

Abstract
Objectives: The aim of the present study was to improve efficiency of isolation and to optimize proliferative potential of human spermatogonial stem cells (SSCs) obtained from obstructive azoospermic (OA) and non‐obstructive azoospermic (NOA) patients, and further, to characterize these cells for potential use in infertility treatment or study of reproductive biology.
Materials and methods: We have applied a cell‐sorting method, using collagen and magnetic activated cell separation to overcome obstacles, developing a collection system, and simple long‐term proliferation system, that yields large numbers of high‐purity SSCs from obstructive OA and NOA patients.
Results: SSCs derived from OA and NOA patients proliferated and maintained their characteristics for more than 12 passages (>6 months) in vitro. Moreover, the population of cells positive for the SSC‐specific markers GFRα‐1 and integrin α6, increased to more than 80% at passage 8.
Conclusion: These finding may support the idea that in vitro propagation of SSCs could be a useful tool for infertility treatment and study of reproductive biology.
Introduction
Spermatogenesis is the process by which undifferentiated germ cells in the testis divide and mature. Continuous function of this process, which produces millions of spermatozoa daily, is required for sustained male fertility. Spermatogonial stem cells (SSCs), precursors of the spermatogonial lineage, have both self‐renewal and differentiation abilities, and are tightly controlled in the stem cell niche (1). However, biology of SSCs is difficult to study due to complexity of the microenvironment and because these cells are rare in the testis, with a frequency estimated at 1 in 3000–4000 cells, in the mouse (2).
Attempts to develop in vitro culture systems that allow mammalian spermatogenesis to be characterized have been reported in the literature since the early 1960s. However, most studies from that period focused on transmeiotic and post‐meiotic differentiation of mammalian, and in particular human, germ cells, and produced limited results (3, 4, 5, 6, 7, 8). The problematic experimental factor in these studies was absence of an in vitro culture system capable of supporting SSC self‐renewal. To overcome this obstacle, Dym et al. have generated telomerase‐immortalized mouse SSC lines and showed that they can be successfully differentiated into post‐meiotic germ cells in the absence of supportive cells. Such immortalized SSC lines may prove to be powerful tools in determining the molecular mechanisms of spermatogenesis (9).
SSCs are generally similar to other stem cells. They are a rare, relatively quiescent population situated in a protected region of the testis among support cells, which may regulate their behaviour. As with haematopoietic stem cells, mammalian SSCs can be transplanted and have the ability to expand the stem cell pool and regenerate an entire depleted spermatogenic lineage. Exploiting these attributes, Kanatsu‐Shinohara et al. have introduced stem cell culture methods involving growth factors and feeder cell co‐cultures, and ultimately succeeded in establishing long‐term cultures of mouse SSCs (10). Developmental potential of these SSCs was confirmed by showing that transplantation into seminiferous tubules restored fertility in congenitally infertile recipient mice (11, 12, 13). This long‐term method for culturing SSCs has been shown to be a useful tool for studying spermatogenetic mechanisms, and has important implications for development of new technologies in transgenesis or otherwise in medicine.
Maintenance of normal spermatogenesis and fertility is dependent on a balance between SSC renewal and differentiation in the adult human testis. These two processes are tightly regulated, both intrinsically, through control of gene expression in the stem cells, and extrinsically, by signals from the surrounding microenvironment, such as from soluble factors and cell adhesion molecules (14). Although several research groups have taken on the challenge of in vitro spermatogenesis, most have focused on improving meiotic completion or post‐meiotic differentiation (15, 16, 17, 18, 19). Adapting techniques first introduced to culture mouse stem cells, Gearhart et al. have demonstrated that human embryonic germ (EG) cells can be successfully established from 5‐ to 9‐weeks post‐fertilization primordial germ cells (20). Our group has reported that SSC‐like cells could be isolated from testicular tissue of non‐obstructive azoospermic (NOA) patients using modified stem cell culture conditions, but could not be maintained in vitro for more than a few passages. However, these same cells differentiated into haploid germ cells and produced embryos, confirming their developmental potential (21). This limited success is likely to reflect culture conditions that are suboptimal for in vitro proliferation and differentiation of human SSCs. Recently, Conrad et al. (22) established conditions for isolating SSCs and culturing multipotent adult germline stem cells (maGSCs) generated from these cells, but their characteristics were changed. These researchers found that maGSC had high proliferative activity. Extracellular matrix (ECM) components included in the isolation step modified the culture system and improved efficiency of isolation and generation of the cells, from normal human testis. The aim of the present study was to improve efficiency of isolation and to optimize proliferative potential of human SSCs obtained from obstructive azoospermic (OA) and NOA patients, and further, to characterize these cells for potential use in infertility treatment or for study of reproductive biology. Our results show that large yields of high‐purity SSCs with germ cell developmental potential can be obtained from both types of azoospermic patients using our modifying culture conditions. To our knowledge, this is the first report describing long‐term (>6 months) in vitro proliferation of human SSCs with germ cell developmental potential from both types of azoospermic patients.
Materials and methods
Patient samples
Testicular tissues were obtained from OA and NOA patients undergoing testicular sperm extraction (TESE)–intracytoplasmic sperm injection (ICSI) treatment. Testicular material remaining after clinical requirement, was donated for this investigation with patients’ consent. The study was approved by the Institutional Review Board of the CHA Gangnam Medical Center (Seoul, Korea). Experimental human TESE samples and quality control of the laboratory facility and equipment were also accordingly managed.
Isolation and in vitro culture of SSCs
Testicular constituents of 37 azoospermic patients (18 with OA and 19 with NOA) were dissociated by modified enzymatic digestion as described previously (7). Briefly, tissues were placed in 10 ml of Enzyme Solution A containing 0.5 mg/ml collagenase (Type I; Sigma‐Aldrich, St Louis, MO, USA), 10 μg/ml DNase I, 1 μg/ml soybean trypsin inhibitor (Gibco/Invitrogen, Grand Island, NY, USA) and 1 mg/ml hyaluronidase (Sigma‐Aldrich) in Ca++‐, Mg++‐free phosphate‐buffered saline (PBS), and incubated for 20 min at room temperature (∼25 °C). After peritubular cells were removed in the washing step, testicular tissues were re‐dissociated in 10 ml of Enzyme Solution B containing 5 mg/ml collagenase (Gibco/Invitrogen), 10 μg/ml DNase I (Sigma‐Aldrich), 1 μg/ml soybean trypsin inhibitor (Gibco/Invitrogen) and 1 mg/ml hyaluronidase (Sigma‐Aldrich) in Ca++‐, Mg++‐free PBS, and incubated for 30 min at 37 °C. Dissociated testicular cells were then plated and incubated on uncoated dishes in selection medium, comprised of Dulbecco’s modified Eagle’s medium (DMEM; Gibco/Invitrogen) containing 20% foetal bovine serum (FBS; Gibco/Invitrogen), 10 μmol/l 2‐mercaptoethanol (Gibco/Invitrogen), 1% non‐essential amino acids (Gibco/Invitrogen) and 10 ng/ml rat glial cell‐derived neurotrophic factor (GDNF; R&D Systems Inc., Minneapolis, MN, USA), at 37 °C under a humidified conditions of 5% CO2 in air. Prior to incubation, testicular spermatozoa from OA patient samples were removed by a modified two‐gradient (35–70%) Percoll method. Over the next 2 days, unattached cells were harvested and re‐plated on collagen‐coated dishes in proliferation medium, comprising StemPro‐34 SFM (Invitrogen) supplemented with 6 mg/ml D(+)‐glucose, 5 × 10−5 mβ‐mercaptoethanol, 1 μm d(L)‐lactic acid, 2 mm l‐glutamine, 30 μm pyruvic acid, 10−4 m ascorbic acid, 60 ng/ml progesterone, 30 ng/ml β‐oestradiol (Sigma‐Aldrich), 0.2% BSA (ICN Chemicals, Costa Mesa, CA, USA), 100 U/ml penicillin, 100 μg/ml streptomycin, 1× insulin–transferrin–selenium (ITS) supplement, 1× MEM vitamin solution, 1× MEM non‐essential amino acids, 20 ng/ml mouse epidermal growth factor (EGF), 10 ng/ml human basic fibroblast growth factor (bFGF; Invitrogen, Carlsbad, CA, USA), 1% KnockoutTM Serum Replacement (KSR, Invitrogen), 10 ng/ml rat glial cell‐derived neurotrophic factor (GDNF; R&D Systems Inc.) and 103 U/ml human leukaemia inhibitory factor (LIF; Chemicon, Temecula, CA, USA) [modified from Kanatsu‐Shinohara et al. (10)]. To obtain highly pure SSCs, unattached cells collected after 4 h incubation were re‐sorted by magnetic activated cell separation (MACS) using anti‐CD9 antibody. Collected cells were cultured on laminin‐coated dishes in proliferation medium. Sorted cells proliferated slowly, then formed loosely attached clump‐like structures (≥10 cells) on the bottom of dishes 2–4 weeks after seeding. During culture, around 80% medium was changed carefully every other day under a stereomicroscope to avoid loss of floating clumps. Clumps were dissociated by trypsinization and re‐plated every 2 weeks using the same medium. Samples of cell clumps were collected for characterization after passages 1, 3 and 8.
Reverse transcriptase‐polymerase chain reaction and real‐time RT‐PCR
Testicular tissue or cultured samples were analysed by reverse transcriptase‐polymerase chain reaction (RT‐PCR) to assess expression of the following stage‐specific marker genes: Oct‐4, for general stem cells; integrin α6 and integrin β1, for spermatogonia; c‐Kit (23) and human testis‐specific histone (TH2B), for differentiating spermatogonia and spermatocytes; synaptonemal complex protein (SCP)3 and transition protein (TP)‐1, for meiotic spermatocytes and spermatids. Total RNA was isolated from patient testicular tissues (∼100 mg), cell clumps (100 clumps) and cultured cells using the TRIzol method (Gibco/Invitrogen). Total RNA (1 μg) was first incubated with 1 IU DNaseI with 5 mmol/l MgCl2 at 37 °C for 30 min, and then reverse transcribed by adding 1 mmol/l dNTP, 2.5 μmol/l oligo‐dT, and 2.5 IU reverse transcriptase (Superscript; Invitrogen) and incubating at 42 °C for 1 h (24). After the reaction was completed, samples were either used directly for RT‐PCR or stored at −20 °C. Amplifications were performed in a reaction mixture containing 10 mmol/l Tris–HCl (pH 8.3), 2 mmol/l MgCl2, 50 mmol/l KCl, 0.25 mmol/l dNTPs, 3‐5 pmol each primer and 1.25 IU Taq polymerase (Invitrogen) in a total volume of 20 μl. The following targets were amplified from cDNA using primers indicated in parentheses: Oct‐4 (F: 5′‐GGA AAG GCT TCC CCC TCA GGG AAA GG‐3′, R: 5′‐AAG AACA TGT GTA AGC TGC GGC CC‐3′; 460 bp; GenBank accession number NM203289); integrin α6 (F: 5′‐GGG AGC CTC TTC GGC TTC TC‐3′, R: CAC ATG TCA CGA CCT TGC CC‐3′; 286 bp; GenBank accession number NM000210); integrin β1 (F: 5′‐CTG CAA GAA CGG GGT GAA TG‐3′, R: CAC AAT GTC TAC CAA CAC GCC C‐3′; 301 bp; GenBank accession number BC020057); TH2B(F: 5′‐GTG CTA CCA TTT CCA AGA AG‐3′, R: 5′‐CTC GCT ATA CGC TCA AAG AT‐3′; 217 bp; GenBank accession number AF397301); c‐Kit (F: 5′‐AAG GAC TTG AGG TTT ATT CCT‐3′, R: 5′‐CTG ACG TTC ATA ATT GAA GTC‐3′; 345 bp; GenBank accession number L04143); SCP3 (F: 5′‐TGT GGA GGA GTT GTG GAG GA‐3′, R: 5′‐CCC ACT GCT GAA ACA AAG TCA‐3′; 492 bp; GenBank accession number L153694);TP‐1 (F: 5′‐AAG GCC TTA AAT ACC CAG AC‐3′, R: 5′‐AGC AAT GTG TGC CTA AGT TT‐3′; 254 bp; Gen Bank accession number M59924); and 18S ribosomal RNA (F: 5′‐TAC CTA CCT GGT TGA TCC TG‐3′, R: 5′‐GGG TTG GTT TTG ATC TGA TA‐3′; 255 bp; GenBank accession number K03432). PCR was initiated by denaturation at 94 °C for 5 min, followed by 35–40 cycles of 30 s at 94 °C, 30 s at 55–60 °C and 30 s at 72 °C. After final extension step for 10 min at 72 °C, products were separated by 1.5% agarose‐gel electrophoresis, and verified by automated nucleotide sequencing. Negative controls included mock transcription without mRNA. For quantification of expression level, amplification products were quantified by DNA Engine 2 fluorescence detection system (MJ research) using the DyNAmo SYBR green qPCR kit (Finnzymes, Espoo, Finland). Reactions were performed in a reaction mixture containing 4 μl DEPC‐treated water, 2 μl forward primer (5 pmol), 2 μl reverse primer (2 pmol), 10 μl premix with SYBR Green and 2 μl cDNA template in a total volume of 20 μl. PCR protocol consisted of initial denaturing step at 95 °C for 10 min, followed by 35–40 cycles denaturation for 30 s at 95 °C, annealing of primers at 55–60 °C for 30 s and extension at 72 °C for 30 s. Fluorescence was measured at the end of each cycle during the 72 °C step. In the final step, a melting curve was generated by raising the temperature from 65 to 95 °C at a rate of 0.1 °C/s, with constant measurement of fluorescence, followed by cooling at 40 °C for 30 s. Relative gene expression was quantified using the 2−ΔΔCT method.
Trypan blue (TB) staining and TUNEL assay for cell viability and apoptosis
SSCs were obtained at passage 0 (P0, immediately after MACS‐sorting) and P3, and washed twice in DPBS. After resuspending in a suitable volume of a DPBS, SSCs (in 50 μl) were transferred to a 1.5 ml Eppendorf tube, and equal volume of 0.4% trypan blue was added. Cells were then gently mixed and allowed to stand for 5 min at room temperature. Stained cells (10 μl) were transferred to a haemocytometer, and number of viable (unstained) and dead (blue‐stained) cells was counted. Average number of blue‐stained (dead) cells in samples from each passage was determined, and percentage of non‐viable cells was calculated by dividing that by number of total cells %.
For terminal dUTP nick‐end labelling (TUNEL) assays, SSCs obtained at P0 and P3 were washed twice in DPBS with 0.1% polyvinylpyrrolidone and fixed in 4% (v/v) paraformaldehyde solution for 24 h. Fixed cells were slide‐mounted using the cytospin method, and cell membranes were permeabilized by incubation in 0.5% Triton X‐100 for 1 h at room temperature. Presence of apoptotic cells was assessed using In Situ Cell Death Detection Kit as described by the manufacturer (Roche Diagnostics, Indianapolis, IN, USA). Slides were washed and transferred to solution containing 10 μg/ml Hoechst 33342 and incubated for 30 min at room temperature in the dark. After washing three times, slides were mounted, and number of apoptotic nuclei and total number of nuclei were determined from optical images obtained using epifluorescence microscopy (Nikon Corp., Tokyo, Japan).
MTT assay for cell proliferation
Proliferation of SSCs was assessed using the MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) assay (CellTiter 96 Non‐Radioactive Cell Proliferation assay kit; Promega, Madison, WI, USA), based on reduction of MTT to its formazan product by mitochondrial dehydrogenase activity of intact cells. Total of 1 × 104 trypsin‐dispersed SSCs in 100 μl of culture medium were seeded into each well of a 96‐well plate. Plates were then cultured for 0, 24, 48, 72 and 96 h prior to addition of 15 μl dye solution (tetrazolium). Plates were then cultured for 4 h at 37 °C in a humidified, 5% CO2 atmosphere, after which 100 μl of solubilization solution/stop mix was added to each well. Plates were allowed to stand overnight and absorbance was determined at 570–630 nm using an ELISA plate reader.
Cell proliferation was also determined by directly counting cells. At each split, 2 × 104 trypsinized cells were plated and then re‐counted at the next passage.
Telomerase activity assay
SSCs were sampled at P2, P5 and P7, and treated with trypsin–EDTA (LS 015‐08, Welgene). Cells (1 × 105) were lysed in 200 μl of cold lysis buffer (10 mm Tris–HCl pH 7.5, 1 mm MgCl2, 0.5 m EGTA, 0.5% CHAPS, 10% glycerol, 0.1 mm AEBSF, 5 mmβ‐mercaptoethanol), incubated on ice for 30 min and microcentrifuged at 5 600 g for 20 min. The supernatant was then transferred to a fresh new tube. Two microlitres of telomerase extract was aliquoted into a new tube, and mixed with 2.5 μl 10X T‐PCR buffer (200 mm Tris–HCl pH 8.3, 15 mm MgCl2, 680 mm KCl, 0.5% Tween‐20, 10 mm EGTA), 0.125 μl 10 mm dNTP, 1 μl TS primer (AATCCGTCGAGCAGAGTT) and 0.2 μl EX TaqTM (RR001A, Takara), then the mixture was incubated at 20 °C for 30 min. After adding 1 μl of CX primer (CCCTTACCCTTACCCTTACCCTAA), PCR reaction was carried out by performing 31 cycles of 45 s at 94 °C, 45 s at 50 °C and 1 min at 72 °C. Samples were separated by electrophoresis on 15% polyacrylamide gels (15 min at 100 V followed by 1 h at 380 V) and then stained with SYBR green at room temperature for 20–30 min.
Immunocytochemical characterization of SSCs obtained from in vitro culture
Alkaline phosphatase activity was assessed by histochemical staining. Collagen‐binding and non‐binding cells were fixed in 4% paraformaldehyde at room temperature for 1 min, washed twice with PBS and stained with alkaline phosphatase substrate solution (10 ml FRV‐Alkaline Solution, 10 ml naphthol AS‐BI alkaline solution; Alkaline Phosphatase kit, Sigma‐Aldrich) for 30 min at room temperature. Alkaline phosphatase activity was detected colorimetrically (red) by light microscopy.
Nuclei in cell colonies were counted by fixing in 4% paraformaldehyde in Dulbecco’s PBS (Gibco) and staining with 1 μg/ml 4,6‐diamidino 2‐phenyindiol (DAPI; Sigma). Expression of GFR α‐1, CD9, integrin α6 in cultured cells was determined immunocytochemically by incubating fixed colonies overnight with primary antibodies (1:100‐500 dilutions) against CD9 (Chemicon), GFR α‐1 (Chemicon) or integrin α6 (BD/Pharmingen, San Diego, CA, US). Primary antibodies were detected using fluorescein isothiocyanate (FITC)‐ or Cy3‐conjugated secondary antibodies (Zymed, San Francisco, CA, USA).
Flow cytometrical analysis of SSCs obtained from in vitro culture
SSCs dissociated in trypsin–EDTA and resuspended in PBS containing 2% FBS were incubated with FITC‐, APC‐ or PE‐conjugated GFR α‐1, integrin α6, integrin β1 (BD/Pharmingen) and c‐Kit (BD/Pharmingen) for 60 min at 4 °C. Stained cells were analysed by flow cytometry using a FACS IV system (Becton Dickinson, San Jose, CA, USA). Cells without antibody staining served as negative controls.
In vitro differentiation of SSCs
At P8, more than 1000 SSC clumps and underlying somatic cells were trypsinized to yield a single‐cell suspension. Dissociated cells were resuspended and then encapsulated with sodium alginate. Alginate‐encapsulated cell aggregates were transferred to 1.0 ml of Germ Cell Differentiation Medium (7) comprising HEPES‐buffered DMEM/F‐12 supplemented with 10 μg/ml ITS solution, 10−4 mol/l vitamin C (Sigma), 10 μg/ml vitamin E (Sigma), 3.3 × 10−7 mol/l retinoic acid (Sigma), 3.3 × 10−7 mol/l retinol (Sigma), 1 mmol/l pyruvate (Sigma), 2.5 × 10−5 IU recombinant human FSH (Gonal‐F; Serono, Geneva, Switzerland), 10−7 mol/l testosterone (Sigma), 1× antibiotic–antimycotic solution (ABAM, containing penicillin, streptomycin and amphotericin B; Gibco) and 10% bovine calf serum (Hyclone, Logan, UT, USA) in a 24‐well dish, and cultured for up to 6 weeks at 32 °C in a humidified atmosphere of 5% CO2 in air. Medium was replaced on alternate days. After 6 weeks in vitro differentiation, differentiated germ cells were analysed by immunocytochemistry and RT‐PCR. For immunocytochemistry, SCP3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), intra‐acrosomal protein (IAP) (25) (Abcam, Cambridge, UK) and tetramethylrhodamine isothiocyanate (TRITC)‐conjugated peanut agglutinin (26) (Sigma) were used for differentiated germ cells. SCP3 and IAP were detected using Cy3‐conjugated secondary antibodies or fluorescein isothiocyanate (FITC) (Zymed).
DNA content analysis
The in vitro differentiated SSCs were dissociated in trypsin–EDTA and resuspended in PBS to release single cells; these were washed in PBS and centrifuged at 1000 rpm for 10 min. Cells were resuspended in PBS, filtered through 80 μm nylon mesh, fixed in cold 70% ethanol and kept at 4 °C until further analysis. For DNA content assay, 1 × 106 cells were washed twice in PBS and incubated in 500 μl of 0.2% pepsin for 10 min at 37 °C. After centrifugation, cells were stained in a solution containing 25 μg/ml propidium iodide (PI) (Sigma), 40 μg/ml RNase (Invitrogen) and 0.3% Tween‐20, in PBS at room temperature for 20 min. Stained cells were analysed using the FACS IV system (Becton Dickinson).
Statistical analysis
Unless otherwise specified, each experiment was carried out using at least three replicates. Data are expressed as mean ± SEM. Statistical significance of differences among treated groups were evaluated by one‐way analysis of variance (ANOVA) using a log‐linear model in the Statistical Analysis System (SAS, Cary, NC, USA). Values of P < 0.05 were considered to be statistically significant.
Results
Isolation and propagation of SSCs from OA and NOA patients
Eighteen OA patients and 19 NOA patients were enrolled in the present study; of the NOA patients, 11 had Sertoli cell‐only syndrome (SCOS) and eight had maturation arrest (MA) (Fig. 1A, Table 1). Testicular tissue from OA patients evaluated before in vitro culture showed positive expression of SSC markers (Oct‐4, integrin α6 and integrin β1 mRNA) and differentiating germ cell markers (c‐Kit mRNA for spermatocytes; TP‐1 mRNA for spermatids). Testicular tissue from NOA patients showed positive or negative expression of differentiating germ‐cell markers (c‐Kit mRNA and TP‐1 mRNA) (Fig. 1D).
Figure 1.
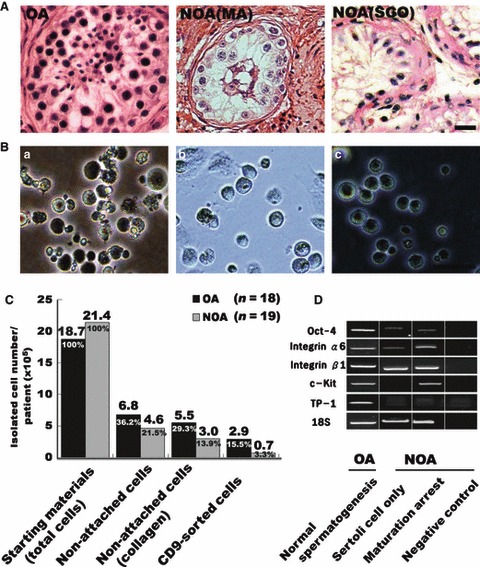
Isolation of spermatogonial stem cells (SSCs) from testicular tissues of obstructive azoospermic (OA) and non‐obstructive azoospermic (NOA) patients. (A) Seminiferous tubules of OA (normal spermatogenesis), NOA (maturation arrest at spermatocyte stage; MA) and NOA (Sertoli cell only; SOC) patients. (B) Testicular cell selection procedure. (a) Primary culture in non‐coated dishes. (b) Non‐attached cells, transferred to collagen‐coated dishes for 4 h. (c) CD9‐positive cells separated using MACS. (C) Separated cell numbers in each step per patient (×105). Testicular biopsy weight per patient; OA = 71 mg and NOA = 81 mg (D). Expression pattern of germ‐cell markers in testicular tissues from OA and NOA, before start of experiment. Oct‐4 (general stem cells); integrin α6 and integrin β1 (spermatogonia); c‐Kit (differentiating spermatogonia and spermatocytes); transition protein (TP)‐1 (spermatids). Bars = 50 μm.
Table 1.
Isolation and viability of cultured spermatogonial stem cells (SSCs)
| Classification | SSC isolation | Cell viability P1 (Day 3) | Cell viability P3 (Day 40) | |||
|---|---|---|---|---|---|---|
| Patients (n=) | n (%) | Trypan blue | TUNEL | Trypan blue | TUNEL | |
| OA | 18 | 15 (83.3) | 93.98 (±1.05) | 7.31 (±1.78) | 91.91 (±2.20) | 5.14 (±1.68) |
| NOA (SCO) | 11 | 4 (36.3) | 95.15 (±1.09) | 6.11 (±1.89) | 94.10 (±1.69) | 6.03 (±1.53) |
| (MA) | 8 | 4 (50.5) | ||||
| Sum | 37 | 23 (62.2) | 94.56 (±1.07) | 6.71 (±1.83) | 93.00 (±1.94) | 5.58 (±1.60) |
Values are mean (±SD). OA, obstructive azoospermia; NOA, non‐obstructive azoospermia; P, passage.
Testicular tissues obtained from OA and NOA patients were grown in non‐coated dishes in selection medium containing bFGF, hLIF and FBS (Fig. 1Ba). After culturing in vitro for 2–4 days, most testicular somatic cells were adherent, but germ cells containing SSCs were not attached.
To increase purity of SSCs, we collected non‐attached cells, transferred them to collagen‐coated dishes in proliferation medium, then incubated them for 4 h (Fig. 1Bb). Of those in collagen‐coated dishes, only a subset of non‐attached cells was strongly positive for stem‐cell marker, alkaline phosphatase (Fig.S1Aa). Also, non‐attached populations were strongly positive for SSC markers GFR α‐1 and CD9 (Fig.S1Ab, B). Non‐attached cells were re‐sorted by MACS using anti‐CD9 antibody (Fig. 1Bc), separated into CD9‐positive and ‐negative cells and then cultured for 2–4 weeks on laminin‐coated dishes in proliferation medium supplemented with GDNF. MACS‐separated CD9‐positive cells (15.3%, 3.3%) (Fig. 1C) from both OA and NOA patients were maintained in culture and proliferated well, ultimately forming small clumps (>10 cells) by attaching to differentiating cells in the culture dish (Fig. 2Aa–j). In contrast, CD9‐negative cells failed to form clumps during culture (Fig. S1C). Cells were passaged by transferring trypsin‐dissociated cells to new six‐well culture dishes in fresh proliferation medium containing GDNF. Some cells attached to the plate after incubation of 2–3 days, but a significant number of germ cells proliferated, re‐forming floating clumps that subsequently became loosely attached to autologous (somatic) feeder cells. Passaging was repeated every 2 weeks. SSCs obtained during early passages could be maintained, and continued proliferating for more than 12 passages (>26 weeks). Using this method, we successfully isolated SSCs and maintained proliferating SSC cultures from more than 83.3% (15/18) of OA patients and 42.1% (8/19) of NOA patients (Table 1). Among the 19 NOA patients, only four of the 11 (36.3%) SCOS patients and four of the eight (50%) MA patients had isolated SSCs.
Figure 2.
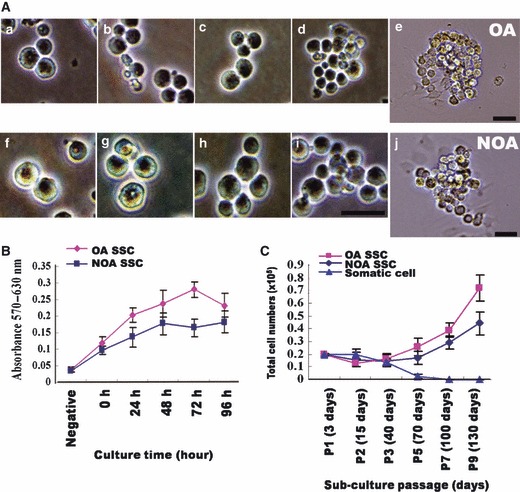
Photomicrograph of spermatogonial stem cells (SSCs) under exogenous feeder‐free culture conditions and their proliferation. (A) In vitro propagation of spermatogonial stem cells (SSCs). SSCs from obstructive azoospermia (OA, a–e) and non‐obstructive azoospermia (NOA, f–j.) (B) MTT assay of proliferating SSCs in in vitro culture. MTT assays to evaluate proliferation of P3 SSCs after culturing in vitro for 5 days. (C) Total cell number SSCs during long‐term in vitro culture. Proliferation of SSCs at each passage of long‐term culture (3∼130 days) Bar = 50 μm.
Comparison of viability, apoptosis and proliferation of SSCs obtained from OA and NOA patients
To analyse viability and apoptosis of SSCs obtained from OA and NOA patients, we performed trypan blue and TUNEL staining. In NOA patients, 95.15 ± 1.09% and 94.10 ± 1.69% of cells were live (not trypan blue‐stained) at P0 and P3, respectively; percentages of live OA patient‐derived cells were not significantly different from those derived from NOA patients at P0 (93.98 ± 1.05%) or P3 (91.91 ± 2.20%). Similar results were obtained using TUNEL assays, which showed that 7.31 ± 1.78% and 5.14 ± 1.68% of OA patient‐derived cells were apoptotic at P0 and P3, respectively; corresponding values for NOA patient‐derived cells were 6.11 ± 1.89% and 6.03 ± 1.53% (Table 1).
To determine whether cultured cells proliferated in medium containing GDNF, we first used MTT assays to evaluate proliferation of P3 SSCs after culture in vitro for 5 days. This analysis showed that the SSCs obtained from both OA and NOA patients were indeed proliferative, and also showed that SSCs obtained from OA patients had greater proliferative activity than those from NOA patients (P < 0.05, Fig. 2B). We also monitored proliferation of SSCs at each passage during long‐term culture. No proliferation was observed until the third or fourth passage, but after P5, proliferation level accelerated. Results from long‐term culture reconfirmed that SSCs obtained from OA patients had higher proliferative activity than those from NOA patients (P < 0.05, Fig. 2C). Despite having relatively lower proliferative activity, NOA patient‐derived SSCs could be maintained, and proliferated for more than 6 months.
We confirmed expression of germ cell markers (integrin α6, c‐Kit, TP‐1 mRNA) from those SSCs collected from both groups at three different passages (P2, P5 and P7) by real‐time RT‐PCR. Integrin α6 mRNA gradually increased during culture; respectively, c‐Kit mRNA gradually decreased. The marker (TP‐1) of differentiated germ cells had low expression level compared to MACS‐negative cells (Fig. 3A). We also analysed telomerase activity during culture. In contrast to results from our previous culture system, in which telomerase activity of SSCs was very high in P2 but slightly decreased before P7, the present culture system supported high levels of telomerase activity [similar to those in human embryonic stem cells (ESCs) and HeLa cells] after P7 in both OA patient‐ and NOA patient‐derived SSCs (Fig. 3B).
Figure 3.
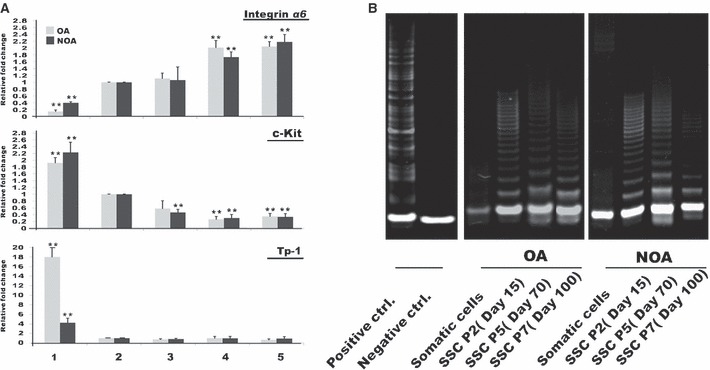
Real‐time RT‐PCR and telomerase assay of spermatogonial stem cell (SSC) from OA and NOA over long‐term culture. (A) Expression level of germ‐cell markers in spermatogonial stem cells (SSC) from OA and NOA during long‐term culture. (1) Non‐selected cells (MACS‐negative), (2) selected cells (MACS‐positive), (3) SSC at P2 (day 15), (4) SSC at P5 (day 70), (5) SSC at P7 (day 100). Integrin α6 (spermatogonia); c‐Kit (differentiating spermatogonia and spermatocytes); transition protein (TP)‐1 (spermatids). (B) Telomerase activity in spermatogonial stem cells (SSC) during long‐term culture. Left panel shows positive and negative controls for telomerase activity. Human embryonic stem cell line (CHA‐hES‐6, P59) used as positive control. Negative control – no template. Middle panel, SSCs from obstructive azoospermia (OA). Right panel, SSCs from non‐obstructive azoospermia (NOA). **Significant difference (P < 0.05).
Immunocytochemical and flow cytometrical characterization of SSCs from OA and NOA patients
Spermatogonial stem cell markers, GFR α‐1 and integrin α6, were detected in some cells of early‐passage (day 7) SSC clumps, obtained from OA and NOA patients, and GFR α‐1 and integrin α6 were strongly detected in P5 (day 70) SSC clumps (Fig. S2). Differentiating germ cell marker c‐Kit was also detected in some cells (data not shown). Frequency and intensity of positive signals did not differ between SSCs obtained from OA and NOA patients.
To quantitatively characterize changes associated with passaging under our specific culture conditions, we analysed SSCs collected from both groups at three different passages (P0, P3 and P8), for reactivity to specific antibodies, using flow cytometry. In SSCs derived from OA and NOA patients, primary cultured cells (P0) showed relatively low percentages of GFR α‐1‐, integrin α6‐ and integrin β1‐positive cells (54.29%, 12.63% and 37.44% in OA patients, and 45.53%, 13.54% and 19.14% in NOA patients, respectively), but high percentages of c‐Kit‐positive cells (78.48% and 85.67% in OA and NOA, respectively). Importantly, the population of cells positive for SSC‐specific markers GFR α‐1, integrin α6 and integrin β1 had increased at P3 to 79.60%, 29.31% and 56.26% in OA patient‐derived cells, and to 67.28%, 24.10% and 33.87% in NOA patient‐derived cells, respectively. These percentages were further increased at P8, at which time 89.68%, 96.04% and 88.91% of cells from OA patients and 91.54% and 82.40%, 82.47% of cells from NOA patients were GFR α‐1, integrin α6‐ and integrin β1‐positive, respectively. In contrast, percentage of differentiating germ cells (c‐Kit–positive) was reduced in both OA patient‐ and NOA patient‐derived cells, decreasing to 57.53% (OA) and 69.54% (NOA) at P3 and further decreasing to 39.16% (OA) and 56.12% (NOA) at P8 (Fig. 4).
Figure 4.
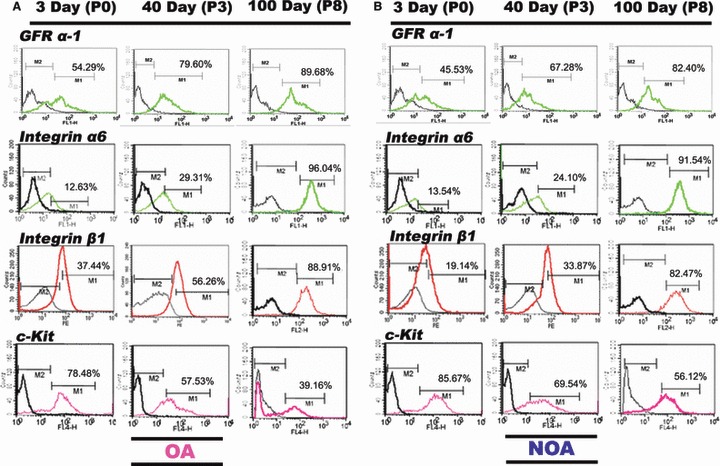
Flow cytometric analysis of germ cell‐specific markers (GFR α‐1, integrin α6, integrin β1 and c‐Kit) during long‐term culture. (A) Obstructive azoospermia (OA). (B) Non‐obstructive azoospermia (NOA); GFR α ‐1, integrin α6 and integrin β1 for spermatogonia; c‐Kit for spermatocyte (differentiated spermatogonial stem cells); green (FITC) or red (APC) or pink (PE) line, specific antibody; black line, control immunoglobulin.
Germ cell production from long‐term proliferated SSCs by in vitro spermatogenesis
To determine function of long‐term cultured SSCs, in vitro differentiation was evaluated by the calcium alginate encapsulation method reported previously (7, 21). At P8, SSC clumps and their underlying somatic cells were detached from plates by trypsin treatment, and then re‐aggregated, encapsulated and cultured for an additional 1–2 weeks. As reported previously, there was no change in integrity or dimension of extruded calcium alginate‐containing SSCs (Fig. 5A) during in vitro culture. However, when encapsulated cells were dissociated, spherical cells with sizes ranging from 7 to 20 μm were observed after 1 or 2 weeks in vitro culture (Fig. 5B). Meiotic or haploid germ cells in differentiation SSCs were determined by SCP3 immunostaining and flow cytometry. We found that meiotic‐ and haploid germ cells (DNA content; N) were present in the in vitro‐differentiated group. Population of positive cells increased during in vitro differentiation, but there were no differences in 1‐week and 2‐week culture groups. Moreover, frequency of positive signals did not differ between SSCs obtained from OA and NOA patients (Fig. 5C,D). In addition, acrosome granules of putative round spermatids with diameters of 7–10 μm provided positive signals after intra‐acrosomal protein(IAP) and TRITC‐peanut agglutinin (PNA) staining (Fig. 6A,B). Differentiated germ cell count using IAP and TRITC‐PNA staining revealed that the population of positive cells increased in size during in vitro differentiation (Fig. 6C,D). By real‐time RT‐PCR analysis, percentage of integrin β1‐positive cells was lower after encapsulation and in vitro culture, whereas expression of differentiating germ cells (c‐Kit‐ TH2B‐, SCP3‐positive and TP‐1) was higher (Fig. S3) in cells from both patient groups.
Figure 5.
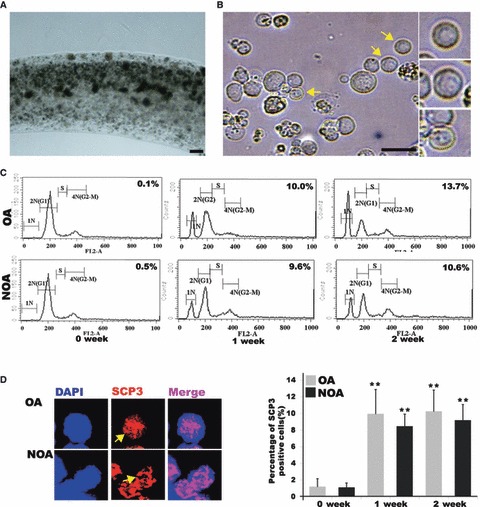
In vitro differentiation into sperm lineage cells. (A) Encapsulation with calcium alginate (B) Putative differentiated germ cells with diameter of 7–20 μm. Putative round spermatids (in small squares); yellow arrows indicate putative round spermatids (C) DNA content of in vitro differentiated SSCs from obstructive azoospermia (OA) and non‐obstructive azoospermia (NOA). 1N represents haploid germ cells, 2N represents diploid cells and 4N represents tetraploid cells. S‐phase represents synthesizing DNA. (D) SCP3 staining in putative meiotic germ cells from in vitro differentiated SSCs. Staining (left figure) and percentage (right figure) of Cy3‐SCP3‐stained cells during in vitro differentiation of SSCs. OA, obstructive azoospermic; NOA, non‐obstructive azoospermia; yellow arrows indicate putative meiotic germ cells. Bar = 50 μm. **Significantly different (P < 0.05).
Figure 6.
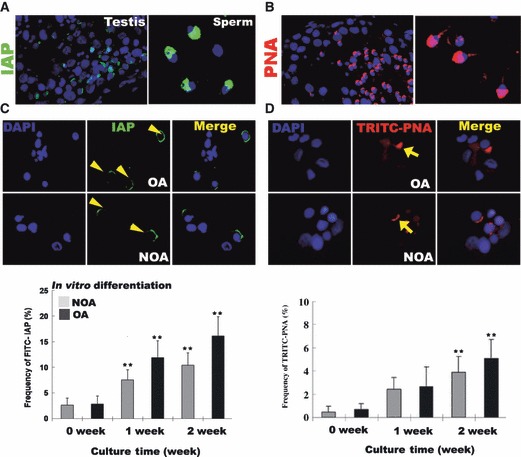
In vitro differentiation into sperm lineage cells. (A) Localization of intra‐acrosomal protein (IAP) in human testis and mature spermatozoa. (B) Localization of peanut agglutinin (acrosomal granule‐specific protein) in human testis and mature spermatozoa. (C) IAP staining in putative round spermatids. Staining (upper figure) and frequency (lower figure) of FITC‐IAP stained cells during in vitro differentiation of SSCs. (D) PNA staining in putative round spermatids. Staining (upper figure) and frequency (lower figure) of TRITC‐PNA‐stained cells during in vitro differentiation of SSCs. OA, obstructive azoospermic; NOA, non‐obstructive azoospermia; yellow arrow head and arrows, indicate positive signal for IAP and PNA. **Significant difference (P < 0.05).
Discussion
In our previous studies, we successfully isolated human testicular SSC‐like cells from some NOA patients and confirmed identity and developmental potential of these cells by chromosome haploidy and embryo production. These cells proliferated slowly as colonies on autologous somatic cells, but could not be maintained for more than five passages. In addition, the number of spermatids produced in vitro from the few SSC‐like cells was extremely small (27, 28, 29), and no progression into spermiogenesis was seen (21). We attributed this limited success to culture conditions that were suboptimal for SSC proliferation in vitro, as well as to possible contributions from competition with somatic cells and adverse environmental influences of NOA patient‐derived material. In the present study, we sought to overcome these limitations by first using cell‐sorting techniques and our standard culture system to SSCs obtained from OA patients. We then developed a collection system and a simple long‐term culture protocol capable of yielding proliferating, high‐purity SSCs from OA and NOA patients.
Germ cells and somatic cells of dissociated testicular tissue come in various shapes and sizes. To obtain a pure population of SSCs, various workers have used cell‐sorting methods based on extracellular matrix (ECM)‐ and surface‐specific antigens. Conrad et al. (22) described a three‐step method for obtaining a pure population of human SSCs. In the first step, single‐cell suspensions of dissociated cells were cultured for 4 days in uncoated dishes in knockout culture medium with GDNF, or in LIF‐supplemented ESC medium. In the second step, unattached cells were collected and pre‐selected with MACS using integrin α6. Finally, an ECM selection procedure involving collagen and laminin was used to obtain a high‐purity SSC population. Similarly, Hamra et al. (30) reported that combination of collagen non‐binding and laminin binding selected for an SSC‐enriched cell population. As only few human SSCs are obtained using the collagen non‐binding/laminin‐binding system, in the present study we have modified the ECM selection procedure. Accordingly, we first collected non‐attached cells in selection medium containing 20% FBS, and then pre‐selected for SSCs by reselecting collagen non‐binding cells. Finally, these cells were sorted by MACS using an antibody against CD9, which is a tetraspanin transmembrane protein that is highly expressed in undifferentiated cells such as SSCs and ESCs (31, 32). When sorted CD9‐positive cells were cultured in proliferation medium, single cells formed small cell clumps that expressed SSC markers within 2 weeks, whereas CD‐9‐negative cells did not.
In our system, isolated SSCs from OA and NOA patients expressed well‐characterized SSC markers, and exhibited high‐survival and low‐apoptosis rates. Interestingly, there was no difference in these parameters between the two types of azoospermic patients (Fig. 2A, Table 1). SSCs from OA and NOA patients shared several other characteristics during long‐term culture; the one exception was proliferative activity, which was lower in SSCs derived from NOA patients than in those derived from OA patients (Fig. 2B,C). Moreover, in contrast to our previous reports (21), we found that SSCs derived from OA and NOA patients using the present method proliferated and maintained their characteristics for more than 12 passages (>6 months) in vitro. We attribute much of the prolongation of NOA SSCs to improvements in the culture system. In fact, using a sequential culture system, in which re‐collected cells are propagated in proliferation medium with GDNF after isolation from the primary culture, may increase proliferation of these human SSCs from testicular cells of NOA patients, extending the culture period by more than five passages (33). As shown in the present study, introduction of a culture system that improved collection of SSCs released the spermatogenesis arrest of SSCs in some NOA patients, and could increase likelihood of successfully treating infertility. In fact, SSCs were isolated and could be proliferated from 42.1% of NOA patients, including those with SCOS and MA (Table 1). Moreover, we confirmed the results of our previous study, namely that these NOA‐patient‐derived SSCs could produce round spermatids in vitro, and we had developed a culture system to increase their number. Further research focusing on in vitro spermiogenesis is needed to obtain functional spermatids or spermatozoa.
Analysis of marker expression revealed presence of various types of SSC and germ cells in SSC clumps during in vitro culture. Expression levels of SSC‐specific markers, GFR α‐1, integrin α6 and integrin β1, gradually increased over time and were particularly high after P8. In contrast, differentiating germ‐cell marker c‐Kit was highly expressed early in culture, but expression levels gradually decreased by later passages. Also, testicular somatic cells remained in cells sorted by ECM and MACS and attached to culture dishes during culture, and finally these cells were observed to gradually decrease after passaging. This result could be estimated by the gradual increase in percentages of GFRα‐1‐ and integrin α6‐positive cells in the clumps after passaging (Fig. S2). We have suggested that some germ cells of SSCs could be differentiated into somatic feeder cells, as feeder cells reformed during culture, even at later passages.
Immunocytochemical and flow cytometric data support the hypothesis that purity of SSCs could be increased using culture conditions that include GDNF (10, 34). Also, interestingly, some pluripotent stem‐cell markers Nanog and SSEA‐4 exhibited limited expression (5–20%) in clumps and that was maintained over long‐term culture (Data not shown). In a co‐localization experiment using SSEA‐4 (marker of pluripotent stem cells) and GFR alpha‐1 (marker of SSCs) in our cultured SSC colonies from P5, we found that these two surface antigens were not co‐localized in most colonies which means that different types of cell (ES‐like and SSC) co‐existed together (Fig. S4). At P8, around 80–90% integrin β1, α6‐positive cells did not form teratomas or other tumours after TP. However, we also found that a small fraction (5–20%) of SSEA‐4‐positive cells appeared during long‐term culture – these sometimes formed small teratomas (1 time/5 times) after collection by MACS and TP (data not shown). This finding was somewhat similar to the report of Conrad et al. (22); the difference in our culture system was that there was no increase in their proportion; also, we sustained them at less than 20%. Hence, we believe that GFR α‐1‐, integrin β1‐, and α6‐positive cells in our culture system were SSCs maintaining germ cell characteristics. These marker expressions in human SSCs were not well matched to those of rodents, but are similar to adult primate testicular cells (35). Although there has been in vivo characterization, formed germ cell colonies after transplantation into seminiferous tubules of immunodeficient mice was not performed in the present study. However, expression of post‐meiotic genes and ploidy analysis have shown maintenance of germ‐line potential of SSCs during long‐term culture (5, 6).
In conclusion, results of the present study indicate that SSCs present in the testes of NOA patients can be isolated and made to propagate in vitro using the highly efficient culture system described here. Furthermore, these derived SSCs were shown to produce differentiating germ cells with developmental potential. This system could be used to diagnose presence of germ cells. When combined with human‐assisted reproductive technology, this could also be useful in treatment of NOA patients.
Supporting information
Figure S1 Characterization of isolating cells during testicular tissues selection procedure. (A) Collagen selection procedure. (a) High levels of alkaline phosphatase activity in slightly attached cells (collagen non‐binding cells) obtained from OA/NOA. (b) Localization of GFR α‐1 in collagen‐isolated testicular cells (collagen non‐binding cells) obtained from OA/NOA. (B) Localization of CD9 in collagen‐isolated testicular cells (collagen non‐binding cells) obtained from OA/NOA. (C) MACS‐separated CD9‐positive cells form clumps (left figure). MACS‐separated CD9‐negative cells do not form clumps (right figure). Bar = 50 μm.
Figure S2 Immunocytochemical analysis of markers for spermatogonial stem cells. The positive signal of GFR α‐1, integrin α6 increased in 70‐day cultured cell clumps compared with 7‐day cultured clump. OA, obstructive azoospermia; NOA, non‐obstructive azoospermia.
Figure S3 Expression of stage‐specific genes (integrin β1, c‐Kit, TH2B, SCP3 and TP‐1) during in vitro differentiation of SSCs obtained from obstructive azoospermia (OA) and non‐obstructive azoospermia (NOA) by real‐time RT‐PCR. Integrin β1 (spermatogonia); c‐Kit and TH2B (differentiating spermatogonia and spermatocytes); SCP3 and TP‐1 (meiotic spermatocytes and spermatids). **Significantly different (P < 0.05).
Figure S4 Co‐localization of specific marker for pluripotent stem cells (SSEA‐4, red colour) and spermatogonial stem cells (GFR α‐1, green colour) in the cells of cultured colony.
Figure S5 Expression of meiotic marker (SCP3, red colour) in testicular tissue obtained from obstructive azoospermia. White circles indicate nuclei of spermatocytes.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This study was supported by a grant from the Stem Cell Research Program (2009‐0084074) of the Ministry of Education, Science, and Technology and National Research Foundation of Korea, Republic of Korea.
References
- 1. Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL (2006) Identifying genes important for spermatogonial stem cell self‐renewal and survival. Proc. Natl. Acad. Sci. USA 103, 9524–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tegelenbosch RA, De Rooij DG (1993) A quantitative study of spermatogonial multiplication and stem cell renewal in the C3H/101 F1 hybrid mouse. Mutat. Res. 290, 193–200. [DOI] [PubMed] [Google Scholar]
- 3. Parvinen M, Wright WW, Phillips DM, Mather JP, Musto NA, Bardin CW (1983) Spermatogenesis in vitro: completion of meiosis and early spermiogenesis. Endocrinology 112, 1150–1152. [DOI] [PubMed] [Google Scholar]
- 4. Moore HD, Akhondi MA (1996) In vitro maturation of mammalian spermatozoa. Rev. Reprod. 1, 54–60. [DOI] [PubMed] [Google Scholar]
- 5. Staub C, Hue D, Nicolle JC, Perrard‐Sapori MH, Segretain D, Durand P (2000) The whole meiotic process can occur in vitro in untransformed rat spermatogenic cells. Exp. Cell Res. 260, 85–95. [DOI] [PubMed] [Google Scholar]
- 6. Tesarik J, Mendoza C, Greco E (2000) In‐vitro maturation of immature human male germ cells. Mol. Cell. Endocrinol. 166, 45–50. [DOI] [PubMed] [Google Scholar]
- 7. Lee DR, Kaproth MT, Parks JE (2001) In vitro production of haploid germ cells from fresh or frozen‐thawed testicular cells of neonatal bulls. Biol. Reprod. 65, 873–878. [DOI] [PubMed] [Google Scholar]
- 8. Wolgemuth DJ, Laurion E, Lele KM (2002) Regulation of the mitotic and meiotic cell cycles in the male germ line. Recent Prog. Horm. Res. 57, 75–101. [DOI] [PubMed] [Google Scholar]
- 9. Feng LX, Chen Y, Dettin L, Pera RA, Herr JC, Goldberg E et al. (2002) Generation and in vitro differentiation of a spermatogonial cell line. Science 297, 392–395. [DOI] [PubMed] [Google Scholar]
- 10. Kanatsu‐Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S et al. (2003) Long‐term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 69, 612–616. [DOI] [PubMed] [Google Scholar]
- 11. Kanatsu‐Shinohara M, Inoue K, Lee J, Yoshimoto M (2004) Generation of pluripotent stem cells from neonatal mouse testis. Cell 119, 1001–1012. [DOI] [PubMed] [Google Scholar]
- 12. Shinohara T, Inoue K, Ogonuki N, Kanatsu‐Shinohara M, Miki H, Nakata K et al. (2002) Birth of offspring following transplantation of cryopreserved immature testicular pieces and in‐vitro microinsemination. Hum. Reprod. 17, 3039–3045. [DOI] [PubMed] [Google Scholar]
- 13. Ryu BY, Kubota H, Avarbock MR, Brinster RL (2005) Conservation of spermatogonial stem cell self‐renewal signaling between mouse and rat. Proc. Natl. Acad. Sci. USA 102, 14302–14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dadoune JP (2007) New insights into male gametogenesis: what about the spermatogonial stem cell niche? Folia Histochem. Cytobiol. 45, 141–147. [PubMed] [Google Scholar]
- 15. Rassoulzadegan M, Paquis‐Flucklinger V, Bertino B, Sage J, Jasin M, Miyagawa K et al. (1993) Transmeiotic differentiation of male germ cells in culture. Cell 75, 997–1006. [DOI] [PubMed] [Google Scholar]
- 16. Tanaka A, Nagayoshi M, Awata S, Mawatari Y, Tanaka I, Kusunoki H (2003) Completion of meiosis in human primary spermatocytes through in vitro coculture with Vero cells. Fertil. Steril. 79, 795–801. [DOI] [PubMed] [Google Scholar]
- 17. Tesarik J, Balaban B, Isiklar A, Alatas C, Urman B, Aksoy S et al. (2000) In‐vitro spermatogenesis resumption in men with maturation arrest: relationship with in‐vivo blocking stage and serum FSH. Hum. Reprod. 15, 1350–1354. [DOI] [PubMed] [Google Scholar]
- 18. Kierszenbaum AL, Tres LL (2002) Bypassing natural sperm selection during fertilization: the azh mutant offspring experience and the alternative of spermiogenesis in vitro. Mol. Cell. Endocrinol. 187, 133–138. [DOI] [PubMed] [Google Scholar]
- 19. Marh J, Tres LL, Yamazaki Y, Yanagimachi R, Kierszenbaum AL (2003) Mouse round spermatids developed in vitro from preexisting spermatocytes can produce normal offspring by nuclear injection into in vivo‐developed mature oocytes. Biol. Reprod. 69, 169–176. [DOI] [PubMed] [Google Scholar]
- 20. Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, Donovan PJ et al. (1998) Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc. Natl. Acad. Sci. USA 95, 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee DR, Kim KS, Yang YH, Oh HS, Lee SH, Chung TG et al. (2006) Isolation of male germ stem cell‐like cells from testicular tissue of non‐obstructive azoospermic patients and differentiation into haploid male germ cells in vitro. Hum. Reprod. 21, 471–476. [DOI] [PubMed] [Google Scholar]
- 22. Conrad S, Renninger M, Hennenlotter J, Wiesner T, Just L, Bonin M et al. (2008) Generation of pluripotent stem cells from adult human testis. Nature 456, 344–439. [DOI] [PubMed] [Google Scholar]
- 23. Robinson LL, Gaskell TL, Saunders PT, Anderson RA (2001) Germ cell specific expression of c‐kit in the human fetal gonad. Mol. Hum. Reprod. 7, 845–852. [DOI] [PubMed] [Google Scholar]
- 24. Huang Z, Fasco MJ, Kaminsky LS (1996) Optimization of Dnase I removal of contaminating DNA from RNA for use in quantitative RNA‐PCR. Biotechniques 20, 1012–1020. [DOI] [PubMed] [Google Scholar]
- 25. Chládek D, Pĕknicová J, Capková J, Geussová G, Teplá O, Madar J (2000) Use of human sperm protein monoclonal antibodies in the diagnosis of sperm pathology and selection of a suitable assisted reproduction method for fertilization. Ceska Gynekol. 65, 28–32. [PubMed] [Google Scholar]
- 26. Kurohmaru M, Kanai Y, Hayashi Y (1991) Lectin‐binding patterns in the spermatogenic cells of the shiba goat testis. J. Vet. Med. Sci. 53, 893–897. [DOI] [PubMed] [Google Scholar]
- 27. Tesarik J, Mendoza C, Testart J (1995) Viable embryos from injection of round spermatids into oocytes. N. Engl. J. Med. 333, 525. [DOI] [PubMed] [Google Scholar]
- 28. Tesarik J, Bahceci M, Ozcan C, Greco E, Mendoza C (1999) Restoration of fertility by in‐vitro spermatogenesis. Lancet 353, 555–556. [DOI] [PubMed] [Google Scholar]
- 29. Tesarik J, Guido M, Mendoza C, Greco E (1998) Human spermatogenesis in vitro: respective effects of follicle‐stimulating hormone and testosterone on meiosis, spermiogenesis, and Sertoli cell apoptosis. Clin. Endocrinol. Metab. 83, 4467–4473. [DOI] [PubMed] [Google Scholar]
- 30. Hamra FK, Chapman KM, Nguyen DM, Williams‐Stephens AA, Hammer RE, Garbers DL (2005) Self renewal, expansion, and transfection of rat spermatogonial stem cells in culture. Proc. Natl. Acad. Sci. USA 102, 17430–17435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oka M, Tagoku K, Russell TL, Nakano Y, Hamazaki T, Meyer EM et al. (2002) CD9 is associated with leukemia inhibitory factor‐mediated maintenance of embryonic stem cells. Mol. Biol. Cell 13, 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanatsu‐Shinohara M, Toyokuni S, Shinohara T (2004) CD9 is a surface marker on mouse and rat male germline stem cells. Biol. Reprod. 70, 70–75. [DOI] [PubMed] [Google Scholar]
- 33. Lim JJ, Lee DR, Kim HJ, Song SH, Yoon TK, Kim KS (2007) Proliferation and differentiation of male germ‐line stem cells from testis of non‐obstructive azoospermia using sequential culture systems. Fertil. Steril. 88(Suppl. 1), 882–882. [Google Scholar]
- 34. Kubota H, Avarbock MR, Brinster RL (2004) Growth factors essential for self‐renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 101, 16489–16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chad BM, Jason P, Thomas R, Michael P, Jane P, Jessie K et al. (2009) Phenotypic and molecular characterization of spermatogonial stem cells in adult primate testes. Hum. Reprod. 24, 1480–1491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Characterization of isolating cells during testicular tissues selection procedure. (A) Collagen selection procedure. (a) High levels of alkaline phosphatase activity in slightly attached cells (collagen non‐binding cells) obtained from OA/NOA. (b) Localization of GFR α‐1 in collagen‐isolated testicular cells (collagen non‐binding cells) obtained from OA/NOA. (B) Localization of CD9 in collagen‐isolated testicular cells (collagen non‐binding cells) obtained from OA/NOA. (C) MACS‐separated CD9‐positive cells form clumps (left figure). MACS‐separated CD9‐negative cells do not form clumps (right figure). Bar = 50 μm.
Figure S2 Immunocytochemical analysis of markers for spermatogonial stem cells. The positive signal of GFR α‐1, integrin α6 increased in 70‐day cultured cell clumps compared with 7‐day cultured clump. OA, obstructive azoospermia; NOA, non‐obstructive azoospermia.
Figure S3 Expression of stage‐specific genes (integrin β1, c‐Kit, TH2B, SCP3 and TP‐1) during in vitro differentiation of SSCs obtained from obstructive azoospermia (OA) and non‐obstructive azoospermia (NOA) by real‐time RT‐PCR. Integrin β1 (spermatogonia); c‐Kit and TH2B (differentiating spermatogonia and spermatocytes); SCP3 and TP‐1 (meiotic spermatocytes and spermatids). **Significantly different (P < 0.05).
Figure S4 Co‐localization of specific marker for pluripotent stem cells (SSEA‐4, red colour) and spermatogonial stem cells (GFR α‐1, green colour) in the cells of cultured colony.
Figure S5 Expression of meiotic marker (SCP3, red colour) in testicular tissue obtained from obstructive azoospermia. White circles indicate nuclei of spermatocytes.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item