

Abstract
Objectives: Previously, we have isolated stem cells (HEAC) from human eyelid adipose tissue and functionally differentiated them into insulin‐secreting cells. In the present study, we examined whether insulin family members might influence insulinogenic differentiation of HEAC.
Materials and methods: Following culture in differentiation media containing insulin family member or not, cells were examined for gene expression, protein expression and, particularly, insulin and C‐peptide secretion, in response to high glucose challenge. Using antibodies against the specific receptor, target receptor mediating effect of the insulin family member was investigated.
Results: Insulin treatment during culture had little effect on either insulin or C‐peptide secretion from HEAC, against high glucose challenge after culture. However, insulin‐like growth factor (IGF) 1 treatment decreased both secretions, and interestingly, IGF2 greatly increased the secretions. HEAC treated with IGF2 had strong expression of Pdx1, Isl1, Pax6 and PC1/3 genes, and distinct staining after insulin and C‐peptide antibodies, and dithizone. IGF2‐enhanced insulinogenic differentiation was totally blocked by antibody against insulin receptor (IR), but not by anti‐IGF1 receptor (IGF1R). Differentiated HEAC expressed both IR and IGF1R genes, whereas they expressed neither IGF2 nor IGF2R genes.
Conclusions: From these results, it is suggested that IGF1 might inhibit insulinogenic differentiation of HEAC, whereas IGF2 enhances differentiation, and that enhancement of IGF2 appeared to be mediated via IR.
Introduction
Insulin and insulin‐like growth factor (IGF) family members have been studied as regulators of β‐cell mass. Exogenous treatment by insulin at low nanomolar concentrations has been shown to reduce apoptosis of rat primary β cells induced by low cell density and low glucose (1) and of primary human and murine islet cells induced by serum withdrawal and low glucose (2, 3). Insulin at high concentration has also been shown to increase proliferation of mouse primary islet cells (4). However, some studies have shown that high doses of insulin do not reduce apoptosis of primary human and mouse islet cells (2), but increase caspase‐3‐mediated cell death in a glucose‐deprived apoptosis model of a mouse pancreatic β‐cell line (5). IGF1 induces proliferation of rat INS‐1 cells in a glucose‐dependent manner (6, 7) and also prevents cytokine‐mediated apoptosis of islet cells from non‐obese diabetic mice (8) and serum withdrawal‐induced apoptosis of both human and rat primary islet cells (3). Several studies, however, have shown that liver‐specific or pancreas‐specific inactivation of IGF1 gene causes pancreatic islets to enlarge, suggesting that IGF1 may serve an inhibitory function on islet growth in vivo (9, 10). Treatment with IGF2 has been shown to protect against cytokine‐mediated apoptosis of neonatal rat primary pancreatic islets in vitro, and the effect diminished when islets were incubated with neutralizing antibody against IGF2 (11). Overexpression of IGF2 during foetal development causes islet hyperplasia and low islet cell apoptosis (12). IGF2 supplementation increases the pool of β cells in pancreatic rudiments isolated from diabetic GK rats in which foetal production of IGF2 was impaired (13, 14). Thus, IGF2 appears to protect islet cells from apoptosis and may serve as a survival factor. However, the above studies used β cells which already were determined or differentiated in vivo, and thus little is known about their roles during differentiation of stem/progenitor cells into β cells.
Various human cells can differentiate into insulin‐secreting cells in vitro. Their transplantation in vitro has been shown to normalize or reduce blood glucose levels in animals of diabetic models (15, 16, 17, 18, 19, 20). Results of some studies are promising for clinical application of insulin‐secreting cells to treatment of human with type 1 diabetes, however, insulin‐secreting cells to be used therapeutically need to secrete insulin in response to high glucose stimulation and to secrete high enough levels of it to regulate blood glucose level of the recipients. Researchers thus have been trying to develop various conditions for differentiation of various cell types into insulin‐secreting cells. Many studies have used insulin or IGFs for insulinogenic differentiation of human embryonic stem cells (16, 17, 21, 22), adult stem cells (18, 23) or further types of cells (24, 25); however, these studies have not clarified whether addition of insulin or IGFs promoted differentiation or not and thus, little is known concerning roles of these factors during differentiation of undetermined cells into insulin‐secreting cells.
Insulin and IGFs function primarily through insulin receptors (IR) including IR‐A and IR‐B, and IGF receptors including IGF1R and IGF2R, respectively (26). However, at high nanomolar concentrations, insulin activates the IGF1R, and IGFs can activate IR (27). In humans, adult pancreatic β cells express both mRNA forms of IR, IR‐A and IR‐B (28, 29). Expression of IGF1R mRNA has been observed in intact tissues and cultured islets of foetal pancreas (30), and in adult pancreatic tissues (31). Expression of IGF2R mRNA and strong immunoreactivity to anti‐IGF2R antibody have been observed in normal pancreatic tissues and in cytoplasm of islet cells, respectively (32).
In the present study, we have examined the effect of insulin and IGFs on differentiation of human eyelid adipose‐derived stem cells (HEAC) into insulin‐secreting cells. During insulinogenic differentiation culture, insulin treatment exhibited little effect and IGF1 had inhibitory effects; in contrast, IGF2 greatly enhanced differentiation. Interestingly, when cells were treated with IGF2 and anti‐IR antibody together, their differentiation drastically decreased. These results suggest that IGF2 could enhance insulinogenic differentiation of HEAC through IR signalling.
Materials and methods
Reagents
All reagents used were purchased from Sigma (St Louis, MO, USA) unless specified otherwise. DMEM media and type I collagenase were supplied by the Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS) was supplied by Hyclone (Logan, UT, USA) and Activin A, basic fibroblast growth factor (bFGF), IGF1 and IGF2 were supplied by Peprotech (Rocky Hill, NJ, USA). Mouse monoclonal antibodies to human proinsulin, insulin, IR and IGF1R were purchased from Abcam (Cambridge, MA, USA). Mouse monoclonal antibody to human C‐peptide was supplied by Monosan (AM Uden, Netherland) and ELISA kits for human insulin and C‐peptide were supplied by Mercodia (Winston Salem, NC, USA). Biotinylated goat anti‐mouse IgG, horseradish peroxidase‐conjugated streptavidin and 3,3′‐diaminobenzidine tetrahydrochloride were purchased from DakoCytomation (Glostrup, Denmark). Human adult pancreatic mRNA (hPan) and human HepG2 mRNA were purchased from Ambion (Austin, TX, USA).
HEAC isolation and culture
Human eyelid adipose tissue was obtained from three subjects aged between 28 and 42 years (mean age: 36.2 years) undergoing cosmetic surgery, after providing informed consent. Fat tissues were surgically dissected from the subcutaneous zone and adipose‐derived stem cells (HEAC) were obtained as previously described (20). Briefly, fat tissue was minced and treated with 0.075% type I collagenase in phosphate‐buffered saline (PBS) for 30 min at 37 °C with gentle stirring. Following centrifugation at 1250 g for 10 min, cell pellets were isolated and washed twice in DMEM‐low glucose type (5.5 mm; DMEM‐LG). Cell suspensions were cultured in DMEM‐LG supplemented with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 3.7 mg/ml sodium bicarbonate at 5% CO2, 37 °C. After 2 weeks in culture, adherent cells were obtained and subjected to passage culturing; afterwards, medium was changed twice a week. All experiments were approved by Institutional Review Boards of Seoul Women’s University.
Differentiation of HEAC into insulin‐secreting cells in vitro
HEAC at passage (p) 3 to p5 were divided into five groups and plated on 48‐well dishes (Nunc, Rochester, NY, USA) at 5 × 103 density cells/well and grown for 3 weeks. For control groups (C), cells were cultured in DMEM‐high glucose type (25 mm; DMEM‐HG) containing 10% FBS for 1 week, and then in DMEM‐LG containing FBS for additional 2 weeks. For other experimental groups, cells were grown for 1 week in DMEM‐HG containing 10% FBS, 4 nm activin A, 20 ng/ml bFGF, and 10 nm glucagon‐like peptide (GLP)‐1. During the subsequent 2 weeks, a group of cells (N group) was grown in DMEM‐LG containing 10% FBS, 10 mm nicotinamide, 4 nm activin A, and 10 nm GLP‐1 (NAG). Cells of other groups were grown in the same medium as N group, but supplemented with 50 ng/ml human insulin (Insulin group), IGF1 (IGF1 group) or IGF2 (IGF2 group).
Receptor blocking
To identify IGF2 target receptor molecule, cells were initially cultured for 1 week in DMEM‐HG containing 10% FBS, 4 nm activin A, 20 ng/ml bFGF and 10 nm GLP‐1. Then they were cultured for 2 weeks, for further differentiation in DMEM‐LG containing 10% FBS, 10 mm nicotinamide, 4 nm activin A, 10 nm GLP‐1 and 50 ng/ml IGF2. During the last 2 weeks in culture, 1 nm IR antibody, 1 μg/ml IGF‐1R antibody or both antibodies were added to culture media. After 3 weeks differentiation culture, amounts of insulin and C‐peptide released into culture media were measured using ELISA, as described below.
Measurement of insulin and C‐peptide
To measure amounts of insulin and C‐peptide released in vitro, cells after differentiation culture were incubated in DMEM‐LG containing 0.5% bovine serum albumin (BSA) for 12 h. They were washed in PBS, and subsequently stimulated with DMEM containing 5.5, 15.0 or 25.0 mm glucose for 2 h at 37 °C. Amounts of insulin and C‐peptide released into these stimulating media were measured using ultrasensitive human insulin and C‐peptide ELISA kit according to the manufacturer’s instructions.
RT‐PCR
RNA was isolated using Tri‐reagent according to the manufacturer’s instructions. Purity of RNA was assessed by determining ratio of absorbance at 260 nm to that at 280 nm (>1.8) and RT‐PCR was performed using a GeneAmp PCR system 2400 (Perkin Elmer, Boston, MS, USA). Subsequent PCR reactions were performed using cDNA, primer pairs (Table 1) and PCR mixture (Fermentas, St Leon‐Rot, Germany) according to manufacturers’ instructions. Amplified products were separated on 1.5% gel and visualized with ethidium bromide under a gel scanner (Ultima; Hoefer, Holliston, MA, USA). Transcript levels of genes were standardized to corresponding GAPDH level and relative mRNA levels were obtained.
Table 1.
Primers used for the RT‐PCR analysis and expected size of PCR products
| Gene | Primer sequence | Size (bp) | Annealing temp. (°C) | Reference |
|---|---|---|---|---|
| GAPDH | 5′‐aca act ttg gta tcg tgg aa‐3′ 5′‐aaa ttc gtt gtc ata cca gg‐3′ | 456 | 53 | NM 002046 |
| Insulin | 5′‐aac caa cac ctg tgc ggc tc‐3′ 5′‐aag ggc ttt att cca tct ctc tcg‐3′ | 320 | 59 | J00265 |
| NeuroG3 | 5′‐cgt gaa ctc ctt gaa ctg agc ag‐3′ 5′‐tgg cac tcc tgg gac gag ttt c‐3′ | 211 | 61 | AF 234829 |
| pdx1 | 5′‐ccc atg gat gaa gtc tac g‐3′ 5′‐gtc ctc ctc ctt ttt cca c‐3′ | 262 | 54 | NM 000209 |
| Glucokinase | 5′‐gaa tac ccc cca gag acc ttt tc‐3′ 5′‐ggt ttc ttc ctg agc cag cg‐3′ | 182 | 61 | M90299 |
| Islet1 | 5′‐agcatcaatgtcctctcaacttcc‐3′ 5′‐tgtttggcaaggcaatgacc‐3′ | 493 | 57 | NM 002202 |
| Nkx6.1 | 5′‐acacgagacccactttttccg‐3′ 5′‐tgctggacttgtgcttcttcaac‐3′ | 336 | 59 | NM 006168 |
| Glut 2 | 5′‐agc ttt gca gtt ggt gga at‐3′ 5′‐aat aag aat gcc cgt gac ga‐3′ | 300 | 57 | NM 000340 |
| NeuroD1 | 5′‐cag aac cag gac atg ccc‐3′ 5′‐atc aaa gga agg gct ggt g‐3′ | 216 | 60 | NM 002500 |
| PC1/3 | 5′‐ttg gct gaa aga gaa cgg gat aca tct‐3′ 5′‐act tct ttg gtg att gct ttg gcg gtg‐3′ | 457 | 60 | NM 000493.3 |
| PC2 | 5′‐gca tca agc aca gac cta cac tcg‐3′ 5′‐gag aca caa cca ccc ttc atc ctt c‐3′ | 309 | 63 | NM 003594.2 |
| GLUT1 | 5′‐ctc act gct caa gaa gac atg g‐3′ 5′‐ctg ggt aac agg gat caa aca g‐3′ | 366 | 60 | NM 006516.1 |
| Nkx 2.2 | 5′‐ttc tac gac agc agc gac aac c‐3′ 5′‐cgt cac ctc cat acc ttt ctc g‐3′ | 393 | 56 | NM 002509.2 |
| IGF I | 5′‐cac tgt cac tgc taa att ca‐3′ 5′‐tga aat aaa agc ccc tgt gtc t‐3′ | 386 | 55 | NM 00618.3 |
| IGF II | 5′‐ctg gag acg tac tgt gct ac‐3′ 5′‐ggt gtt taa agc caa tcg att‐3′ | 547 | 55 | NM 000612.4 |
| IR | 5′‐cat caa ggg tga ggc aga ga‐3′ 5′‐ttg gcc tca tct tgg ggt tg‐3′ | 710 | 59 | NM 000208.1 |
| IGF IR | 5′‐tgg agt gct gta tgc ctc tg‐3′ 5′‐tga tga cca gtg ttg gct gg‐3′ | 329 | 59 | NM 000875.3 |
| IGF IIR | 5′‐tca aca tct gtg gaa gtg tgg a‐3′ 5′‐gaa tag aga agt gtc cgg atc gga gtc‐3′ | 430 | 59 | NM 000876.2 |
Immunocytochemistry
After culture in Lab‐Tek chamber slides (Nunc), HEAC were washed in PBS then permeabilized in 0.5% Triton X‐100 for 10 min. Endogenous peroxidase activity was inactivated using 3% hydrogen peroxide for 15 min at room temperature (RT). Slides were incubated in blocking solution, consisting of 2% BSA in PBS for 1 h at RT. Between each step, cells were washed in 0.1% BSA in PBS. Cells were incubated with primary antibodies overnight at 4 °C; the following primary antibodies against human proteins were used: mouse monoclonal antibodies to C‐peptide (1:200), proinsulin (1:200) and insulin (1:200). After labelling with primary antibodies, cells were incubated in biotinylated goat anti‐mouse IgG followed by horseradish peroxidase‐conjugated streptavidin for 20 min, respectively, at RT. Immunoreactivity was visualized using 3,3′‐diaminobenzidine tetrahydrochloride counterstained with Mayer’s haematoxylin. Cells were photographed using bright‐field microscope illumination (Axioskop2+; Carl Zeiss, Jena, Germany).
Dithizone staining
Cultured cells were incubated in 100 μg/ml dithizone (DTZ) solution at 37 °C for 15 min.
Statistical analysis
Data were expressed as mean ± SEM. Statistical significance was analysed by one‐way ANOVA test using SPSS 18.0 (Somers, NY, USA). A P‐value <0.05 or 0.01 was considered statistically significant.
Results
During maintenance culture up to p5 in DMEM containing FBS alone, morphology of HEAC remained unchanged. After culture in the five different differentiation culture conditions (Fig. 1A), amounts of insulin and C‐peptide released by HEAC in response to 25.0 mm glucose was analysed using ELISA. As shown in Fig. 1B, HEAC of the control group demonstrated little (1.4 ± 0.7 pg/ml) insulin secretion, whereas those of N group secreted 113.9 ± 10.7 pg/ml. Addition of insulin to the N conditions, however, resulted in 288.3 ± 30.6 pg/ml insulin secretion. IGF1 addition instead of insulin somewhat lowered insulin secretion to 53.6 ± 20.2 pg/ml. Strikingly, IGF2 addition greatly increased insulin secretion to 950.1 ± 151.5 pg/ml. Amounts of C‐peptide secreted from each HEAC group exhibited similar patterns, 14.8 ± 3.9 pg/ml by controls, 268.1 ± 60.3 pg/ml by N group, 167.0 ± 33.2 pg/ml by Insulin group, 55.4 ± 24.1 pg/ml by IGF1 group and 1257.5 ± 94.0 pg/ml by IGF2 group (Fig. 1C). These results demonstrated that IGF2 could strongly enhance differentiation of HEAC into insulin‐secreting cells and that IGF1 might be an inhibitor of insulinogenic differentiation. Insulin did not appear to affect differentiation.
Figure 1.
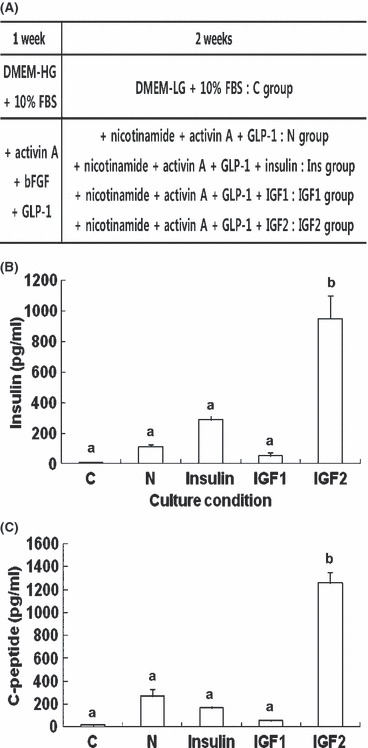
Effects of insulin families on differentiation of HEAC into insulin‐secreting cells. (A) Schematic diagram of experimental procedure. (B, C) ELISA analyses of insulin and C‐peptide release from various adult stem cells after culture in control or insulinogenic differentiation media. Insulin (B) and C‐peptide (C) released from HEAC after culture in various differentiation media for 3 weeks as described in the Materials and methods section. C, control group; N, NAG group; Insulin, Insulin group; IGF1, IGF1 group; IGF2, IGF2 group. Values were obtained from three independent experiments. a and b, P < 0.01 compared to C group, by ANOVA testing.
To examine whether secretion of insulin and C‐peptide from differentiated HEAC might occur in a glucose‐dependent manner, cells incubated in presence of 5.5, 15 or 25 mm glucose concentration for 2 h then cultured in medium, were analysed. HEAC of the control group secreted 7.5 ± 3.4, 15.3 ± 3.0 and 11.4 ± 2.9 pg/ml insulin in response to the above glucose concentrations, respectively. However, cells of the N group secreted 15.3 ± 3.0, 52.2 ± 2.8 and 127.3 ± 7.8 pg/ml insulin; cells of IGF2 group secreted 25.7 ± 2.6, 113.9 ± 1.0 and 687.8 ± 58.1 pg/ml insulin, respectively (Fig. 2A). Amounts of C‐peptide released into the medium were 9.4 ± 1.5, 12.4 ± 5.1 and 9.4 ± 11.1 pg/ml for control group, and 6.7 ± 4.0, 96.6 ± 36.3 and 242.9 ± 40.0 pg/ml N group, and 17.1 ± 7.0, 407.1 ± 17.4 and 1147.1 ± 220.0 pg/ml IGF2 group, respectively (Fig. 2B).
Figure 2.
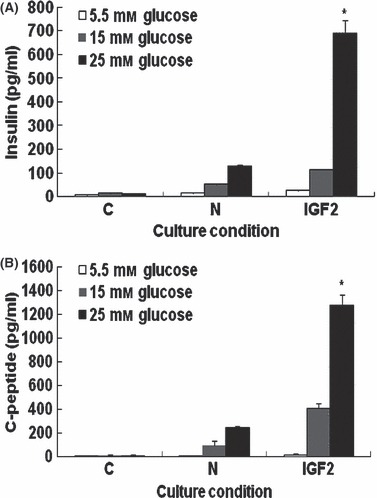
ELISA analyses of insulin and C‐peptide release from HEAC in response to different concentrations of glucose. HEAC cultured in either DMEM containing FBS (control), DMEM containing FBS and NAG (N), or DMEM containing FBS, NAG and IGF2 (IGF2). Then cells were incubated in DMEM containing 5.5, 15.0 or 25.0 mm glucose concentration. Amounts of insulin (A) and C‐peptide (B) released into each medium was measured. Values were obtained from three independent experiments. a and b, P < 0.01 compared to C group, by ANOVA testing.
RT‐PCR analyses showed that control group HEAC expressed genes coding for glucokinase, NeuroD1, and Isl1, but did not express other β‐cell‐related genes such as Ngn3, Nkx2.2, Pdx1, glut2, Pax6, PC1/3, Nkx6.1 and Ins (Fig. 3A). In contrast, HEAC of IGF2 group began to express Ngn3, Nkx2.2, Pdx1 and glut2 genes during the first week of insulinogenic differentiation, Pax6 gene during the second week, and PC1/3, Nkx6.1 and insulin genes during the (final) third week. Although HEAC of N group also showed a similar expression pattern, IGF2 group HEAC had a greater gene expression of Pdx1, Isl1, Pax6 and PC1/3. Compared to human pancreatic tissue (hPAN), IGF2 group HEAC of had weaker expression of glut2 gene, but enhanced expression of Isl1 and Pdx1. Immunocytochemical staining showed that IGF2 group HEAC were strongly immunoreactive using antibodies to human proinsulin, insulin and C‐peptide, compared to controls (Fig. 3B). DTZ, known to discriminate β cells from others, intensely stained cells of colony‐forming area of IGF2 group (Fig. 3C). These results suggest that IGF2 group differentiated cells could functionally resemble β cells.
Figure 3.
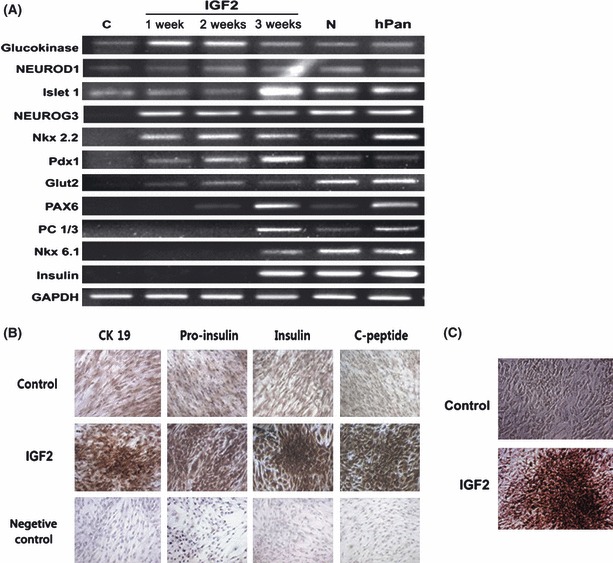
Gene expression profile of HEAC after culture in various differentiating media. C, HEAC cultured in DMEM containing FBS alone for 3 weeks; IGF2, HEAC cultured in DMEM containing FBS, NAG and IGF2 for 1 week (1 w), 2 weeks (2 w) or 3 weeks (3 w); N, HEAC cultured in DMEM containing FBS and NAG only; hPAN, human pancreatic cell mRNA.
We also examined whether increased insulin and C‐peptide secretion from IGF2 group HEAC was due to either enhanced differentiation or proliferation by IGF2. Cells were divided into four groups and then cultured. At the end of culture time, cell number of each group increased from the initial 5 × 103 to 5.0 ± 1.1 × 104 control C group, 6.6 ± 1.2 × 104 N group, 6.7 ± 1.2 × 104 Insulin group and to 7.5 ± 1.0 × 104 IGF2 group. To make satisfactory comparison, cell number of all groups was normalized to 5 × 104 cells and amounts of insulin and C‐peptide secretion were analysed. When undifferentiated cells of C group secreted insulin of 9.6 ± 5.1 pg/ml/5 × 104 cells and C‐peptide of 21.1 ± 2.0 pg/ml/5 × 104 cells at 25 mm glucose concentration, differentiated cells of N group secreted 94.5 ± 26.6 pg of insulin and 293.2 ± 50.9 pg of C‐peptide under the same conditions. In contrast, differentiated cells of the Insulin group secreted 196.5 ± 43.3 pg insulin and 261.2 ± 26.2 pg C‐peptide, and cells of IGF2 group secreted 504.3 ± 76.2 pg insulin and 812.2 ± 174.2 pg C‐peptide (Fig. 4A,B). Based on secretions of both insulin and C‐peptide, the results show that insulin treatment did not enhance insulinogenic differentiation of HEAC, whereas IGF2 greatly enhanced it.
Figure 4.
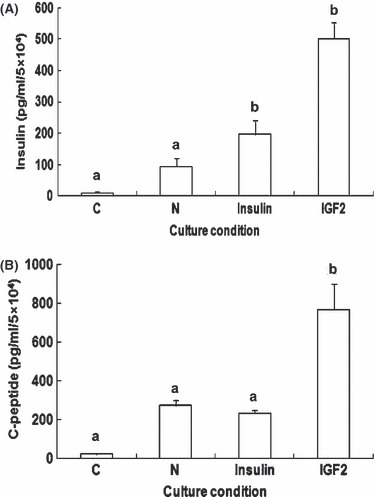
ELISA analyses of insulin and C‐peptide release from various adult stem cells after differentiation. Cell number of all groups was normalized to 5 × 104 cells and amounts of insulin (A) and C‐peptide (B) secretion in response to 25 mm glucose, compared to each other. C, control group; N, NAG group; Insulin, Insulin group; IGF2, IGF2 group. Values were obtained from three independent experiments. a and b, P < 0.01 compared to C group, by ANOVA testing.
To find target receptor molecules that mediate the IGF2 effect, antibody to IR and/or IGF‐1R were added to culture medium of IGF2 group during differentiation culture. After 3 weeks, amounts of insulin and C‐peptide released by HEAC into the medium were analysed (Fig. 5). IGF2 group HEAC without antibody treatment demonstrated greatly enhanced insulin and C‐peptide secretion in a glucose‐dependent manner. In contrast, those treated with anti‐IR antibody mostly had diminished secretion of both insulin and C‐peptide such that amounts of both proteins were near quantities of the control group. In contrast, both secretions from HEAC treated with anti‐IGF1R antibody were not affected. Combined treatment of both anti‐IR and anti‐IGF1R antibodies resulted in diminished secretion similar to anti‐IR antibody treatment alone. These results demonstrate that IGF2 signalling might be mediated via IR in HEAC during insulinogenic differentiation in vitro.
Figure 5.
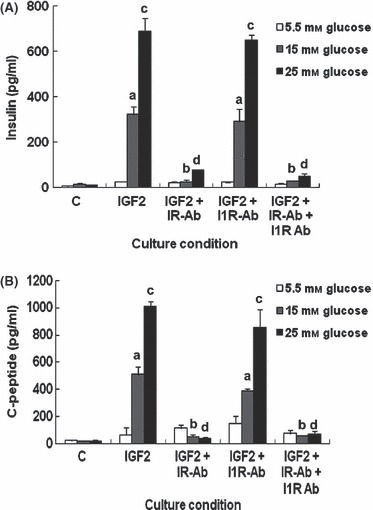
Effects of antibodies to IR and IGF1R on secretion of insulin and C‐peptide by HEAC in the presence of IGF2. During the last 2 weeks of culture, for insulinogenic differentiation, 1 nm IR antibody, 1 μg/ml IGF1R antibody or both antibodies were added to culture media. After culture, amounts of insulin and C‐peptide release from cell into media were measured using ELISA. Values were obtained from three independent experiments. a and b, P < 0.05, c and d, P < 0.01 compared to IGF2 group, by ANOVA testing.
Finally, we investigated expression profiles of insulin‐ and its receptor‐related genes including IGF1, IR‐A, IR‐B and IGF1R in IGF2 group HEAC, before and after differentiation (Fig. 6). Undifferentiated HEAC strongly expressed IR‐A gene and showed weak expression of IR‐B, IGF1R and IGF2R. They expressed neither IGF1 nor IGF2 genes. During the first week of differentiation, HEAC began to express IGF1 and increased expression of IR‐B, IGF1R and IGF2R. During second and third weeks of culture, interestingly, HEAC exhibited little expression of IGF2R and diminished expression of IR‐B, IGF1 and IGF1R, whereas they consistently exhibited strong expression of IR‐A. HEAC never expressed IGF2 gene whether differentiated or not. The human HepG2 hepatocyte cell line showed a distinct expression of all these genes. These results demonstrate that IGF2’s effect cannot be mediated via IGF2R in HEAC during insulinogenic differentiation in vitro.
Figure 6.
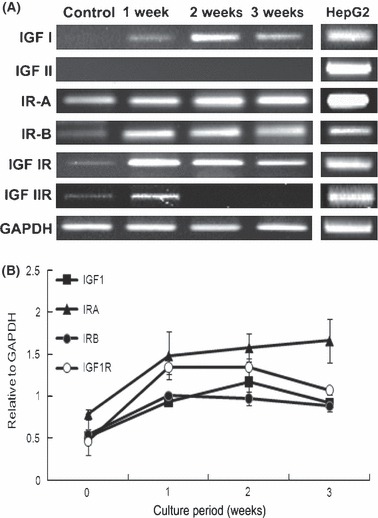
Insulin‐related gene expression profile of HEAC during insulinogenic differentiation culture. (A) Gene expression profile of insulin and IR receptor families by HEAC during insulinogenic differentiation culture. (B) Semiquantitative densitometric analysis of selected products in (A). Values standardized to corresponding GAPDH. HEAC were prepared as shown in Fig. 3. For comparison, human HepG2 mRNA was used as human pancreatic cell mRNA library was devoid of both IGF2 and IGF2R mRNAs.
Discussion
In this study, we observed that IGF2 in vitro greatly enhanced differentiation of human somatic stem cells derived from eyelid adipose tissues, into insulin‐secreting cells, while insulin had little effect and IGF1 inhibited their differentiation. Furthermore, IGF2‐enhanced differentiation appeared to be mediated via IR of HEAC.
Many studies have shown that IGF2 has anti‐apoptotic activity and/or increases survival of pancreatic β cells. IGF2 protects against cytokine‐mediated apoptosis in primary pancreatic islets of neonatal rat (11), and increases the pool of β cells in pancreatic rudiments isolated from diabetic GK rats (13, 14). Its overexpression during foetal development results in islet hyperplasia and reduced islet cell apoptosis (12). In the present study, IGF2 treatment caused marked increase in insulin secretion by HEAC following differentiation culture. Compared to our N group in which IGF2 was omitted, amounts of insulin and C‐peptide secreted in response to high glucose increased more than 3‐fold of each. Cell numbers in the same culture, however, increased only slightly. These observations demonstrate that the enhancing effect of IGF2 on insulinogenic differentiation of HEAC is caused mainly by its differentiation‐inducing ability and little by cell proliferation‐promoting activity.
HEAC differentiated in the presence of IGF2 showed no expression of either IGF2 or IGF2R, but distinctly expressed mRNAs of insulin, IGF1 and their receptor genes including IR‐A, IR‐B and IGF1R. Thus, it is not likely that IGF2 elicited its differentiation‐enhancing effect via IGF2’s receptor, which serves as a clearance receptor that can modulate availability of extracellular molecule (33, 34). IGF2 is more likely to act via other types of receptor. Insulin and IGFs act through their homo‐receptors including IR‐A, IR‐B, IGF1R and IGF2R, or hybrid receptors including IR‐A/IR‐B, IR‐A/IGF1R and IR‐B/IGF1R. Of these receptors, IGF2 could bind IR‐A, IGF1R and hybrid receptor of IR‐A/IGF1R, but not bind IR‐B and hybrid receptor of IR‐B/IGF1R (35, 36). However, antibody treatment experiments showed that anti‐IGF1R antibody did not affect IGF2‐induced differentiation, whereas anti‐IR antibody mostly inhibited IGF2‐induced differentiation. These results suggest that the IGF2 effect might be mediated by IR‐A rather than by IR‐B, IGF1R or hybrid receptors. IGF2 also plays a role in other differentiation processes; in mouse embryonic fibroblasts, interference of IGF2 production can inhibit production of both muscle‐specific structural proteins and of myocyte fusion to form multinucleate myotubes, suggesting that IGF2 plays a key role in satellite‐cell differentiation (37). In this model of myogenic differentiation, however, IGF2 signals through IGF1R.
Unlike IGF2, insulin treatment did not elicit significant enhancing effects on insulinogenic differentiation of HEAC even though it has higher affinity to IR‐A than IGF2 (36). One explanation is that insulin may act on HEAC in other ways from that of IGF2. An earlier study using IR− mouse embryonic fibroblasts transfected with IR‐A gene indicated that IGF2 was more effective than insulin in stimulating cell proliferation, while insulin was more potent than IGF2 in stimulating glucose uptake (35). Moreover, two types of IR have been proposed to play different roles in pancreatic β cells of βIRKO mice. Insulin secreted by pancreatic β cells on glucose stimulation activates its own insulin gene via IR‐A, while it simultaneously stimulates transcription of ß‐cell glucokinase gene via IR‐B (38). Microarray studies of R−/IR‐A cells has revealed that while 214 transcripts were similarly regulated by either insulin or IGF2, 45 genes were differentially transcribed in response to only one of the two molecules (39). Differential roles of IR isoforms have also been demonstrated in other types of cells. Data obtained using murine 32D haematopoietic cells has indicated that IR‐A responded to IGF2 and preferentially sent mitogenic, anti‐apoptotic signals, whereas IR‐B, poorly responsive to IGF2, tended to send differentiation signals (40). Immortalized neonatal hepatocytes from IR‐knockout mice have shown that hepatocytes reconstituted with IR‐A could fully restore glucose uptake up to levels observed in wild‐type hepatocytes, whereas those with IR‐B failed to do so (41). From these observations, it is suggested that binding of insulin to both IR‐A and IR‐B receptors during differentiation of HEAC might elicit different responses, unlike IGF2, which acts mainly through the IR‐A receptor.
Insulin or IGFs have been frequently used in insulinogenic differentiation media for various human cells, in vitro (21, 22, 24, 26, 28). These studies demonstrated generation of insulin‐secreting cells, as revealed by secretion of insulin or C‐peptide from differentiated cells. However, these studies did not examine effects of removal of insulin or IGFs from medium upon differentiation. Thus, it is not clear whether such molecules actually promoted insulinogenic differentiation or not. In the present study, we have added a control group whose medium did not contain insulin or IGFs. Compared to this control group, we observed that insulin was hardly effective, and IGF1 was inhibitory in promoting differentiation in contrast, IGF2 markedly enhanced differentiation. Further studies will clarify whether these differential effects of insulin and IGFs on insulinogenic differentiation are limited to HEAC or exist in other types of cells too.
While a variety of human cell types have been examined for ability of differentiating into insulin‐secreting cells, fewer studies have been conducted using human normal somatic cells. As use of adult stem cells is free from ethical issue associated with embryonic stem cells, and are not tumorigenic in vivo, insulinogenic cells produced from them seem to be plausible for therapeutic applications. Some of these studies involve normalized or reduced blood glucose level following transplantation to diabetic animal recipients, and we have observed presence of only human insulin and C‐peptide in blood of normalized mice, suggesting that human insulin was solely responsible for the normoglycemia (20). However, even in this study, success rate was limited to 50%, which was possibly due to insufficient amounts of insulin secretion from the differentiated cells compared to normal human islet cells (42). In the present study, using the same cells and culture conditions, we have observed that IGF2 treatment resulted in more than 3‐fold increase in insulin secretion after insulinogenic differentiation of HEAC. Further studies will verify whether these in vitro differentiated cells could provide better results with respect to treatment of diabetic animals.
In conclusion, we have shown that IGF2 treatment greatly enhanced functional differentiation of HEAC into insulin‐secreting cells in vitro, as seen in expression of β‐cell‐specific genes, β‐cell‐specific staining of cells, and particularly, markedly increased, high glucose‐dependent secretion of both insulin and C‐peptide from the differentiated cells. The effect of IGF2 appeared to be mediated via IR‐A alone, as anti‐IR antibody treatment almost ablated the enhancement effect of IGF2 while anti‐IGF1R antibody treatment did not, and IGF2R gene was not expressed. These findings may be used not only for preparation of therapeutic cells applicable to type 1 diabetes but also for research into β‐cell differentiation.
Author disclosure statement
Authors confirm no competing financial interests exist.
Acknowledgements
This work was supported by a special research grant from Seoul Women’s University (2010).
References
- 1. Navarro‐Tableros V, Sánchez‐Soto MC, García S, Hiriart M (2004) Autocrine regulation of single pancreatic beta‐cell survival. Diabetes 53, 2018–2023. [DOI] [PubMed] [Google Scholar]
- 2. Johnson JD, Bernal‐Mizrachi E, Alejandro EU, Han Z, Kalynyak TB, Li H et al. (2006) Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc. Natl. Acad. Sci. USA 103, 19575–19580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cahuana GM, Tejedo JR, Hmadcha A, Ramírez R, Cuestal AL, Soria B et al. (2008) Nitric oxide mediates the survival action of IGF‐1 and insulin in pancreatic beta cells. Cell. Signal. 20, 301–310. [DOI] [PubMed] [Google Scholar]
- 4. Beith JL, Alejandro EU, Johnson JD (2008) Insulin stimulates primary β‐cell proliferation via Raf‐1 kinase. Endocrinology 149, 2251–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guillen C, Bartolomé A, Nevado C, Benito M (2008) Biphasic effect of insulin on beta cell apoptosis depending on glucose deprivation. FEBS Lett. 582, 3855–3860. [DOI] [PubMed] [Google Scholar]
- 6. Hugl SR, White MF, Rhodes CJ (1998) Insulin‐like growth factor I (IGF‐I)‐stimulated pancreatic beta‐cell growth is glucose‐dependent. Synergistic activation of insulin receptor substrate‐mediated signal transduction pathways by glucose and IGF‐I in INS‐1 cells. J. Biol. Chem. 273, 17771–17779. [DOI] [PubMed] [Google Scholar]
- 7. Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ (2002) Activation of IRS‐2‐mediated signal transduction by IGF‐1, but not TGF‐alpha or EGF, augments pancreatic beta‐cell proliferation. Diabetes 51, 966–976. [DOI] [PubMed] [Google Scholar]
- 8. Hill DJ, Petrik J, Arany E, McDonald TJ, Delovitch TL (1999) Insulin‐like growth factors prevent cytokine‐mediated cell death in isolated islets of Langerhans from pre‐diabetic non‐obese diabetic mice. J. Endocrinol. 161, 153–165. [DOI] [PubMed] [Google Scholar]
- 9. Yakar S, Liu JL, Fernandez AM, Wu Y, Schally AV, Frystyk J et al. (2001) Liver‐specific igf‐1 gene deletion leads to muscle insulin insensitivity. Diabetes 50, 1110–1118. [DOI] [PubMed] [Google Scholar]
- 10. Lu Y, Herrera PL, Guo Y, Sun D, Tang Z, LeRoith D et al. (2004) Pancreatic‐specific inactivation of IGF‐I gene causes enlarged pancreatic islets and significant resistance to diabetes. Diabetes 53, 3131–3141. [DOI] [PubMed] [Google Scholar]
- 11. Petrik J, Arany E, McDonald TJ, Hill DJ (1998) Apoptosis in the pancreatic islet cells of the neonatal rat is associated with a reduced expression of insulin‐like growth factor II that may act as a survival factor. Endocrinology 139, 2994–3004. [DOI] [PubMed] [Google Scholar]
- 12. Petrik J, Pell JM, Arany E, McDonald TJ, Dean WL, Reik W et al. (1999) Overexpression of insulin‐like growth factor‐II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology 140, 2353–2363. [DOI] [PubMed] [Google Scholar]
- 13. Serradas P, Goya L, Lacorne M, Gangnerau MN, Ramos S, Alvarez C et al. (2002) Fetal insulin‐like growth factor‐2 production is impaired in the GK rat model of type 2 diabetes. Diabetes 51, 392–397. [DOI] [PubMed] [Google Scholar]
- 14. Calderari S, Gananerau MN, Thibault M, Meile MJ, Kassis N, Alvarez C et al. (2007) Defective IGF2 and IGF1R protein production in embryonic pancreas precedes beta cell mass anomaly in the Goto‐Kakizaki rat moder of type 2 diabetes. Diabetologia 50, 1463–1471. [DOI] [PubMed] [Google Scholar]
- 15. Chang CM, Kao CL, Chang YL, Yang MJ, Chen YC, Sung BL et al. (2007) Placenta‐derived multipotent stem cells induced to differentiate into insulin‐positive cells. Biochem. Biophys. Res. Commun. 357, 414–420. [DOI] [PubMed] [Google Scholar]
- 16. Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G et al. (2007) Generation of insulin‐producing islet‐like clusters from human embryonic stem cells. Stem Cells 25, 1940–1953. [DOI] [PubMed] [Google Scholar]
- 17. Jiang W, Shi Y, Zhao D, Chen S, Yong J, Zhang J et al. (2007) In vitro derivation of functional insulin‐producing cells from human embryonic stem cells . Cell Res. 17, 333–344. [DOI] [PubMed] [Google Scholar]
- 18. Chao KC, Chao KF, Fu YS, Liu SH (2008) Islet‐like clusters derived from mesenchymal stem cells in Wharton’s Jelly of the human umbilical cord for transplantation to control type 1 diabetes. PLoS One 3, e1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao AZ, Zhao H, Teague J, Fujimoto W, Beavo JA (1997) Attenuation of insulin secretion by insulin‐like growth factor 1 is mediated through activation of phosphodiesterase 3B. Proc. Natl. Acad. Sci. USA 94, 3223–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang HM, Kim J, Park S, Kim J, Kim H, Kim KS et al. (2009) Insulin‐secreting cells from human eyelid‐derived stem cells alleviate type I diabetes in immunocompetent mice. Stem Cells 27, 1999–2008. [DOI] [PubMed] [Google Scholar]
- 21. D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG et al. (2006) Production of pancreatic hormone‐expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 24, 1392–1401. [DOI] [PubMed] [Google Scholar]
- 22. Phillips BW, Hentze H, Rust WL, Chen QP, Chipperfield H, Tan EK et al. (2007) Directed differentiation of human embryonic stem cells into the pancreatic endocrine lineage. Stem Cells Dev. 16, 561–578. [DOI] [PubMed] [Google Scholar]
- 23. Lin HY, Tsai CC, Chen LL, Chiou SH, Wang YJ, Hung SC (2010) Fibronectin and laminin promote differentiation of human mesenchymal stem cells into insulin producing cells through activating Akt and ERK. J. Biomed. Sci. 17, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hori Y, Gu X, Xie X, Kim SK (2005) Differentiation of insulin‐producing cells from human neural progenitor cells. PLoS Med. 2, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tateishi K, He J, Taranova O, Liang G, D’Alessio AC, Zhang Y (2008) Generation of insulin‐secreting islet‐like clusters from human skin fibroblasts. J. Biol. Chem. 283, 31601–31607. [DOI] [PubMed] [Google Scholar]
- 26. Li G, Barrett EJ, Wang H, Chai W, Liu Z (2005) Insulin at physiological concentrations selectively activates insulin but not insulin‐like growth factor I (IGF‐I) or insulin/IGF‐I hybrid receptors in endothelial cells. Endocrinology 146, 4690–4696. [DOI] [PubMed] [Google Scholar]
- 27. De Meyts P, Whittaker J (2002) Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1, 769–783. [DOI] [PubMed] [Google Scholar]
- 28. Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, McClain DA (1990) Functionally distinct insulin receptors generated by tissue‐specific alternative splicing. EMBO J. 9, 2409–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muller D, Huang GC, Amiel S, Jones PM, Persaud SJ (2006) Identification of insulin signaling elements in human beta‐cells: autocrine regulation of insulin gene expression. Diabetes 55, 2835–2842. [DOI] [PubMed] [Google Scholar]
- 30. Miettinen PJ, Otonkoski T, Voutilainen R (1993) Insulin‐like growth factor‐II and transforming growth factor‐alpha in developing human fetal pancreatic islets. J. Endocrinol. 138, 127–136. [DOI] [PubMed] [Google Scholar]
- 31. Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M (1995) Insulin‐like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 55, 2007–2011. [PubMed] [Google Scholar]
- 32. Ishiwata T, Bergmann U, Kornmann M, Lopez M, Beger HG, Korc M (1997) Altered expression of insulin‐like growth factor II receptor in human pancreatic cancer. Pancreas 15, 367–373. [DOI] [PubMed] [Google Scholar]
- 33. Hawkes C, Kar S (2004) The insulin‐like growth factor‐II/mannose‐6‐phosphate receptor: structure, distribution and function in the central nervous system. Brain Res. Rev. 44, 117–140. [DOI] [PubMed] [Google Scholar]
- 34. Scott CD, Firth SM (2004) The role of the M6P/IGF‐II receptor in cancer: tumor suppression or garbage disposal? Horm. Metab. Res. 36, 261–271. [DOI] [PubMed] [Google Scholar]
- 35. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A et al. (1999) Insulin receptor isoform A, a newly recognized, high‐affinity insulin‐like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Belfiore A (2007) The role of insulin receptor isoforms and hybrid insulin/IGF‐I receptors in human cancer. Curr. Pharm. Des. 13, 671–686. [DOI] [PubMed] [Google Scholar]
- 37. Wilson EM, Hsieh MM, Rotwein P (2003) Autocrine growth factor signaling by insulin‐like growth factor‐II mediates MyoD‐stimulated myocyte maturation. J. Biol. Chem. 278, 41109–41113. [DOI] [PubMed] [Google Scholar]
- 38. Leibiger B, Leibiger IB, Moede T, Kemper S, Kulkarni RN (2001) Selective signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic β cells. Mol. Cell 7, 559–570. [DOI] [PubMed] [Google Scholar]
- 39. Pandini G, Medico E, Conte E, Sciacca L, Vigneri R, Belfiore A (2003) Differential gene expression induced by insulin and insulin‐like growth factor‐II through the insulin receptor isoform A. J. Biol. Chem. 278, 42178–42189. [DOI] [PubMed] [Google Scholar]
- 40. Sciacca L, Prisco M, Wu A, Belfiore A, Vigneri R, Baserga R (2003) Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate‐1. Endocrinology 144, 2650–2658. [DOI] [PubMed] [Google Scholar]
- 41. Nevado C, Valverde AM, Benito M (2006) Role of insulin receptor in the regulation of glucose uptake in neonatal hepatocytes. Endocrinology 147, 3709–3718. [DOI] [PubMed] [Google Scholar]
- 42. Johnson D, Shepherd RM, Gill D, Gorman T, Smith DM, Dunne MJ (2007) Glucose‐dependent modulation of insulin secretion and intracellular calcium ions by GKA50, a glucokinase activator. Diabetes 56, 1694–1702. [DOI] [PubMed] [Google Scholar]