

Abstract
Abstract. We tested the effect of iron deprivation on cell death induction in human Raji cells pre‐adapted to differing availability of extracellular iron. Iron deprivation was achieved by incubation in a defined iron‐free medium. Original Raji cells have previously been adapted to long‐term culture in a defined medium with 5 µg/ml of iron‐saturated human transferrin as a source of iron. Raji/lowFe cells were derived from original Raji cells by subsequent adaptation to culture in the medium with 50 µm ferric citrate as a source of iron. Raji/lowFe‐re cells were derived from Raji/lowFe cells by re‐adaptation to the transferrin‐containing (5 µg/ml) medium. Iron deprivation induced cell death in both Raji cells and Raji/lowFe‐re cells; that is, cells pre‐adapted to a near optimum source of extracellular iron (5 µg/ml of transferrin). However, Raji/lowFe cells preadapted to a limited source of extracellular iron (50 µm ferric citrate) became resistant to the induction of cell death by iron deprivation. We demonstrated that cell death induction by iron deprivation in Raji cells correlates with the activation of executioner caspase‐3 and the cleavage of caspase‐3 substrate, poly‐ADP ribose polymerase. Two other executioner caspases, caspase‐7 and caspase‐6, were not activated. Taken together, we suggest that in human Raji cells, iron deprivation induces apoptotic cell death related to caspase‐3 activation. However, the sensitivity of the cells to death induction by iron deprivation can be reversibly changed by extracellular iron availability. The cells pre‐adapted to a limited source of extracellular iron became resistant.
INTRODUCTION
Apoptosis represents an evolutionarily conserved program of cell self‐destruction by which cells are eliminated from a multicellular organism (Jacobson et al. 1997; Raff 1998). Apoptosis is the type of programmed cell death that is dependent on the activation of executioner caspase(s), particularly on the activation of caspase‐3. The activation of executioner caspase(s) results in cleavage of proteins known as ‘death substrates’. This cleavage represents the start of cell autodestruction (Earnshaw et al. 1999; Jaattela & Tschopp 2003; Fink & Cookson 2005; Ghorbrial et al. 2005; Kim 2005).
It has been shown that iron is essential for cell proliferation and also for maintaining cell viability (Le & Richardson 2002). Iron represents an irreplaceable component of some key enzymes, such as enzymes of the mitochondrial respiratory chain and ribonucleotide reductase, responsible for deoxyribonucleotide synthesis (Thelander et al. 1983; Lill et al. 1999). It is therefore not surprising that iron deficiency could lead to impaired function of the mitochondrial respiratory chain or DNA damage, resulting in the inhibition of cell proliferation or apoptosis induction (Rakba et al. 2000; Simonart et al. 2002). Enhanced requirement for iron supply of proliferating tumour cells is supposed to be a prerequisite for cancer therapy based on apoptosis induction by iron deprivation (Shterman et al. 1991). Various types of tumour cells, especially those of haematopoietic origin and neuroblastomas, have been shown to be sensitive to iron deprivation (Taetle et al. 1989; White et al. 1990; Seligman & Crawford 1991; Kemp et al. 1992; Kovar et al. 1995; Donfrancesco et al. 1996) and in some tumour cells, iron deprivation can induce apoptosis (Fukuchi et al. 1994; Haq et al. 1995; Hileti et al. 1995; 1997a, 2001; Ido et al. 1999; Rakba et al. 2000; Simonart et al. 2000).
We have demonstrated previously that iron deprivation can specifically induce apoptosis (Kovar et al. 1997a). Some types of cells are sensitive to apoptosis induction by iron deprivation, whereas other types of cells are resistant. It seems that most of the cells sensitive to apoptosis induction by iron deprivation are of haematopoietic origin, particularly of B‐cell origin. On the other hand, cells of solid tumours seem to be resistant (Kovar et al. 2001). Recently, we have demonstrated that apoptosis induced by iron deprivation in mouse B‐cell lymphoma 38C13 is independent of the p53 pathway (Truksa et al. 2003) and dependent on the activation of executioner caspase‐3 (Koc et al. 2005).
Human Raji cells (Burkitt's lymphoma) have been previously found to be sensitive to apoptosis induction by iron deprivation (Kovar et al. 2001). In the present study, we tested the effect of iron deprivation on cell‐death induction in Raji cells pre‐adapted to differing availability of external iron. We demonstrated that iron deprivation induces caspase‐3 activation‐related death in Raji cells previously cultured in medium containing a near‐optimum level of the iron source. However, surprisingly, Raji cells pre‐adapted to culture in low levels of external iron became resistant to the induction of cell death by iron deprivation. Furthermore, these resistant cells again became sensitive to iron deprivation when readapted to the original more favourable availability of external iron.
MATERIALS AND METHODS
Materials
Human transferrin (apotransferrin) from Sigma‐Aldrich (St. Louis, MO, USA) was rendered iron saturated as described previously (Kovar & Franek 1989). Primary antibodies used for Western blot analysis: rabbit polyclonal cleaved caspase‐3 (Asp175) antibody against human caspase‐3 and rabbit polyclonal caspase‐6 antibody against human caspase‐6 from Cell Signalling Technology (Danvers, MA, USA), mouse monoclonal antihuman/mouse caspase‐7 antibody (MCH3101.62) against human caspase‐7 from RD System (Minneapolis, MN, USA), mouse monoclonal anti‐poly‐ADP ribose polymerase (PARP) antibody (CZ‐10) against human PARP from BD Pharmingen (San Diego, CA, USA), rabbit polyclonal lamin A (H‐102) antibody against human lamin A from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and mouse monoclonal TU‐01 antibody against human α‐tubulin from Exbio (Prague, Czech Republic). Goat antimouse IgG and antirabbit IgG horseradish peroxidase‐conjugated secondary antibodies from Santa Cruz Biotechnology were used.
Cell lines
We used the human Burkitt's lymphoma cell line Raji and its Raji/lowFe and Raji/lowFe‐re derivatives. Raji/lowFe cells were derived from Raji cells, normally cultured in the medium containing transferrin (5 µg/ml) as a source of iron, by culture in the medium with decreasing concentration of ferric citrate (from 500 µm to 50 µm) as a source of iron. Raji/lowFe‐re cells were derived from Raji/lowFe cells by re‐adaption to culture in the medium containing transferrin (5 µg/ml) as a source of iron. Raji cells, as described previously (Kovar et al. 2001), and readapted Raji/lowFe‐re cells are sensitive to death induced by iron deprivation. Derived Raji/lowFe cells are resistant. Approximately 30–50% of sensitive Raji cells display cell death features after 48 h under iron deprivation conditions and they die more or less completely within 96 h. Resistant Raji/lowFe cells do not show any sign of cell death during 96 h of incubation under iron deprivation. Cells were routinely tested for mycoplasma contamination using the fluorescent Hoechst 33258 staining method (Chen 1997).
Culture media and culture conditions
Defined serum‐free media were used. Transferrin medium was a basic medium supplemented with 5 µg/ml of iron‐saturated human transferrin and similarly low‐iron medium was supplemented with 50 µm ferric citrate as a source of iron. Iron‐free medium was a basic medium without any iron compound added. The basic medium was RPMI 1640 containing extra l‐glutamine (300 µg/ml), sodium pyruvate (110 µg/ml), penicillin (100 U/ml), streptomycin (100 µg/ml), Hepes (15 mm), ethanolamine (20 µm), ascorbic acid (20 µm), hydrocortisone (5 nm), 11 trace elements, as described previously (Kovar 1988; Kovar & Franek 1989), and 2‐mercaptoethanol (50 µm). In these experiments, media with an iron source represented control culture conditions and iron‐free medium represented iron‐depriving conditions. Cells were incubated at 37 °C in a humidified atmosphere of 5% CO2.
Cell growth and survival experiments
Cells maintained in the relevant culture medium were harvested by low‐speed centrifugation, washed with iron‐free medium and then seeded at 1 × 104 cells/100 µl of relevant medium into wells of a 96‐well plastic plate. Cell growth and survival were evaluated after 24, 48, 72 and 96 h of incubation. The number of living cells was determined by haemacytometer counting after staining with trypan blue, or were assessed employing the MTT method (Kovar et al. 1997b).
Iron deprivation experiments
Cells previously maintained in iron‐containing medium were harvested by low‐speed centrifugation, washed with iron‐free medium and seeded at 2 × 105 cells/ml of iron‐free medium (iron deprivation) and relevant iron‐containing medium (control) into plastic culture flasks. The effect of iron deprivation on the tested characteristics was determined after 48 h of incubation. It has been shown previously that a significant portion (up to 50%) of the sensitive Raji cells undergo cell death within 48‐h incubation under iron deprivation.
Measurement of caspase‐3 and caspase‐6 activities
Commercial colourimetric kits Caspase‐3 Assay Kit from Sigma (St. Louis, MO, USA) and Caspase‐6 Colorimetric Assay Kit from Alexis Biochemicals (Nottingham, UK) were used. Cells (approximately 5 × 106 cells per sample) were harvested by low‐speed centrifugation and washed with phosphate‐buffered saline (PBS). The cell pellet was lysed in 50 µl of the lysis buffer from the relevant kit for 10 min at 4 °C. Crude cell lysate was centrifuged and the supernatant was collected into a fresh, new Eppendorf tube. Total protein content was determined by the bicinchoninic acid assay (Smith et al. 1985) and samples were then frozen at −80 °C until further analysis. Samples (80 µg of total protein per sample) were incubated in 96‐well microplates for 2 h at 37 °C. The activity of caspases was determined as the absorbance of a sample at 405 nm using a Sunrise enzyme‐linked immunosorbent assay Reader (Tecan, Maennedorf, Switzerland).
Whole cell lysate preparation
Cells (approximately 107 cells per sample) were harvested by low‐speed centrifugation and washed twice with PBS. The cell pellet was lysed in 50 µl of Triton lysis buffer (1% Triton X‐100, 0.15 m NaCl, 1 mm ethylenediaminetetraacetic acid, 30 mm NaF, 20 µm leupeptin, 1 mm phenylmethanesulfonyl fluoride, pH 7.6) for 10 min on ice. Crude cell lysate was centrifuged (20 000 g, 15 min, 4 °C). The supernatant was collected into a fresh new Eppendorf tube and frozen at −80 °C until further analysis. Total protein content was determined by the bicinchoninic acid assay (Smith et al. 1985).
SDS‐PAGE and Western blot analysis
Sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blot analysis were performed similarly as described previously (Koc et al. 2005). Briefly, protein samples (100 µg) were heated for 5 min at 100 °C in the sample loading buffer (62.5 mm Tris, 10% glycerol, 2% SDS, 5% 2‐mercaptoethanol, 0.05% bromphenol blue). Samples were run at a constant current of 0.03 A for 1 h, employing mini‐Protean 3 (Bio‐Rad Laboratories, Hercules, CA, USA), on 12% polyacrylamide gel (4% polyacrylamide stacking gel). Proteins separated by SDS‐PAGE were blotted on to 0.2 µm Protran nitrocellulose membrane from Schleicher & Schuell BioScience (Dassel, Germany) for 1 h at a constant current of 0.25 A, using Mini Trans‐Blot Cell from Bio‐Rad Laboratories (Hercules, CA, USA). The membrane was blocked with 5% nonfat milk in Tris‐buffered saline (TBS) for 30 min and then was washed with 0.1% Tween/TBS three times. The washed membrane was incubated overnight at 4 °C with the relevant primary antibody (0.5 µg/ml of TBS containing 1% non‐fat milk and 0.1% Tween). After the incubation, the membrane was washed with 0.1% Tween/TBS three times. The washed membrane was incubated for 1 h with corresponding horseradish peroxidase‐conjugated secondary antibody (1 : 10 000 in TBS containing 1% non‐fat milk and 0.1% Tween). The membrane was then washed with 0.1% Tween/TBS three times. Horseradish peroxidase‐conjugated antibody was detected by enhanced chemiluminescence using the enhanced chemiluminescent (ECL) chemiluminescence reagent from Pierce Biotechnology (Rockford, IL, USA) and CCD camera LAS1000 (Fuji, Shizuoka, Japan).
RESULTS
Raji/lowFe and Raji/lowFe‐re cells
Raji cells were previously adapted to long‐term culture in a chemically defined culture medium with 5 µg/ml of iron‐saturated human transferrin (transferrin medium) as a source of iron (Kovar et al. 1994). In the next step, these Raji cells were adapted during eight passages to long‐term culture in the medium with 500 µm ferric citrate as a source of iron instead of transferrin. We have previously demonstrated that proliferation characteristics of cells cultured in transferrin medium (5 µg/ml of transferrin) and in the medium with 500 µm ferric citrate are very similar and near optimum (Kovar et al. 1997b).
In order to derive Raji cells capable of growing with a limited supply of iron, we started with Raji cells adapted to the medium with 500 µm ferric citrate, and employed subsequent adaptation (eight passages) via 200 µm and 100 µm ferric citrate. Finally, we derived Raji cells adapted to long‐term culture in the medium with 50 µm ferric citrate as a source of iron (low‐iron medium). These Raji cells were called Raji/lowFe. Raji/lowFe cells were again readapted during eight passages to long‐term culture in transferrin medium. These readapted Raji/lowFe cells were called Raji/lowFe‐re (Fig. 1).
Figure 1.
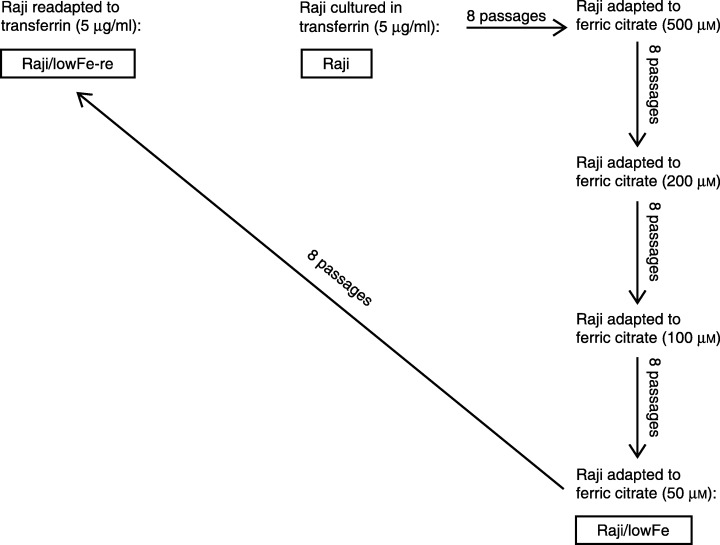
Relationships among Raji, Raji/lowFe and Raji/lowFe‐re cells. Sources of iron in relevant culture media are shown. Original Raji cells were cultured in a defined culture medium with 5 µg/ml of iron‐saturated human transferrin as a source of iron. Raji/lowFe cells were derived from original Raji cells by subsequent adaptation to the culture medium with 50 µm ferric citrate as a source of iron. Raji/lowFe‐re cells were derived from Raji/lowFe cells by re‐adaptation to the transferrin‐containing (5 µg/ml) medium.
Growth and survival under iron deprivation
We tested the growth and survival of Raji cells (routinely maintained in transferrin medium) in iron‐free medium. Transferrin medium was used as a control medium. Raji cells started to die within 48–72 h of incubation in iron‐free medium. After 72 h, a significant fraction of the cells were dead, and after 96 h, only a few of Raji cells were alive. The number of living cells in control transferrin medium increased approximately threefold within 96 h of incubation (Fig. 2a).
Figure 2.
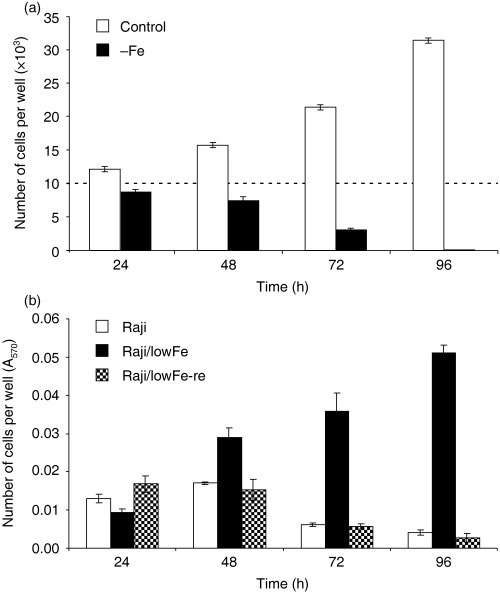
(a) Effect of iron deprivation on the growth and survival of Raji cells. Cells were incubated in iron‐free medium (– Fe) and cells of the control in transferrin (5 µg/ml) medium. Cells were seeded at 10 × 103 cells/100 µl of medium in the well. The number of living cells was determined by counting (see Materials and methods). The number of cells of the inoculum is shown as a dotted line. Each column represents the mean of eight separate cultures ± SEM. (b) Effect of iron deprivation on the growth and survival of Raji, Raji/lowFe and Raji/lowFe‐re cells. Cells were incubated in iron‐free medium. They were seeded at 10 × 103 cells/100 µl of medium in the well. The number of living cells was determined employing the MTT method (see Materials and methods). Each column represents the mean of eight separate cultures ± SEM.
In the next step, we compared the growth and survival of Raji cells (maintained in transferrin medium), Raji/lowFe cells (adapted to low‐iron medium) and Raji/lowFe‐re cells (re‐adapted to transferrin medium) in iron‐free medium. Both Raji cells and Raji/lowFe‐re cells were dying in a similar manner during 96‐h incubation in iron‐free medium. In contrast, the number of living Raji/lowFe cells in iron‐free medium increased several times within 96 h of incubation (Fig. 2b).
Effect of iron deprivation on caspase‐3/caspase‐7 activity
In order to characterize cell death induced by iron deprivation in Raji and in Raji/lowFe‐re cells, we tested the activation of executioner caspases. First, we tested the effect of iron deprivation on caspase‐3 and caspase‐7 activity.
The employed colourimetric assay showed that the activity of caspase‐3 in both sensitive Raji and sensitive Raji/lowFe‐re cells increased significantly (approximately two‐ to threefold) within 48‐h incubation in iron‐free medium when compared with the incubation in control transferrin (5 µg/ml) medium. On the other hand, we did not find any significant increase of caspase‐3 activity in resistant Raji/lowFe cells under iron deprivation (Fig. 3a). Western blot analysis (Fig. 3b) confirmed the activation of caspase‐3 in sensitive Raji cells incubated 48 h under iron deprivation. The large subunit (17 kDa) of active caspase‐3 was detected in sensitive Raji cells. Only the inactive precursor (30 kDa) of caspase‐3 was found in sensitive Raji cells and resistant Raji/lowFe cells under control conditions as well as in resistant Raji/lowFe cells under iron deprivation. Western blot analysis also showed that caspase‐7 was not activated in both cell types under iron deprivation. The large subunit (20 kDa) of active caspase‐7 was not detected. Western blot analysis (Fig. 3c) of caspase‐3/caspase‐7 substrate PARP showed that PARP was cleaved only in the case of sensitive Raji cells under iron deprivation when caspase‐3 was activated and cell death was induced.
Figure 3.
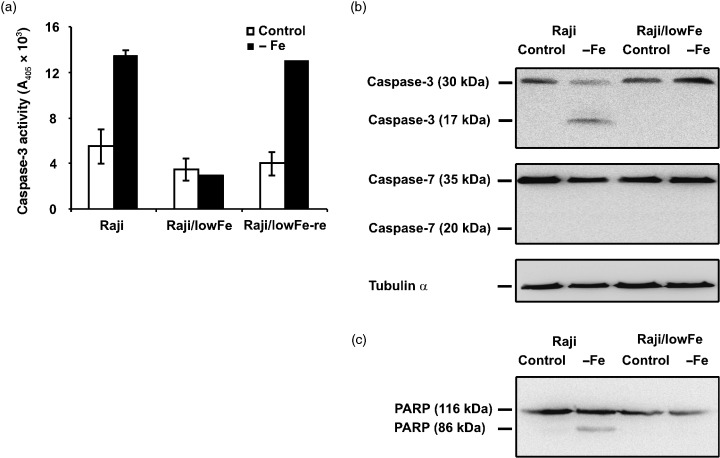
Effect of iron deprivation on caspase‐3/caspase‐7 activity in sensitive Raji cells, resistant Raji/lowFe cells and sensitive Raji/lowFe‐re cells. Cells were incubated in iron‐free medium (– Fe) for 48 h. Raji and Raji/lowFe‐re cells of the control were incubated in transferrin (5 µg/ml) medium and Raji/lowFe cells of the control in low‐iron (50 µm ferric citrate) medium. (a) The activity of caspase‐3 was measured as the absorbance of the cleaved product of a caspase‐3 chromogenic substrate at 405 nm employing the commercial colourimetric assay kit (see Materials and methods). Each column represents the mean of two experimental values ± SEM. The data shown were obtained in one representative experiment of three independent experiments. (b) The activation of caspase‐3 and caspase‐7 were assessed by Western blot analysis, employing specific rabbit polyclonal antibody against human caspase‐3 and specific mouse monoclonal antibody against human caspase‐7 (see Materials and methods). Locations of the 17‐kDa fragment of active caspase‐3 and of the 30‐kDa inactive precursor of caspase‐3 are shown. Similarly, locations of the 20‐kDa fragment of active caspase‐7 and of the 35‐kDa inactive precursor of caspase‐7 are shown. Specific mouse monoclonal antibody against α‐tubulin (see Materials and methods) was used to confirm equal protein loading. The data shown were obtained in one representative experiment of three independent experiments. (c) The activity of caspase‐3 was also assessed by Western blot analysis employing specific mouse monoclonal antibody against specific caspase‐3/caspase‐7 substrate PARP (see Materials and methods). Locations of the 86‐kDa cleaved fragment of PARP and of the 116‐kDa intact PARP are shown. The data shown were obtained in one representative experiment of two independent experiments.
Effect of iron deprivation on caspase‐6 activity
We also tested the effect of iron deprivation on the activity of executioner caspase‐6. The employed colourimetric assay showed that the activity of caspase‐6 did not increase significantly in sensitive Raji cells as well as in resistant Raji/lowFe cells within 48‐h incubation in iron‐free medium (Fig. 4a). Western blot analysis (Fig. 4b) confirmed that caspase‐6 was not activated. The small subunit (15 kDa) of active caspase‐6 was not detected and only the inactive precursor (35 kDa) of caspase‐6 was found in sensitive Raji and resistant Raji/lowFe cells under control conditions and under iron deprivation. Western blot analysis (Fig. 4c) of caspase‐6 substrate lamin A showed that lamin A was not cleaved in either cell type under iron deprivation conditions.
Figure 4.
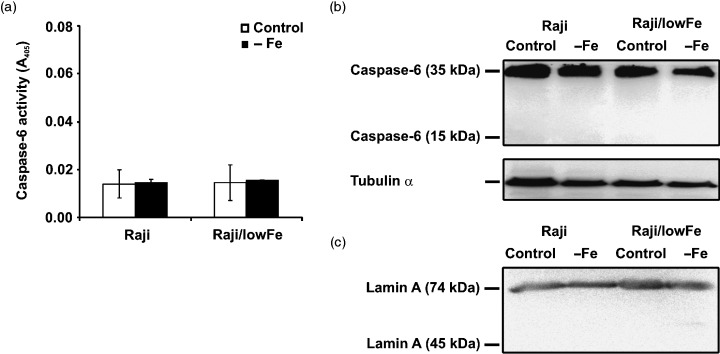
Effect of iron deprivation on caspase‐6 activity in sensitive Raji and resistant Raji/lowFe cells. Cells were incubated in iron‐free medium (– Fe) for 48 h. Raji cells of the control were incubated in transferrin (5 µg/ml) medium and Raji/lowFe cells of the control in low‐iron (50 µm ferric citrate) medium. (a) The activity of caspase‐6 was measured as the absorbance of the cleaved product of a caspase‐6 chromogenic substrate at 405 nm employing the commercial colourimetric assay kit (see Materials and methods). Each column represents the mean of two experimental values ± SEM. The data shown were obtained in one representative experiment of two independent experiments. (b) The activation of caspase‐6 was assessed by Western blot analysis employing specific rabbit polyclonal antibody against human caspase‐6 (see Materials and methods). Locations of the 15‐kDa fragment of active caspase‐6 and of the 35‐kDa inactive precursor of caspase‐6 are shown. Specific mouse monoclonal antibody against α‐tubulin (see Materials and methods) was used to confirm equal protein loading. The data shown were obtained in one representative experiment of three independent experiments. (c) The activity of caspase‐6 was also assessed by Western blot analysis employing specific rabbit polyclonal antibody against specific caspase‐6 substrate lamin A (see Materials and methods). Locations of the 45‐kDa cleaved fragment of lamin A and of the 74‐kDa intact lamin A are shown. The data shown were obtained in one representative experiment of three independent experiments.
DISCUSSION
This team has previously shown that iron deprivation specifically induces cell death (Kovar et al. 1997a). We have also demonstrated that some types of cells are sensitive whereas other types are resistant to cell death induced by iron deprivation. Cells of haematopoietic origin and particularly those of B cell origin seem to be sensitive (Kovar et al. 2001). Human Raji cells (Burkitt's lymphoma) and similarly mouse 38C13 cells (B cell lymphoma) have been found to be sensitive too (Kovar et al. 2001; Truksa et al. 2003). Cell death induced by iron deprivation in mouse 38C13 cells has been described as apoptotic cell death related to caspase‐3 activation (Koc et al. 2005).
Raji cells sensitive to cell death induction by iron deprivation and newly derived Raji/lowFe cells, which are resistant to cell‐death induction by iron deprivation (see Fig. 1), represent a unique experimental model for studies concerning the molecular mechanisms involved in cell death induced by iron deprivation. Both cell types are of the same origin and they differ only in their sensitivity to iron deprivation. This model enables discrimination between changes coupled with cell‐death induction by iron deprivation and changes resulting just from iron deprivation but unrelated to cell‐death induction (Truksa et al. 2003; Koc et al. 2005).
We demonstrated that cell death induced by iron deprivation in sensitive Raji cells correlated with the activation of key executioner caspase‐3. The cleavage of caspase‐3 substrate PARP was also shown (see Fig. 3). On the other hand, two other executioner caspases, caspase‐7 and caspase‐6, were not activated in sensitive Raji cells under iron deprivation (see 3, 4). Thus, we can characterize cell death induced by iron deprivation in Raji cells as apoptotic cell death related to caspase‐3 activation, similar to cell death induced by iron deprivation in mouse 38C13 cells. Detailed study has shown that cell death induced by iron deprivation in 38C13 cells represents apoptosis induced via the mitochondrial pathway (Koc et al. 2005).
When sensitive Raji cells were preadapted to long‐term culture in medium with a limited source of iron (50 µm ferric citrate), the cells (referred to as Raji/lowFe) became resistant to induction of cell death by iron deprivation (see Fig. 2). This implies that adaptation to the changed availability of an external iron source resulted in changed sensitivity to death induction by iron deprivation. More interestingly, when resistant Raji/lowFe cells were re‐adapted to long‐term culture in medium with the near optimum source of iron (5 µg/ml of iron‐saturated transferrin) (Kovar et al. 1997b), the cells (referred to as Raji/lowFe‐re) again became sensitive to death induction by iron deprivation (see Fig. 2) and vice versa, sensitive Raji/lowFe‐re cells readapted to low‐iron medium (50 µm ferric citrate) again became resistant (data not shown). Sensitivity to death induction by iron deprivation can thus be changed by the availability of external iron, reversibly. It can therefore be said that sensitivity of Raji cells to apoptotic cell‐death induction by iron deprivation reflects the particular metabolic status of the cells, concerning iron utilization, and its change does not involve cell genetic change and subsequent selection of the adapted clone(s). Similar findings, showing that the sensitivity of cells to the tested death stimulus reflects the status of the cells, have been presented by several other authors recently (Hahn & Mayne 2004; Yao et al. 2005). Such findings have probably more general meaning when considering the sensitivity of certain cells to a specific cell‐death stimulus.
We can suggest two possible mechanisms to explain our findings. First, the more general mechanism is related to the proliferative activity of the cells. One can assume that the faster‐growing cells, that is, cells adapted to growth in the medium with a near optimum source of iron (5 µg/ml of transferrin), exhaust the internal reserves of required iron under iron deprivation more rapidly than slowly growing cells; that is, cells adapted to growth in the medium with a limited source of iron (50 µm ferric citrate). Thus, the cells adapted to the near optimum source of iron are more sensitive to iron deprivation than those adapted to the limited source of iron. However, Raji and Raji/lowFe‐re cells adapted to near optimum sources of iron do not seem to grow significantly faster than Raji/lowFe cells adapted to the limited source of iron. The second, more specific mechanism is related to the possible function of heat shock protein 90 (Hsp90). It has been demonstrated that transformed haematopoietic cells with increased expression of Hsp90 are more sensitive to apoptosis induction than cells with decreased expression of Hsp90, which are relatively resistant to apoptosis induction (Galea‐Louri et al. 1996). On the other hand, several authors have demonstrated that higher levels of iron lead to enhanced expression of Hsp90 (Fukuda et al. 1996; Nardai et al. 2000). However, the suggested mechanisms are only speculative and further studies are required to elucidate the real situation.
Taken together, we can conclude that iron deprivation induces apoptotic cell death in human Raji cells. The cell death is related to caspase‐3 activation but not to caspase‐7 and/or caspase‐6 activation. However, the sensitivity of Raji cells to death induction by iron deprivation can be changed reversibly by external iron availability. Raji cells adapted to a near optimum source of external iron (5 µg/ml of transferrin) are sensitive to death induction, whereas Raji cells adapted to a limited source of external iron (50 µm ferric citrate) are resistant. This finding shows that the sensitivity of cells to a certain cell death stimulus can depend on the particular metabolic status of the cells.
ACKNOWLEDGEMENTS
This work was supported by grant KJB5052401 from the Grant Agency of the Academy of Sciences of the Czech Republic.
REFERENCES
- Chen TR (1977) In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 104, 255–262. [DOI] [PubMed] [Google Scholar]
- Donfrancesco A, Deb G, De Sio L, Cozza R, Castellano A (1996) Role of deferoxamine in tumor therapy. Acta Haematol. 95, 66–69. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68, 383–424. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi K, Tomoyasu S, Tsuruoka N, Gomi K (1994) Iron deprivation‐induced apoptosis in HL‐60 cells. FEBS Lett. 350, 139–142. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Osawa T, Oda H, Tanaka T, Toyokuni S, Uchida K (1996) Oxidative stress response in iron‐induced acute nephrotoxicity: enhanced expression of heat shock protein 90. Biochem. Biophys. Res. Commun. 219, 76–81. [DOI] [PubMed] [Google Scholar]
- Galea‐Louri J, Richardson AJ, Latchman DS, Katz. DR (1996) Increased heat shock protein 90 (hsp90) expression leads to increased apoptosis in the monoblastoid cell line U937 following induction with TNF‐alpha and cycloheximide: a possible role in immunopathology. J. Immunol. 157, 4109–4118. [PubMed] [Google Scholar]
- Ghorbrial IM, Wiltzig TE, Adjei AA (2005) Targeting apoptosis pathway in cancer therapy. CA Cancer J. Clin. 55, 178–194. [DOI] [PubMed] [Google Scholar]
- Hahn MA, Mayne GC (2004) Phorbol ester‐induced cell death in PC‐12 cells overexpressing Bcl‐2 is dependent on the time at which cells are treated. Cell Biol. Int. 28, 345–359. [DOI] [PubMed] [Google Scholar]
- Haq RU, Wereley JP, Chitambar CR (1995) Induction of apoptosis by iron deprivation in human leukemic CCRF‐CEM cells. Exp. Hematol. 23, 428–432. [PubMed] [Google Scholar]
- Hileti D, Panayiotidis P, Hoffbrand AV (1995) Iron chelators induce apoptosis in proliferating cells. Br. J. Haematol. 89, 181–187. [DOI] [PubMed] [Google Scholar]
- Ido Y, Muto N, Inada A, Kohroki J, Mano M, Odani T, Itoh N, Yamamoto K, Tanaka K (1999) Induction of apoptosis by hinokitiol, a potent iron chelator, in teratocarcinoma F9 cells is mediated through the activation of caspase‐3. Cell Prolif. 32, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaattela M, Tschopp M (2003) Caspase‐independent cell death in T lymphocytes. Nat. Immunol. 4, 416–423. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development. Cell 88, 347–354. [DOI] [PubMed] [Google Scholar]
- Kemp JD, Thorson JA, Stewart BC, Naumann PW (1992) Inhibition of hematopoietic tumor growth by combined treatment with deferoxamine and an IgG monoclonal antibody against the transferrin receptor: evidence for a threshold model of iron deprivation toxicity. Cancer Res. 52, 4144–4148. [PubMed] [Google Scholar]
- Kim R. (2005) Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 103, 1551–1560. [DOI] [PubMed] [Google Scholar]
- Koc M, Nadova Z, Truksa J, Ehrlichova M, Kovar J (2005) Iron deprivation induces apoptosis via mitochondrial changes related to Bax translocation. Apoptosis 10, 381–393. [DOI] [PubMed] [Google Scholar]
- Kovar J (1988) Growth‐stimulating effect of ferric citrate on hybridoma cells: characterization and relation to transferrin function. Hybridoma 7, 255–263. [DOI] [PubMed] [Google Scholar]
- Kovar J, Franek F (1989) Growth‐stimulating effect of transferrin on a hybridoma cell line: relation to transferrin iron‐transporting function. Exp. Cell Res. 182, 358–369. [DOI] [PubMed] [Google Scholar]
- Kovar J, Seligman P, Gelfand EW (1994) Lymphocyte lines under iron‐depriving conditions: transferrin receptor expression related to various growth responses. Immunol. Lett. 42, 123–127. [DOI] [PubMed] [Google Scholar]
- Kovar J, Naumann PW, Stewart BC, Kemp JD (1995) Differing sensitivity of non‐hematopoietic human tumors to synergistic anti‐transferrin receptor monoclonal antibodies and deferoxamine in vitro . Pathobiology 63, 65–70. [DOI] [PubMed] [Google Scholar]
- Kovar J, Stunz. LL, Stewart BC, Kriegerbeckova K, Ashman RF, Kemp JD (1997a) Direct evidence that iron deprivation induces apoptosis in murine lymphoma 38C13. Pathobiology 65, 61–68. [DOI] [PubMed] [Google Scholar]
- Kovar J, Kuhn LC, Richardson V, Seiser C, Kriegerbeckova K, Musilkova J (1997b) The inability of cells to grow in low iron correlates with increasing activity of their iron regulatory protein (IRP). In Vitro Cell Dev. Biol. Anim. 33, 633–639. [DOI] [PubMed] [Google Scholar]
- Kovar J, Valenta T, Stybrova H (2001) Differing sensitivity of tumor cells to apoptosis induced by iron deprivation in vitro . In Vitro Cell Dev. Biol. Anim. 37, 450–458. [DOI] [PubMed] [Google Scholar]
- Le NT, Richardson DR (2002) The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim. Biophys. Acta 1603, 31–46. [DOI] [PubMed] [Google Scholar]
- Lill R, Diekert K, Kaut A, Lange H, Pelzer W, Prohl C, Kispal G (1999) The essential role of mitochondria in the biogenesis of cellular iron‐sulfur proteins. Biol. Chem. 380, 1157–1166. [DOI] [PubMed] [Google Scholar]
- Nardai G, Sass B, Eber J, Orosz. G, Csermely P (2000) Reactive cysteines of the 90‐kDa heat shock protein, Hsp90. Arch. Biophys. 384, 59–67. [DOI] [PubMed] [Google Scholar]
- Raff M (1998) Cell suicide for beginners. Nature 396, 119–122. [DOI] [PubMed] [Google Scholar]
- Rakba N, Loyer P, Gilot D, Delcros JG, Glaise D, Baret P, Pierre JL, Brissot P, Lescoat G (2000) Antiproliferative and apoptotic effects of O‐Trensox, a new synthetic iron chelator, on differentiated human hepatoma cell lines. Carcinogenesis 21, 943–951. [DOI] [PubMed] [Google Scholar]
- Seligman PA, Crawford ED (1991) Treatment of advanced transitional cell carcinoma of the bladder with continuous‐infusion gallium nitrate. J. Natl. Cancer Inst. 83, 1582–1584. [DOI] [PubMed] [Google Scholar]
- Shterman N, Kupfer B, Moroz C (1991) Comparison of transferrin receptors, iron content and isoferritin profile in normal and malignant human breast cell lines. Pathobiology 59, 19–25. [DOI] [PubMed] [Google Scholar]
- Simonart T, Boelaert JR, Mosselmans R. (2002) Antiproliferative and apoptotic effect of iron chelators on human cervical carcinoma cells. Gynecol. Oncol. 85, 95–102. [DOI] [PubMed] [Google Scholar]
- Simonart T, Degraef C, Andrei G, Mosselmans R, Hermans P, Van Vooren JP, Noel JC, Boelaert JR, Snoeck R, Heenen M (2000) Iron chelators inhibit the growth and induce the apoptosis of Kaposi's sarcoma cells and of their putative endothelial precursors. J. Invest. Dermatol. 115, 893–900. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Taetle R, Honeysett JM, Bergeron R. (1989) Combination iron depletion therapy. J. Natl. Cancer Inst. 81, 1229–1235. [DOI] [PubMed] [Google Scholar]
- Thelander L, Graslund A, Thelander M (1983) Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: possible regulation mechanism. Biochem. Biophys. Res. Commun. 110, 859–865. [DOI] [PubMed] [Google Scholar]
- Truksa J, Kovar J, Valenta T, Ehrlichova M, Polak J, Naumann PW (2003) Iron deprivation induces apoptosis independently of p53 in human and murine tumour cells. Cell Prolif. 36, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S, Taetle R, Seligman PA, Rutherford M, Trowbridge IS (1990) Combinations of anti‐transferrin receptor monoclonal antibodies inhibit human tumor cell growth in vitro and in vivo: evidence for synergistic antiproliferative effects. Cancer Res. 50, 6295–6301. [PubMed] [Google Scholar]
- Yao K, Gietema JA, Shida S, Selvakumaran M, Fonrose X, Haas NB, Testa J, O'Dwyer PJ (2005) In vitro hypoxia‐conditioned colon cancer cell lines derived from HCT116 and HT29 exhibit altered apoptosis susceptibility and a more angiogenic profile in vivo . Br. J. Cancer 93, 1356–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]