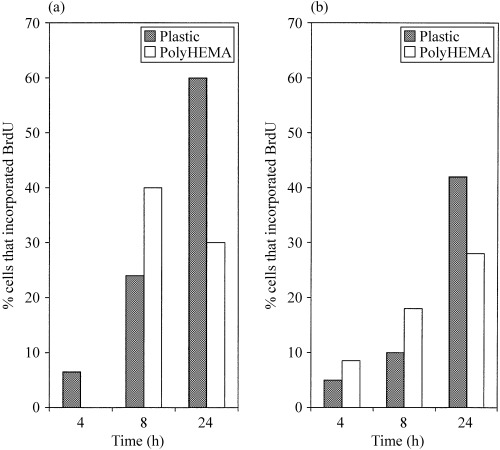
Graphical representation of the percentage incorporating BrdU cells in (a) HaCaT and (b) SW742. Where no value appears (HaCaT 4 h on polyHEMA) the percentage BrdU incorporation is zero.
On polyHEMA SW742 cell numbers did not alter substantially between 8 and 24 h although the rate of apoptosis was high. The number of apoptotic and mitotic events during this time must therefore have been similar. The percentage of BrdU‐positive cells also increased (Fig. 4b), providing additional evidence that at least a proportion of the cells were in active cell cycle and suggesting that, unlike in HaCaT cells, cell death was not occurring exclusively in the cycling cell population. Nevertheless, the flow cytometry results indicated that surviving SW742 cells on polyHEMA accumulated in G0/G1 in comparison to the control cells grown on plastic.
Thus, in cultures of HaCaT cells, the cycling cells are being preferentially deleted, while those that remain undergo cell cycle arrest. In contrast, SW742 cells on polyHEMA are growth‐arresting in G0/G1, though cells that have passed the G1/S restriction point at the time of plating enter S‐phase and progress through the cycle, where they either arrest in the subsequent G0/G1 phase or undergo apoptosis during the intervening S and G2/M phases.
Effects of kinase inhibitors
The cells were plated into polyHEMA‐coated wells in culture medium supplemented with protein kinase inhibitors at the concentrations indicated in order to investigate whether inhibition of kinase function could abrogate the induction of anoikis. An additional polyHEMA well containing 1 µL/ml DMSO was included as a control. Cell numbers were again determined by haemocytometer and the percentage apoptosis by DAPI staining.
The number of viable HaCaT cells remaining after 24 h on polyHEMA was increased relative to controls in wells containing AG490 or Ro 31–8220 (Fig. 5a). In most cases, SW742 cell numbers decreased relative to the control wells in response to the kinase inhibitors (Fig. 5b). Our data in contrast also suggest that wortmannin at 50 nm and Ro 31–8220 at 5 µm caused slight increases in cell numbers.
Figure 5.
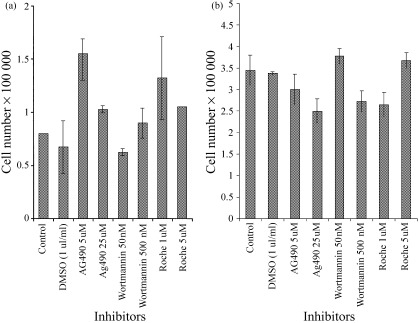
Graphical representation of (a) HaCaT and (b) SW742 cell numbers after 24 h on polyHEMA in the presence of kinase inhibitors. Bards represent means and whiskers standard deviations.
At the lower concentrations used, AG490 and Ro 31–8220 caused a dramatic rise in the percentage of BrdU‐incorporating HaCaT cells at 24 h after plating on polyHEMA (Table 1). In each case this was accompanied by a rise in the proportion of apoptotic cells. This suggests that these two compounds are overriding the cell cycle arrest observed in surviving HaCaT cells under these conditions, allowing them to continue in active cell cycle and thus rendering them vulnerable to anoikis. However, the possibility that AG490 and Ro 31–8220 may directly induce apoptosis must not be ignored. At higher concentrations the stimulation of BrdU incorporation was markedly decreased or abolished, and apoptotic rates were yet higher, suggesting that at these levels AG490 and Ro 31–8220 did indeed induce apoptosis directly, presumably at the G1/S checkpoint. It would appear that there was a dose response effect to wortmannin, with the higher concentration causing apoptosis. Neither concentration affected BrdU labelling, implying that wortmannin at 500 nm may also cause apoptosis directly.
Table 1.
Percentage of apoptosis and BrdU incorporation in HaCaT and SW742 after 24 h on polyHEMA in the presence of kinase inhibitors. The results shown are from a representative experiment and are means of duplicate samples
| Inhibitor concentration | HaCaT | SW742 | ||
|---|---|---|---|---|
| % BrdU incorporation | % Apoptosis | % BrdU Incorporation | % Apoptosis | |
| Control | 28.6 | 0 | 28.6 | 7.1 |
| DMSO | 25 | 0 | 32 | 8 |
| AG490 (5 µm) | 78.3 | 4.3 | 25 | 16 |
| AG490 (25 µm) | 33.3 | 6.6 | 25 | 12.5 |
| Wortmannin (50 nm) | 33.3 | 0 | 20.8 | 8.3 |
| Wortmannin (500 nm) | 20 | 6.6 | 77.8 | 33.3 |
| Ro31–8220 (1 µm) | 66.7 | 9.5 | 100 | 0 |
| Ro31–8220 (5 µm) | 25 | 12.5 | 41.7 | 8.3 |
The figures in bold show positive effects of the inhibitor on BrdU incorporation and apoptosis.
In SW742 cells, AG490 had little apparent effect on BrdU incorporation (Table 1), but apoptotic rates were markedly raised relative to controls, indicating that in this cell line AG490 could activate apoptosis directly, rather than through reactivation of the cell cycle and subsequent anoikis. In contrast, wortmannin at high concentration stimulated both cell cycling and apoptosis, while Ro 31–8220 at the lower concentration stimulated cycling but inhibited apoptosis, relative to controls. The higher concentration of Ro‐31–8220 used produced less dramatic effects: firstly reduced cell cycle stimulation and secondly, loss of apoptotic inhibition. This supports the conclusion that cycling and apoptosis are processes that are independently regulated in these cells. In addition, the effects of Ro 31–8220 suggest that PKC inhibition in SW742 tumour cells may suppress the anoikis effect as well as stimulating cell proliferation or relieving cell cycle arrest.
DISCUSSION
The phenomenon of anoikis has been proposed as a protective mechanism which operates to prevent growth of displaced epithelial and endothelial cells in inappropriate environments. Loss of anoikis has been identified as one of the key steps in progression towards invasiveness and metastasis in epithelial tumour progression. The protective effect of anoikis may be of particular importance in the epithelia of the lining of the gut, where senescent cells are constantly displaced from the basement membrane or are sloughed off by the passage of material through the gut lumen. Cells abraded in this manner may include cells which, unless prevented through apoptosis, might reattach and eventually establish growth in a distant, inappropriate environment.
HaCaT cells have previously been shown to undergo anoikis when cultured for 20 h on polyHEMA‐coated plastic (Frisch & Francis 1994), but few data exist on the susceptibility of colon epithelial cell lines to this process. Indeed, the establishment of nontransformed colonic epithelial cell lines has been hindered by the sensitivity of normal colonic cells to loss of adhesion: normal colonic cells in resuspended crypts will die rapidly by anoikis. The effects are exacerbated when the crypts are preincubated with antibody to β1 integrin prior to cell resuspension, causing a temporary loss of adhesion to matrix protein (Strater et al. 1996). The relative ease by which colonic tumour cell lines can be developed thus provides support for the proposition that anoikis is suppressed in colon cancer.
The present study confirms that a colon tumour cell line exhibit a greater capacity to survive in the absence of cell‐matrix contact in comparison with the nontransformed cell line HaCaT. Both cell lines underwent apoptosis in response to lost cell‐matrix contact. However, in HaCaT cells this apoptosis predominantly affected cycling cells and spared cells remaining in cell cycle arrest, indicating that the apoptotic process was initiated at one or more of the cycle checkpoints. In contrast, in the tumour cell line a large enough proportion of the cells remaining in cycle were able to evade anoikis so that cell numbers remained approximately constant over 24 h in the absence of adhesion (levels of apoptosis were typically 15% at 24 h and BrdU labelling was 28%). This ability would provide a significant advantage in the processes of invasion and metastasis to these tumour cells in vivo.
The intracellular mechanisms by which loss of adhesion induces cell death and/or cycle arrest remains to be fully elucidated. Signalling through integrins and other transmembrane adhesion‐linked receptors is likely to be central to these processes. Using specific inhibitors of three key proteins involved in signalling cascades, we investigated some of the possible intracellular pathways involved in anoikis in HaCaT and SW742 cells.
Protein kinase C (PKC) is activated through G‐proteins in response to growth factor‐receptor binding. Activated PKC may phosphorylate a variety of downstream signalling components, including MAP kinase, which in turn may induce the activity of the transcription factor component jun (Pulverer et al. 1991; Wood et al. 1992; Kribben et al. 1993; Bogoyevitch et al. 1994). In the present study, the effects of inhibiting PKC activity using Ro 31–8220 suggest that PKC may partially protect HaCaT cells from anoikis through contributing to the cell cycle arrest undergone by surviving cells under nonadherent conditions. Ro 31–8220 similarly increases cycling in detached SW742 cells, but here it inhibits apoptosis, indicating that PKC plays an altered role in the regulation of proliferation and apoptosis in these tumour cells.
Phosphoinositide‐3‐kinase (PI3K) is activated by interacting directly with receptor tyrosine kinases following phosphorylation of the receptor in response to ligand binding. PI3K is involved in inducing cell motility, vesicle trafficking and secretion, and apoptosis (Malarkey et al. 1995; Carpenter & Cantley 1996) and is thought to regulate susceptibility to apoptosis through PKB/Akt and phosphorylation of Bad10. In our study, inhibition of PI3K by wortmannin did not induce cell cycling in HaCaT cells prevented from adhesion, although at high concentration it induced apoptosis, perhaps through its effects on the PKB/Akt‐Bad pathway. Wortmannin promoted both cell cycling and apoptosis in adhesion‐prevented SW742 cells, suggesting that PI3K may play a cycle‐ and survival‐regulating function in these cells which differs from its role in HaCaT. Activation of p70(s6k) results in ribosomal protein phosphorylation and G1 progression (Lane et al. 1993). PI3K is thought to mediate growth factor signalling to p70(s6k) independently of PKC (Chung et al. 1994).
The Janus kinases or JAKs are a family of cytoplasmic protein tyrosine kinases activated by the phosphorylation of tyrosine residues on receptors in response to ligand/receptor binding (Silvennoinen et al. 1993; Ihle et al. 1994; Witthuhn et al. 1994; Malarkey et al. 1995). Activated JAKs link cytokine receptors to transcriptional regulators and ultimately to mitogenesis via the signal transducers and activators of transcription (STAT) family of cytoplasmic proteins (Ihle et al. 1994; Quelle et al. 1994). JAK2 has been identified as being activated by growth hormone receptor (Argetsinger et al. 1993), IL‐3 (Silvennoinen et al. 1993), and interferon‐γ (Watling et al. 1993). In erythroid cells, JAK2 is associated with the erythropoietin receptor and couples erythropoietin binding to tyrosine phosphorylation and mitogenesis (Witthuhn et al. 1993).
Abnormal JAK/STAT signalling and growth regulation is believed to occur in tumour cells (Nielsen et al. 1997). Current studies have observed the effects of cytokine binding on phosphorylation of JAKs, but few data exist detailing the responses of JAKs to activation of other receptors such as integrins. In our system, inhibition of JAK2 by AG490 released HaCaT cells cultured on polyHEMA from growth inhibition and increased their apoptotic rate. Thus, like PKC, JAK2 may partially protect HaCaT cells from anoikis through contributing to the cell cycle arrest undergone by surviving cells under nonadherent conditions. However, the tumour cell line SW742 underwent apoptosis without concomitant increase in cell cycling in response to AG490, suggesting that inhibition of JAK2 may induce apoptosis directly in these tumour cells.
The susceptibility of the HaCaT cell line to cell cycle arrest and apoptosis in response to prevention of adhesion may reflect the way in which nontransformed epithelial cells are prevented from ectopic growth in vivo. The divergent behaviour of SW742 cells under similar conditions provides insight into the mechanisms by which tumour cells survive during the processes of invasion and metastasis, when they are no longer in contact with native adhesion substrate. The different roles of PKC, PI3K and JAK2 in the two cell lines indicate that these protein kinases are involved in signalling pathways which regulate cycling and survival in response to adhesion. Disruption of these pathways may occur in tumour cells. It remains to be seen which of the complex web of intracellular signalling cascades is critical to the development of adhesion‐independent survival during tumourigenesis in vivo. Such pathways would provide a powerful target for drug therapy against tumour invasion and metastasis.
Acknowledgements
We would like to thank Mrs. Olivia Smith (Institute for Cancer Studies, Sheffield) for her help with flow cytometry.
This work was funded by Yorkshire Cancer Research and The University of Sheffield.
References
- Argetsinger LS, Campbell GS, Yang XN et al. (1993) Identification of JAK2 as a growth‐hormone receptor‐associated tyrosine kinase. Cell 74, 237. [DOI] [PubMed] [Google Scholar]
- Bates RC, Buret A, Van Helden DF, Horton MA, Burns GF (1994) Apoptosis induced by inhibition of intercellular contact. J. Cell Biol. 125, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, Mareel MM, Van Roy FM, Birchmeier W (1989) Dissecting tumour cell invasion. epithelial cells acquire invasive properties after the loss of uvomorulin‐mediated cell‐cell adhesion. J. Cell Biol. 108, 2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Glennon PE, Andersson MB et al. (1994) Endothelian‐1 and fibroblast growth factors stimulate the mitogen activated protein kinase signalling cascade in cardiac myocytes‐the potential role of the cascade in the integration of 2 signalling pathways leading to myocyte hypertrophy. J. Biol. Chem. 269, 1110. [PubMed] [Google Scholar]
- Carpenter CL & Cantley LC (1996) Phosphoinositide kinases. Curr. Opin. Cell. Biol. 8, 153. [DOI] [PubMed] [Google Scholar]
- Chung JK, Grammer TC, Lemon KP, Kazlaukas AAND, Blenis J (1994) PDGF‐dependent and insulin‐dependent PP. 70 (S6K) activation mediated by phosphatidylinositol‐3OH kinase. Nature 370, 71. [DOI] [PubMed] [Google Scholar]
- Davies MA, Lu Y, Sano T et al. (1998) Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits Akt activation and induces anoikis. Cancer Res. 58, 5285. [PubMed] [Google Scholar]
- Folkman J & Moscona A (1978) Role of cell shape in growth control. Nature 273, 345. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A (1997) Direct regulation of the Akt proto‐oncogene by phosphatidylinositol‐3,4‐bisphosphate. Science 275, 665DOI: 10.1126/science.275.5300.665 [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO et al. (1995) The protein kinase encoded by the Akt proto‐oncogene is a target of the PDGF‐activated phosphatidylinositol 3‐kinase. Cell 81, 727. [DOI] [PubMed] [Google Scholar]
- Frisch SM & Francis H (1994) Disruption of epithelial cell–matrix interactions induces apoptosis. J.Cell Biol. 124, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM & Ruoslahti E (1997) Integrins and anoikis. Curr. Opin. Cell. Biol. 9, 701. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Kelaita Sicks S (1996b) A role for jun‐N‐terminal kinase in anoikis; suppression by bcl‐2 and crmA. J. Cell Biol. 135, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chanhui PY (1996a) Control of adhesion‐dependent cell survival by focal adhesion kinase. J. Cell Biol. 134, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle JN, Withuhn BA, Quelle FW et al. (1994) Signalling by the cytokine receptor superfamily‐JAKS and STATA. TIBS 19, 222. [DOI] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez‐Viciana P, Wennstrom S, Warne PH, Downward J (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3‐OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16, 2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kribben A, Wieder ED, Li XM et al. (1993) AVP‐induced activation of MAP kinase in vascular smooth‐muscle cells is mediated through protein kinase C. Am. J. Physiol. 265, 939. [DOI] [PubMed] [Google Scholar]
- Lane HA, Fernandez A, Lamb NJC, Thomas G (1993) p70 (s6k) function is essential for G1‐progression. Nature 363, 170. [DOI] [PubMed] [Google Scholar]
- Malarkey K, Belham CM, Paul A, Graham A, McLees A, Scott PH, Plevin R (1995) The regulation of tyrosine kinase signalling pathways by growth factor and G‐protein‐coupled receptors. Biochem. J. 309, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Kaltoft K, Nordahl M et al. (1997) Constitutive activation of a slowly migrating isofrom of STAT3 in mycosis fungoides. tyrphostin AG490 inhibits STAT3 activation and growth of mycosis fungoides tumour cell lines. Proc. Natl. Acad. Sci. USA. 94, 6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulverer BJ, Kyriakis JM, Avruch J, Nikolakai E, Woodgett JR (1991) Phosphorylation of c‐jun mediated by MAP kinases. Nature 353, 670. [DOI] [PubMed] [Google Scholar]
- Quelle FW, Sato N, Witthuhn BA, Inhorn RC, Eder M, Miyajima A (1994) JAK2 associates with the β (c) chain of the receptor for granulocyte‐macrophage colony stimulating factor and its activation requires the membrane proximal region. Mol. Cell. Biol. 14, 4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E & Reed JC (1994) Anchorage dependence, integrins and apoptosis. Cell 77, 477. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O, Witthuhn BA, Quelle FW, Cleveland JL, Yi TL, Ihle JN (1993) Structure of the murine JAK2 protein‐tyrosine kinase and its role in interleukin‐3 signal transduction. Proc. Natl. Acad. Sci. USA. 90, 8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T et al. (1997) Dual role of phosphatidylinositol 3,4,5‐triphosphate in the activation of protein kinase B. Science 277, 567DOI: 10.1126/science.277.5325.567 [DOI] [PubMed] [Google Scholar]
- Strater J, Wedding U, Barth TF, Koretz K, Elsing C, Moller P (1996) Rapid onset of apoptosis in vitro follows disruption of beta 1‐integrin/matrix interactios in human colonic crypt cells. Gastroenterol. 10, 1776. [DOI] [PubMed] [Google Scholar]
- Streuli M (1996) Protein tyrosine phosphatases in signalling. Curr. Opinion Cell Biol. 8, 182. [DOI] [PubMed] [Google Scholar]
- Stryer L (1993). Biochemistry. 3rd edn p. 830 New York. W.H. Freeman. [Google Scholar]
- Watling D, Guschin D, Muller M et al. (1993) Complementation by the protein‐tyrosine kinase JAK2 of a mutant cell line defective in the interferon‐γ signal transduction pathway. Nature 366, 166. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O et al. (1993) JAK2 associates with the erythropoietin receptor and is tyrosine‐phosphorylated and activated following stimulation with erythropoietin. Cell 74, 237. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Silvennoinen O, Miura O et al. (1994) Involvement of the JAK‐3 Janus Kinase in signalling by interleukin‐2 and interleukin‐4 in lymphoid and myeloid cells. Nature 370, 153. [DOI] [PubMed] [Google Scholar]
- Wood KW, Sarnecki C, Roberts TM, Blenis J (1992) Ras mediates nerve growth factor receptor modulation of 3 signal transducing protein kinases‐MAP kinase, RAF‐1 and RSK. Cell 68, 1041. [DOI] [PubMed] [Google Scholar]