

Abstract
Background and Objectives: Mesenchymal stem cells (MSC) are multipotent progenitor cells that are have found use in regenerative medicine. We have previously observed that aspirin, a widely used anti‐inflammatory drug, inhibits MSC proliferation. Here we have aimed to elucidate whether aspirin induces MSC apoptosis and whether this is modulated through the Wnt/β‐catenin pathway.
Materials and methods: Apoptosis of MSCs was assessed using Hoechst 33342 dye and an Annexin V–FITC/PI Apoptosis Kit. Expression of protein and protein phosphorylation were investigated using Western blot analysis. Caspase‐3 activity was detected by applying a caspase‐3/CPP32 Colorimetric Assay Kit.
Results: In these MSCs, aspirin induced morphological changes characteristic of apoptosis, cytochrome c release from mitochondria, and caspase‐3 activation. Stimulating the Wnt/β‐catenin pathway by both Wnt 3a and GSK‐3β inhibitors (LiCl and SB 216763), blocked aspirin‐induced apoptosis and protected mitochondrial function, as demonstrated by decreased cytochrome c release and caspase‐3 activity. Aspirin initially caused a time‐dependent decrease in COX‐2 expression but subsequently, and unexpectedly, elevated the latter. Stimulation of COX‐2 expression by aspirin was further enhanced following stimulation of the Wnt/β‐catenin pathway. Application of the COX‐2 inhibitor NS‐398 suppressed elevated COX‐2 expression and promoted aspirin‐induced apoptosis.
Conclusion: These results demonstrate that the Wnt/β‐catenin pathway is a key modulator of aspirin‐induced apoptosis in MSCs by regulation of mitochrondrial/caspase‐3 function. More importantly, our findings suggest that aspirin may influence MSC survival under certain conditions; therefore, it should be used with caution when considering regenerative MSC transplantation in patients with concomitant chronic inflammatory diseases such as arthritis.
Introduction
Mesenchymal stem cells (MSC), which reside predominantly in the bone marrow, are multipotent progenitor cells with many different properties, which include the ability to suppress immune responses, to exert anti‐inflammatory effects, and to generate paracrine factors that may enhance angiogenesis and survival of cells (1, 2). MSCs are also capable of differentiating into numerous lineages, including endothelial (3), neural (4), chondrocyte, bone marrow stromal (5), and cardiac (6). Moreover, MSCs can be invasive, potentially providing the opportunity for their exploitation in cell‐mediated gene therapy or even promoting tissue regeneration (7). In this regard, studies on various arthritic conditions and myocardial infarction have demonstrated that MSC transplantation could generate replacement tissues and repair damaged structures (5, 8).
However, survival of transplanted MSCs still remains a major limitation that significantly hampers their potential use as cell therapy in regenerative medicine (9). Thus, identifying factors that hinder MSC survival and understanding the mechanisms through which such actions are mediated would have a significant impact on promoting the use of MSC‐based therapy in regenerative medicine. In this regard, we have previously demonstrated that serum deprivation and hypoxia induce MSC apoptosis (10). Furthermore, we have also reported recently that aspirin, a drug used in treating a variety of inflammatory diseases, including rheumatoid arthritis, cardiovascular events and even tumours (11, 12, 13), inhibits MSC proliferation (14).
The recognized action of aspirin is inhibition of activity of cyclooxygenase (COX) enzymes (13). However, aspirin has recently been shown to exert other effects that target cell signalling events, such as those mediated by the Wnt/β‐catenin pathway (15, 16, 17). This signalling plays a critical role in self‐renewal, differentiation and survival of MSCs (18, 19), and has been reported to be involved in aspirin‐induced inhibition of MSC proliferation (14). Contributing to this increasing array of novel actions of aspirin, we now report, and for the first time, that aspirin is also capable of inducing apoptosis in MSCs through inhibition of Wnt/β‐catenin signalling and activation of the mitochondrial apoptotic pathway. Moreover, we found that aspirin caused an unexpected enhancement of COX‐2 expression, which may be a compensatory response to prolonged aspirin treatment of MSCs. These novel findings may raise concerns over the clinical exploitation of MSCs for regenerative medicine in patients with concomitant chronic inflammatory diseases, such as arthritis, who may be on substantial and sustained doses of aspirin therapy.
Materials and Methods
Materials
Iscove’s modified Dulbecco’s medium (IMDM) and foetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). Hoechst 33342, aspirin, LiCl, SB216763, and mouse monoclonal anti‐rat β‐actin and anti‐rat β‐catenin (p ser33/p ser37) antibodies were from Sigma‐Aldrich (St Louis, MO, USA). The Annexin V–FITC Apoptosis Detection Kit was purchased from Oncogene (San Diego, CA, USA), and Wnt‐3a was from R&D Systems (Minneapolis, MN, USA). Rabbit polyclonal anti‐rat caspase‐3, Bax, GSK‐3β (p ser9), and β‐catenin antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Mouse polyclonal anti‐rat cyclin D1, anti‐rat Bcl‐2, and horseradish peroxidase‐conjugated secondary anti‐rabbit and anti‐mouse antibodies, and Chemiluminescence Detection Kit, were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse polyclonal anti‐rat GSK‐3β was purchased from Kangchen Bio‐tech (Shanghai, China). The Caspase‐3/CPP32 Colorimetric Assay Kit, the Cytochrome c Releasing Apoptosis Assay Kit, and the mouse monoclonal anti‐rat cytochrome c antibody were from BioVision (Palo Alto, CA, USA). Nitrocellulose membrane was purchased from Amersham (Piscataway, NJ, USA). Cytoplasm and Nuclear Protein Extraction Kit and Bradford Protein Assay Kit were purchased from Beyotime Institute of Biotechnology (Shanghai, China).
Cell culture and treatment
Mesenchymal stem cells were isolated from Sprague‐Dawley rats (Vital River Laboratory Animal Inc., Beijing, China) as previously described (10). All procedures in the present study were approved by the Animal Care Committee of the Cardiovascular Institute & Fu Wai Hospital, Chinese Academy of Medical Science & Peking Union Medical College. MSCs were cultured in IMDM supplemented with 10% inactivated FBS and 100 units/ml penicillin/streptomycin. For assays of aspirin effects, MSCs were cultured in 1% heat‐inactivated FBS for 12 h prior to incubation with aspirin (5 mm). In inhibition studies, MSCs were pre‐incubated with Wnt‐3a (0.1 μg/ml), and the GSK‐3β inhibitors SB216763 (5 μm) or LiCl (0.5 mm) or COX‐2 inhibitor NS‐398 (5 μm, 10 μm) for 30 min before addition of aspirin (5 mm) for 24 h.
Assessment of morphological changes of apoptosis
Mesenchymal stem cells treated with various agents were studied by staining with Hoechst 33342 as previously described, for nuclear condensation and fragmentation (10). Briefly, MSCs were washed with phosphate‐buffered saline (PBS) and stained with Hoechst 33342 (5 μg/ml), and morphological changes were observed using fluorescence microscopy (Olympus IX70 Microscope, Olympus Corp., Tokyo, Japan; RS Image Express processing software, version 4.5). Apoptotic cells were defined as having condensed, fragmented nuclei and undergoing cell shrinkage.
Flow cytometry analysis of cell apoptosis
Phosphatidylserine exposure on the surface of plasma membranes was detected using the Annexin V–FITC Apoptosis Detection Kit, according to the manufacturer’s protocols (10). Briefly, cells were harvested, washed in ice‐cold PBS and resuspended in 200 μl binding buffer before being incubated with 10 μl of annexin V–FITC solution (30 min, 4 °C) in the dark. Cells were then incubated with 5 μl of propidium iodide for 5 min, and were immediately analysed by bivariate flow cytometry using a FACScan‐LSR flow cytometer equipped with CellQuest software (BD Biosciences, San Jose, CA, USA). At least 1 × 104 cells per sample were acquired and analysed.
Protein extraction, Western blot analysis and measurement of caspase‐3 activation
Preparation of cell extracts from MSCs for Western blot analysis was carried out as previously described (20). To prepare nuclear protein extraction, cells were lysed using a Cytoplasm and Nuclear Protein Extraction Kit, according to the manufacturer’s instructions (Beyotime Institute of Biotechnology). Lysates were centrifuged at 12 000 g for 10 min at 4 °C. Nuclear extracts were prepared by resuspending the pellet with 50 μl nuclear extraction buffer. Resuspended pellets were vortexed at high speed for 10 min at 4 °C and centrifuged for 10 min in a microcentrifuge at 4 °C. Supernatants were collected and assayed. Caspase‐3 activity was detected by using the Caspase‐3/CPP32 Colorimetric Assay Kit (Biovision, Palo Alto, CA, USA) according to the manufacturer’s instructions. Briefly, 150 μg of protein, in a total volume of 50 μl, was added to 50 μl of 2× reaction buffer and 5 μl of DEVD‐pNA substrate (200 μm final concentrations). After incubation (1–2 h, 37 °C), DEVD‐pNA cleavage was monitored by detecting enzyme‐catalysed release of pNA at 405 nm using a microplate reader (Thermo electron corporation, MULTISKAW MK3).
Statistical analysis
Data are expressed as mean ± standard error of the mean. Differences among groups were tested by one‐way analysis of variance. Comparisons between two groups were evaluated using Student’s t‐test with a value of P < 0.05.
Results
Aspirin‐induced MSC apoptosis
To determine whether aspirin had a pro‐apoptotic effect on MSCs, cells were exposed to the drug following a 12 h incubation in media containing 1% heat‐inactivated FBS. Hoechst 33342 staining showed that most cells in the control group had large regular nuclei, with only a few showing apoptotic changes with condensed chromatin. In contrast, cells exposed to aspirin showed clear evidence of apoptotic fragmented nuclei with condensed chromatin. More importantly, this effect was evident and statistically significant at 1 mm (P < 0.020), further increasing concentration‐dependently thereafter (Fig. 1a,b). Aspirin‐induced apoptosis was further confirmed by flow cytometry, with analysis carried out showing concentration‐dependent increase in positively stained annexin V–FITC cells (Fig. 1c,d) when exposed to aspirin. This effect was again evident and statistically significant at ≥1 mm (P < 0.038), confirming the pro‐apoptotic action of aspirin in MSCs.
Figure 1.

Aspirin induced apoptosis in mesenchymal stem cells (MSC). MSCs were incubated with the indicated concentrations of aspirin for 24 h. In panels (a) and (b), induction of apoptosis was determined by apoptotic nuclear condensation using a fluorescence microscope, following Hoechst 33342 staining. Panels (c) and (d) represent FACScan flow cytometry analysis of apoptotic cells after annexin V–FITC/propidium iodide (PI) staining. Viable cells are annexin V−/PI−. Annexin V+/PI−, annexin V+/PI+ and annexin V−/PI+ are in early apoptotic, late apoptotic and necrotic phases, respectively. Each column represents mean ± standard error of the mean of three independent experiments (*P < 0.05 vs. control).
Aspirin‐induced activation of mitochondrial caspase‐3 pathway
To evaluate whether aspirin‐induced apoptosis was mediated via the mitochondrial pathway, we determined changes in Bcl‐2 and Bax expression, two proteins that, respectively, 1. protect and 2. enhance mitochondrial damage and, hence, cytochrome c release. Data obtained revealed that levels of Bcl‐2 dose‐dependently decreased with no apparent change in Bax after aspirin treatment (Fig. 2a). In parallel studies, cytochrome c was detected in the cytosolic fraction of cells treated with 5 mm aspirin (Fig. 2b). As release of mitochondrial cytochrome c leads to caspase‐3 activation, Western blot analysis for caspase‐3 was carried out using anti‐caspase‐3 selective antibody. Results in Fig. 2c and 2d show that aspirin converted 35 kDa procaspase‐3 to cleaved 17 kDa active caspase‐3 (CL.caspase‐3). These observations are consistent with the parallel increase in caspase‐3 activity determined colorimetrically using the Caspase‐3/CPP32 assay and showed concentration‐dependent increase in caspase‐3 activity (Fig. 2e).
Figure 2.
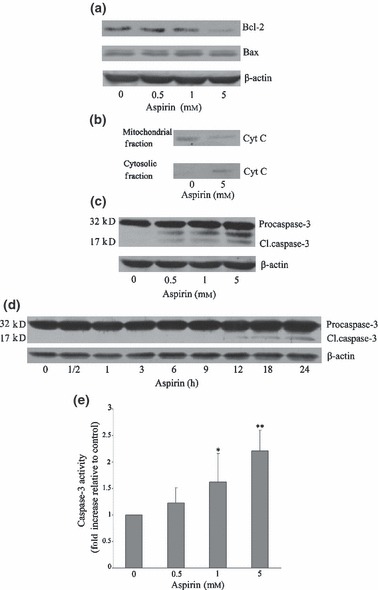
Aspirin stimulated the mitochondrial/caspase‐3 pathway. Mesenchymal stem cells (MSC) were incubated with the indicated concentrations of aspirin for 24 h. Panels (a) to (c) represent expression of Bcl‐2, Bax, mitochondrial and cytosolic cytochrome c, and capase‐3 as detected by Western blotting. In panel (d), MSCs were exposed to aspirin (5 mm) for the indicated time and changes in caspase‐3 expression detected by Western blotting. Panel (e) represents activity of caspase‐3, determined colorimetrically. Blots shown are representative of at least three independent experiments and data in the graphs are mean ± standard error of the mean of at least three independent experiments (*P < 0.05 vs. control; **P < 0.01 vs. control).
Role of Wnt/β‐catenin pathway in aspirin‐induced apoptosis in MSCs
As highlighted earlier, the Wnt/β‐catenin pathway has been indicated to play an important role in the effect of aspirin in some cell systems. We therefore examined whether this pathway was involved in aspirin‐induced apoptosis in MSCs. There was a clear trend showing increase in levels of β‐catenin phosphorylation accompanied by parallel decrease in phospho‐GSK‐3β and cyclin D1 expression. These changes were apparent at concentrations of 0.5–1 mm, becoming more pronounced at 5 mm (Fig. 3a). The increase in β‐catenin phosphorylation was initiated at 3 h and sustained up to 24 h after exposure to aspirin (Fig. 3b), indicating that aspirin is capable of inhibiting the activity of Wnt/β‐catenin pathway in MSCs.
Figure 3.

Wnt/β‐catenin pathway mediated aspirin‐induced mesenchymal stem cell (MSC) apoptosis. For data in panel (a), MSCs were incubated with the indicated concentrations of aspirin for 24 h, and the expressions of GSK‐3β, phosphor‐GSK‐3β, β‐catenin, phosphor‐β‐catenin and cyclin D1 detected by Western blotting. Panel (b) represents expression of phosphor‐β‐catenin in MSCs incubated at the indicated time points with aspirin at a concentration of 5 mm. In panels (c) to (f), MSCs were pre‐incubated for 30 min with or without SB 216763 (5 μm), LiCl (0.5 mm) or Wnt 3a (0.1 μg/ml), respectively, before exposure to aspirin (5 mm). Panels (c) and (d), Western blot analysis of the expressions of β‐catenin, nuclear β‐catenin, phosphor‐β‐catenin and cyclin D1. In panels (e) and (f), induction of apoptosis was determined by Hoechst 33342 staining. Panel (g) represents FACScan flow cytometry analysis of apoptotic cells pre‐incubated with or without Wnt 3a (0.1 μg/ml) before exposure to aspirin (5 mm). Blots shown are representative of at least three independent experiments and data in the graphs are mean ± standard error of the mean of at least three independent experiments (*P < 0.05 vs. control; **P < 0.01 vs. control; # P < 0.05 vs. aspirin; ## P < 0.01 vs. aspirin).
To further elucidate the effects of aspirin on this pathway, the cells were treated with SB 216763 or LiCl. Western blot analysis revealed that both SB 216763 and LiCl prevented aspirin‐stimulated β‐catenin phosphorylation, and enhanced cyclin D1 expression. Likewise, activation of the Wnt/β‐catenin pathway with Wnt 3a also prevented aspirin‐induced β‐catenin phosphorylation and promoted cyclin D1 expression (Fig. 3c). In addition, pre‐treatment of cells with LiCl or Wnt 3a increased β‐catenin in nuclei, confirming activation of the Wnt/β‐catenin pathway by these Wnt mimetics (Fig. 3d).
Hoechst 33342 staining showed that activating Wnt/β‐catenin pathway with SB 216763, LiCl, or Wnt 3a, protected cell nuclei. This contrasts with apoptotic nuclei and condensed chromatin (Fig. 3e,f) seen in cells treated with aspirin alone. Results from flow cytometry of positive annexin V–FITC‐stained cells treated with Wnt 3a also supported the observation that activating the Wnt/β‐catenin pathway blocks aspirin‐induced MSC apoptosis (Fig. 3g).
Further studies investigated whether aspirin‐induced activation of the mitochondria/caspase‐3 pathway in MSCs would be mediated via Wnt/β‐catenin. As shown in Fig. 4a and 4b, activation of this reversed aspirin‐induced inhibition of Bcl‐2 expression and induced cytochrome c release from mitochondria. These changes correlated well with the reduction of procaspase‐3 cleavage and caspase‐3 activation (Fig. 4c,d). The above data demonstrate that aspirin‐induced apoptosis in MSCs is dependent on Wnt/β‐catenin, which acts upstream of mitchondria/caspase‐3 pathway.
Figure 4.
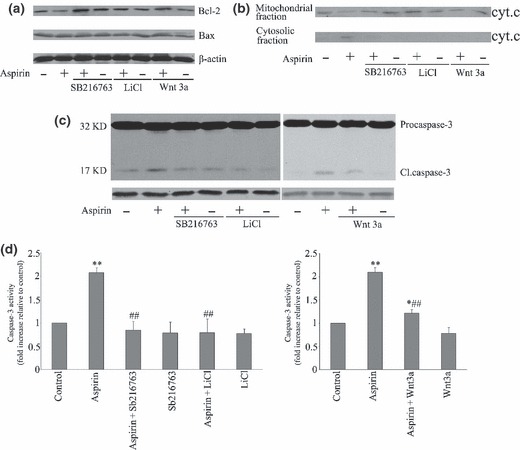
Wnt/β‐catenin pathway mediation of aspirin‐induced mesenchymal stem cell (MSC) apoptosis by mitchondrial/caspase‐3 pathway. MSCs were pre‐incubated for 30 min with or without SB 216763 (5 μm), LiCl (0.5 mm) or Wnt 3a (0.1 μg/ml), respectively, before exposure to aspirin (5 mm) for a further 24 h. Changes in expression of Bcl‐2 and Bax (a), mitochondrial and cytosolic cytochrome c (b) and capase‐3 (c) were determined by Western blot analysis. Changes in activity of caspase‐3 (d) were determined colorimetrically. Blots shown are representative of at least three independent experiments and data in the graphs are mean ± standard error of the mean of at least three independent experiments (*P < 0.05 vs. control; **P < 0.01 vs. control; ## P < 0.01 vs. aspirin).
Regulation of COX expression in aspirin‐treated MSCs
Since the therapeutic benefit of aspirin in many cases is ascribed to its ability to inhibit cyclooxygenase (COX‐1/COX‐2), we assessed the expression of COX‐1/COX‐2 in aspirin‐treated MSCs, using Western blot analysis. Aspirin treatment did not affect COX‐1 expression, but dose‐dependently increased COX‐2 expression in cells exposed to aspirin (0–5 mm) when examined at a fixed time point of 24 h (Fig. 5a). Pre‐incubation of cells with SB 216763, LiCl, or Wnt 3a prior to aspirin led to further increase of COX‐2 expression over this period (Fig. 5b). In parallel studies, aspirin time‐dependently inhibited COX‐2 expression, virtually abolishing the protein after 12 h of exposure (Fig. 5c).
Figure 5.
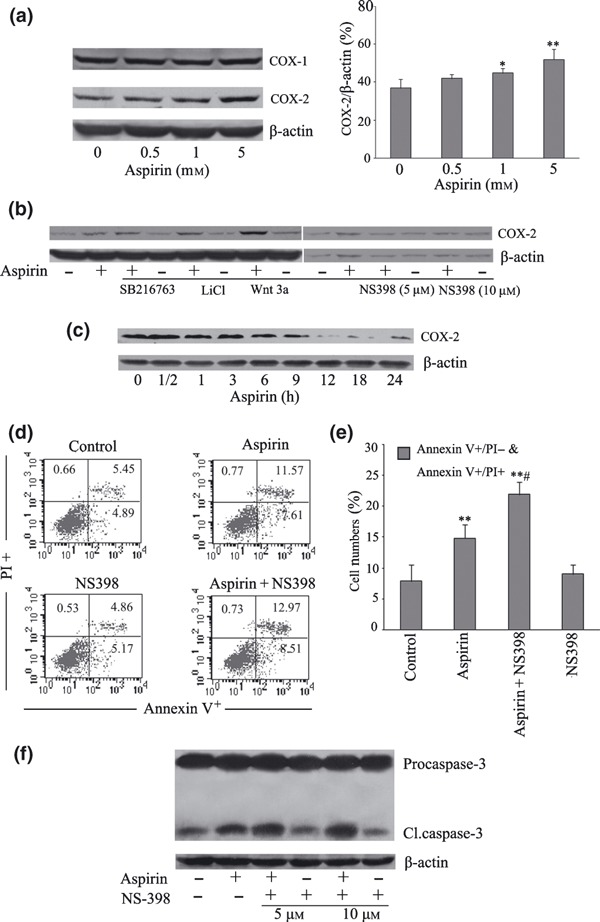
Regulation of cyclooxygenase (COX) expression in aspirin‐treated mesenchymal stem cells (MSC). Changes in expressions of COX‐1/COX‐2 in cells treated with increasing concentrations of aspirin for 24 h were detected by Western blot analysis using isoform selective antibodies (a). Panel (b) represents changes in expression of COX‐2 in MSCs pre‐treated with or without SB 216763 (5 μm), LiCl (0.5 mm), Wnt 3a (0.1 μg/ml) or NS‐398 (5 μm or 10 μm) for 30 min prior to exposure to aspirin (5 mm) for 24 h. Panel (c) represents expression of COX‐2 in MSCs incubated at the indicated time points with aspirin at a concentration of 5 mm. Panels (d) and (e) are FACScan flow cytometry analysis of apoptosis in MSCs pre‐incubated with or without NS‐398 (10 μm) prior to exposure to aspirin (5 mm) for 24 h (**P < 0.01 vs. control; # P < 0.05 vs. aspirin). (f) Level of pro‐caspase‐3 cleavages in MSCs pre‐incubated with or without NS‐398 (5 or 10 μm), respectively, before exposure to aspirin (5 mm) for 24 h is detected. Blots shown are representative of at least three independent experiments.
To further explore the role of COX‐2 in pro‐apoptotic activity of aspirin, we treated cells with the COX‐2 inhibitor, NS‐398. This compound decreased aspirin‐stimulated COX‐2 expression (Fig. 5b), but slightly increased the percentage of positive annexin V–FITC‐stained cells (Fig. 5d,e) and increased precaspase‐3 cleavage (Fig. 5f), as compared to cells treated with aspirin alone.
Discussion
Survival of MSCs in transplanted tissues is a critical factor that determines their therapeutic potential as cell‐based therapy in regenerative medicine (9). However, factors that regulate survival of transplanted MSCs in vivo are poorly defined at the moment and could be dependent not only on the local microenvironment but potentially also on existing medication(s) used in treating any underlying pathology of the prospective patient. One such medication is the non‐steroidal anti‐inflammatory drug aspirin, which is widely used for its anti‐platelet‐, antipyretic‐, analgesic and anti‐inflammatory actions (13), and for treating cardiovascular disease (12) and even cancers (21). This drug is of particular interest because of its ability to inhibit MSC proliferation in vitro (14), an action that we have speculated could be disadvantageous and limiting for MSC‐based therapies.
To further identify any additional effects that aspirin may exert on MSCs, we have extended our previous studies (14) by investigating its ability to influence MSC survival. The data obtained demonstrate pro‐apoptotic action that was evident at concentrations of aspirin that could be achieved during in vivo treatmenr, specially in patients with chronic inflammatory conditions. In such cases, the daily dose of aspirin may reach 1–5 mm (22, 23, 24, 25, 26) and this is within the concentration range that could possibly induce MSC apoptosis as demonstrated in our studies. For such patients, combined use of aspirin and MSC‐based therapy may prove futile.
Experiments aimed at unravelling underlying mechanism(s) that could mediate actions of aspirin, revealed that suppression of the Wnt/β‐catenin pathway may be a critical step in induction of apoptosis by aspirin. This finding fits well with the fact that activation of Wnt/β‐catenin is known to play a central role in regulating cell proliferation, differentiation and survival of MSCs (18, 19, 27).
Under normal conditions, β‐catenin is phosphorylated by a β‐catenin destruction complex (CK 1, GSK‐β, APC, and Axin) in the absence of Wnt stimulation. This prevents β‐catenin translocation to the cell nucleus and instead promotes its ubiquitination and degradation through the proteosome pathway (28). In contrast, when present, Wnt promotes dissociation of the destruction complex from β‐catenin, maintaining the latter in the non‐phosphorylated state, which in turn results in cytosolic accumulation of β‐catenin. β‐catenin may subsequently translocate to the nucleus, form a complex with members of the LEF/TCF family of transcription factors, and activate expression of multiple Wnt target genes including those for cyclin D1 and COX‐2 (18, 29, 30, 31).
Our observation shows inhibition of Wnt/β‐catenin by aspirin, and confirms previous studies which also have demonstrated the ability of this compound to decreases activity of the same pathway in MSCs (14) and colorectal cancer cells leading to inhibition of proliferation (16, 17). In the present study, we have further demonstrated that aspirin increased GSK‐3β activity by decreasing its phosphorylation on ser9 residue, and that inhibition of GSK‐3β activity by LiCl and SB216763 reduced aspirin‐induced apoptosis. This suggests that GSK‐3β may be central in the process of aspirin‐induced apoptosis in MSCs.
Inhibition of GSK‐3β activity by LiCl in MSCs is consistent with studies in human colon cancer cell line SW948 where LiCl was also capable of inhibiting GSK‐3β and reducing aspirin‐mediated increase of phosphorylation β‐catenin level (17). However, these findings, together with ours, contrast with those on another colon cancer cell line SW480, where GSK‐3β phosphorylation on ser9 was not changed by aspirin and pre‐treatment with LiCl failed to reverse aspirin‐induced phosphorylation of β‐catenin (16, 17). The reasons for this disparity are currently unclear but may reflect cell‐type differences and perhaps highlight different mechanism through which phosphorylation of β‐catenin may be induced by aspirin.
In our studies, further evidence in support of action of aspirin through the Wnt/‐β‐catenin pathway was obtained using Wnt mimetic, Wnt3a, which reversed aspirin‐induced MSC apoptosis. Moreover, Wnt 3a (and LiCl, the inhibitor of GSK‐3β), increased accumulation of nuclear β‐catenin under our experimental conditions. Taken together, our current findings therefore support our hypothesis that aspirin‐induced apoptosis in MSCs may be mediated by suppression of Wnt/β‐catenin signalling. Thus, if a patient was to receive MSCs through transplantation, and critically requires aspirin, then agents such as lithium (used clinically to inhibit GSK‐3β (32)), could be applied to protect MSCs and enhance their survival. Such a strategy may not only improve survival but may also facilitate cell proliferation through activation of cyclin D1, which we have now shown to be induced following stimulation of Wnt signalling in these cells. Cyclin D1 is a potential target gene of the canonical Wnt pathway (29, 33) that regulates both apoptosis (34) and cell cycle progression (35). Thus, its induction through Wnt/β‐catenin activation may improve MSC survival, promoting proliferation and potentially inhibiting apoptosis induced by aspirin.
It is worth noting that transcription studies have suggested that cyclin D1 may not be an immediate downstream target of Wnt/β‐catenin (36, 37). In addition, in vitro studies on colon carcinoma cell lines have revealed that endogenous levels of cyclin D1 are not affected by antagonism of the Wnt pathway. Thus, in these systems, cyclin D1 may not be a bona fide target gene for Wnt/‐β‐catenin signalling (30). This contrasts with our current data which clearly show up‐regulation in cyclin D1 expression that parallels nuclear location of β‐catenin. Although the reasons for the discrepancy remain to be addressed, that one other study has demonstrated direct regulation of cyclin D1 by β‐catenin in MSCs (27) implies that the disparity reported above may, at least in part, reflect cell type differences.
Gao et al. (38) have demonstrated that nitric oxide‐donating aspirin induced β‐catenin cleavage through activation of caspase‐3 in colon adenocarcinoma cells (38). In our studies, we could not show any decrease in β‐catenin expression in aspirin‐treated MSCs. In addition, our results showed that phosphorylated β‐catenin preceded caspase‐3 activation (2, 3) and Wnt/β‐catenin pathway mediated aspirin‐induced activation of caspase‐3. Thus, caspase‐3 could be down‐stream of Wnt/β‐catenin in our system.
Therapeutic actions of aspirin are mediated largely through its anti‐platelet and anti‐inflammatory actions (12). Aspirin’s anti‐platelet effects have been attributed to inhibition of platelet COX‐1 activity, while its anti‐inflammatory actions are mediated by inhibition of COX‐2 (39). Our present data reveal that COX‐1 expression was not altered but COX‐2 expression unexpectedly increased when MSCs were exposed to aspirin for 24 h. This period correlated with the time point at which apoptosis was induced by aspirin.
Although aspirin is generally known to inhibit COX‐2 expression (40), there are reports suggesting that it may cause the reverse effect in certain cell types. This is the case in myofibroblasts (41), stomach epithelial cells (42) and monocytes/macrophages (43), where aspirin treatment has resulted in enhanced expression of COX‐2. We speculate that elevated COX‐2 expression may be a compensatory response to prolonged aspirin treatment and that this may occur as a survival mechanism through which cells could be protected against induced apoptosis. Our current data aimed at addressing this and strongly support this notion. In the first case, increase of COX‐2 expression was preceded by a time‐dependent decrease in detected levels of the protein, induced by aspirin. Secondly, pre‐treatment of cells with Wnt 3a and the other Wnt agonists (LiCl and SB216763) enhanced COX‐2 expression above that seen with aspirin alone, and perhaps more importantly, reversed aspirin‐induced apoptosis. Furthermore, COX‐2 specific inhibitor NS‐398 accelerated aspirin‐induced apoptosis while causing a marginal increase in caspase‐3. Thus, consistent with other reports (44, 45), our experiments support that enhanced COX‐2 expression may protect cells against programmed cell death and it is feasible that increase caused by aspirin alone may be a compensatory response by MSCs to pro‐apoptotic action of this drug.
In conclusion, we have demonstrated for the first time, that aspirin can induce apoptosis in MSCs and mediate this effect through the mitochondrial/caspase‐3 pathway, which in turn requires activation of Wnt/β‐catenin. Moreover, parallel enhancement of cyclin D1 and COX‐2 expression by agonists of the Wnt pathway may provide the link through which activation of the latter protects cells against apoptosis. Perhaps of more significance is that we have generated, to our knowledge, the first piece of evidence that may specify a contraindication for aspirin in MSC‐based therapy. This may be particularly relevant to patients with diseases such as rheumatic fever, rheumatoid arthritis or other chronic inflammatory disorders, where high concentrations of aspirin may be required daily, but its therapeutic plasma concentrations could be in the range of 1.5–2.5 mm for patients with arthritis and up to 5.0 mm for those with other inflammatory conditions (22, 23, 25, 26). Thus, for these patients, aspirin should be used with caution when MSC transplantation is being considered. Where this is deemed necessary, co‐administration of therapeutic doses of compounds such as lithium may be necessary to protect and enhance MSC survival. In addition, although beyond the scope of this report, MSCs have been shown to originate malignancy in a chronic inflammation environment and to promote growth and metastasis of tumour cells (46, 47, 48), which suggests that MSCs may be a potential target of cancer therapy. Our results hold implication for partly explaining the anti‐cancer effects of aspirin (11, 49), which may also lead to designing novel therapeutic approaches against cancer.
Acknowledgements
This study was supported by the National Basic Research Program of China (973 Program, 2007CB512100 & 2010CB529508).
References
- 1. Kinnaird T, Stabile E, Burnett MS, Lee CW, Barr S, Fuchs S, Epstein SE (2004) Marrow‐derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ. Res. 94, 678–685. [DOI] [PubMed] [Google Scholar]
- 2. Nagaya N, Kangawa K, Itoh T, Iwase T, Murakami S, Miyahara Y, Fujii T, Uematsu M, Ohgushi H, Yamagishi M, Tokudome T, Mori H, Miyatake K, Kitamura S (2005) Transplantation of mesenchymal stem cells improves cardiac function in a rat model of dilated cardiomyopathy. Circulation 112, 1128–1135. [DOI] [PubMed] [Google Scholar]
- 3. Oswald J, Boxberger S, Jorgensen B, Feldmann S, Ehninger G, Bornhauser M, Werner C (2004) Mesenchymal stem cells can be differentiated into endothelial cells in vitro . Stem Cells 22, 377–384. [DOI] [PubMed] [Google Scholar]
- 4. Kohyama J, Abe H, Shimazaki T, Koizumi A, Nakashima K, Gojo S, Taga T, Okano H, Hata J, Umezawa A (2001) Brain from bone: efficient ‘meta‐differentiation’ of marrow stroma‐derived mature osteoblasts to neurons with Noggin or a demethylating agent. Differentiation 68, 235–244. [DOI] [PubMed] [Google Scholar]
- 5. Chen FH, Tuan RS (2008) Mesenchymal stem cells in arthritic diseases. Arthritis Res. Ther. 10, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, Sano M, Takahashi T, Hori S, Abe H, Hata J, Umezawa A, Ogawa S (1999) Cardiomyocytes can be generated from marrow stromal cells in vitro . J. Clin. Invest. 103, 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kawada H, Fujita J, Kinjo K, Matsuzaki Y, Tsuma M, Miyatake H, Muguruma Y, Tsuboi K, Itabashi Y, Ikeda Y, Ogawa S, Okano H, Hotta T, Ando K, Fukuda K (2004) Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood 104, 3581–3587. [DOI] [PubMed] [Google Scholar]
- 8. Pittenger MF, Martin BJ (2004) Mesenchymal stem cells and their potential as cardiac therapeutics. Circ. Res. 95, 9–20. [DOI] [PubMed] [Google Scholar]
- 9. Copland IB, Lord‐Dufour S, Cuerquis J, Legault‐Coutu D, Annabi B, Wang E, Galipeau J (2008) Improved autograft survival of MSCs by PAI‐1 inhibition. Stem Cells 27(2):467–77. [DOI] [PubMed] [Google Scholar]
- 10. Zhu W, Chen J, Cong X, Hu S, Chen X (2006) Hypoxia and serum deprivation‐induced apoptosis in mesenchymal stem cells. Stem Cells 24, 416–425. [DOI] [PubMed] [Google Scholar]
- 11. Mahdi JG, Mahdi AJ, Mahdi AJ, Bowen ID (2006) The historical analysis of aspirin discovery, its relation to the willow tree and antiproliferative and anticancer potential. Cell Prolif. 39, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Awtry EH, Loscalzo J (2000) Aspirin. Circulation 101, 1206–1218. [DOI] [PubMed] [Google Scholar]
- 13. Vane JR, Botting RM (2003) The mechanism of action of aspirin. Thromb. Res. 110, 255–258. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Chen X, Zhu W, Zhang H, Hu S, Cong X (2006) Growth inhibition of mesenchymal stem cells by aspirin: involvement of the WNT/β‐catenin signal pathway. Clin. Exp. Pharmacol. Physiol. 33, 696–701. [DOI] [PubMed] [Google Scholar]
- 15. Lu W, Tinsley HN, Keeton A, Qu Z, Piazza GA, Li Y (2009) Suppression of Wnt/β‐catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 602, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bos CL, Kodach LL, Van Den Brink GR, Diks SH, Van Santen MM, Richel DJ, Peppelenbosch MP, Hardwick JC (2006) Effect of aspirin on the Wnt/β‐catenin pathway is mediated via protein phosphatase 2A. Oncogene 25, 6447–6456. [DOI] [PubMed] [Google Scholar]
- 17. Dihlmann S, Klein S, Doeberitz Mv MK (2003) Reduction of β‐catenin/T‐cell transcription factor signaling by aspirin and indomethacin is caused by an increased stabilization of phosphorylated β‐catenin. Mol. Cancer Ther. 2, 509–516. [PubMed] [Google Scholar]
- 18. Ling L, Nurcombe V, Cool SM (2009) Wnt signaling controls the fate of mesenchymal stem cells. Gene 433, 1–7. [DOI] [PubMed] [Google Scholar]
- 19. Boland GM, Perkins G, Hall DJ, Tuan RS (2004) Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell. Biochem. 93, 1210–1230. [DOI] [PubMed] [Google Scholar]
- 20. Chen J, Baydoun AR, Xu R, Deng L, Liu X, Zhu W, Shi L, Cong X, Hu S, Chen X (2008) Lysophosphatidic acid protects mesenchymal stem cells against hypoxia and serum deprivation‐induced apoptosis. Stem Cells 26, 135–145. [DOI] [PubMed] [Google Scholar]
- 21. Harris RE, Beebe‐Donk J, Doss H, Burr Doss D (2005) Aspirin, ibuprofen, and other non‐steroidal anti‐inflammatory drugs in cancer prevention: a critical review of non‐selective COX‐2 blockade (review). Oncol. Rep. 13, 559–583. [PubMed] [Google Scholar]
- 22. Farthing D, Gehr L, Karnes HT, Sica D, Gehr T, Larus T, Farthing C, Xi L (2007) Effects of salicylic acid on post‐ischaemic ventricular function and purine efflux in isolated mouse hearts. Biomarkers 12, 623–634. [DOI] [PubMed] [Google Scholar]
- 23. Dikshit P, Chatterjee M, Goswami A, Mishra A, Jana NR (2006) Aspirin induces apoptosis through the inhibition of proteasome function. J. Biol. Chem. 281, 29228–29235. [DOI] [PubMed] [Google Scholar]
- 24. Frantz B, O’Neill EA (1995) The effect of sodium salicylate and aspirin on NF‐κB. Science 270, 2017–2019. [DOI] [PubMed] [Google Scholar]
- 25. Nulton‐Persson AC, Szweda LI, Sadek HA (2004) Inhibition of cardiac mitochondrial respiration by salicylic acid and acetylsalicylate. J. Cardiovasc. Pharmacol. 44, 591–595. [DOI] [PubMed] [Google Scholar]
- 26. Smith MJ, Dawkins PD (1971) Salicylate and enzymes. J. Pharm. Pharmacol. 23, 729–744. [DOI] [PubMed] [Google Scholar]
- 27. Neth P, Ciccarella M, Egea V, Hoelters J, Jochum M, Ries C (2006) Wnt signaling regulates the invasion capacity of human mesenchymal stem cells. Stem Cells 24, 1892–1903. [DOI] [PubMed] [Google Scholar]
- 28. Huang H, He X (2008) Wnt/β‐catenin signaling: new (and old) players and new insights. Curr. Opin. Cell Biol. 20, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tetsu O, McCormick F (1999) β‐Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426. [DOI] [PubMed] [Google Scholar]
- 30. Clevers H (2006) Wnt/β‐catenin signaling in development and disease. Cell 127, 469–480. [DOI] [PubMed] [Google Scholar]
- 31. Araki Y, Okamura S, Hussain SP, Nagashima M, He P, Shiseki M, Miura K, Harris CC (2003) Regulation of cyclooxygenase‐2 expression by the Wnt and ras pathways. Cancer Res. 63, 728–734. [PubMed] [Google Scholar]
- 32. Rowe MK, Chuang DM (2004) Lithium neuroprotection: molecular mechanisms and clinical implications. Expert. Rev. Mol. Med. 6, 1–18. [DOI] [PubMed] [Google Scholar]
- 33. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben‐Ze’ev A (1999) The cyclin D1 gene is a target of the β‐catenin/LEF‐1 pathway. Proc. Natl. Acad. Sci. USA 96, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rieber M, Rieber MS (2006) Cyclin D1 overexpression induces epidermal growth factor‐independent resistance to apoptosis linked to BCL‐2 in human A431 carcinoma. Apoptosis 11, 121–129. [DOI] [PubMed] [Google Scholar]
- 35. Stacey DW (2003) Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 15, 158–163. [DOI] [PubMed] [Google Scholar]
- 36. Sansom OJ, Reed KR, Van De Wetering M, Muncan V, Winton DJ, Clevers H, Clarke AR (2005) Cyclin D1 is not an immediate target of β‐catenin following Apc loss in the intestine. J. Biol. Chem. 280, 28463–28467. [DOI] [PubMed] [Google Scholar]
- 37. Van De Wetering M, Sancho E, Verweij C, De Lau W, Oving I, Hurlstone A, Van Der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon‐Pon‐Fong M, Moerer P, Van Den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H (2002) The β‐catenin/TCF‐4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111, 241–250. [DOI] [PubMed] [Google Scholar]
- 38. Gao J, Liu X, Rigas B (2005) Nitric oxide‐donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc. Natl. Acad. Sci. USA 102, 17207–17212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu KK (2000) Aspirin and salicylate: an old remedy with a new twist. Circulation 102, 2022–2023. [DOI] [PubMed] [Google Scholar]
- 40. Chan AT, Ogino S, Fuchs CS (2007) Aspirin and the risk of colorectal cancer in relation to the expression of COX‐2. N. Engl. J. Med. 356, 2131–2142. [DOI] [PubMed] [Google Scholar]
- 41. Mifflin RC, Saada JI, Di Mari JF, Valentich JD, Adegboyega PA, Powell DW (2004) Aspirin‐mediated COX‐2 transcript stabilization via sustained p38 activation in human intestinal myofibroblasts. Mol. Pharmacol. 65, 470–478. [DOI] [PubMed] [Google Scholar]
- 42. Davies NM, Sharkey KA, Asfaha S, Macnaughton WK, Wallace JL (1997) Aspirin causes rapid up‐regulation of cyclo‐oxygenase‐2 expression in the stomach of rats. Aliment. Pharmacol. Ther. 11, 1101–1108. [DOI] [PubMed] [Google Scholar]
- 43. Simmons DL, Botting RM, Robertson PM, Madsen ML, Vane JR (1999) Induction of an acetaminophen‐sensitive cyclooxygenase with reduced sensitivity to nonsteroid antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 96, 3275–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. You Z, Saims D, Chen S, Zhang Z, Guttridge DC, Guan KL, MacDougald OA, Brown AM, Evan G, Kitajewski J, Wang CY (2002) Wnt signaling promotes oncogenic transformation by inhibiting c‐Myc‐induced apoptosis. J. Cell Biol. 157, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Han JA, Kim JI, Ongusaha PP, Hwang DH, Ballou LR, Mahale A, Aaronson SA, Lee SW (2002) P53‐mediated induction of Cox‐2 counteracts p53‐ or genotoxic stress‐induced apoptosis. EMBO J. 21, 5635–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563. [DOI] [PubMed] [Google Scholar]
- 47. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC (2004) Gastric cancer originating from bone marrow‐derived cells. Science 306, 1568–1571. [DOI] [PubMed] [Google Scholar]
- 48. Cao H, Xu W, Qian H, Zhu W, Yan Y, Zhou H, Zhang X, Xu X, Li J, Chen Z, Xu X (2009) Mesenchymal stem cell‐like cells derived from human gastric cancer tissues. Cancer Lett. 274, 61–71. [DOI] [PubMed] [Google Scholar]
- 49. Thun MJ, Namboodiri MM, Heath CW Jr (1991) Aspirin use and reduced risk of fatal colon cancer. N. Engl. J. Med. 325, 1593–1596. [DOI] [PubMed] [Google Scholar]