

Abstract
Objectives
microRNAs (miRNAs), are non‐coding RNAs that regulate gene expression, and are involved in tumour development. The aim of this study was to investigate microRNA‐497 (miR‐497) expression and its role in development of colorectal cancer (CRC).
Materials and methods
RT‐PCR was performed to detect expression of miR‐497 in CRC cell lines (HCT8, LOVO, Ls‐174, HCT116 and HT29) and in clinical cancer specimens. To further understand its role, we restored expression of miR‐497 in the HCT116 cell line by transfection with miR‐497 mimics or inhibitors. Effects of miR‐497 on cell proliferation, migration and invasion of targets were also determined both in vitro and in vivo.
Results
miR‐497 expression decreased in 34 CRC tissues compared to non‐tumour tissues and in tumour cell lines. Overexpression of miR‐497 did not inhibit cancer cell growth but suppressed metastasis and invasion both in vitro and in vivo. Vascular endothelial growth factor‐A (VEGF‐A) was confirmed to be a target of miR‐497. Furthermore, we found overexpression of miR‐497 altered expression of key molecules of the VEGF‐A/ERK/MMP‐9 signalling pathway.
Conclusions
Thus our results provide evidence that miR‐497 might function as a metastasis suppressor in CRC. Targeting miR‐497 may provide a strategy for blocking its metastasis.
Introduction
CRC is one of the most common causes of cancer death worldwide 1. Approximately half the patients with colorectal cancer (CRC) develop metastases during the course of the disease 2. Formation of metastases is a multistep process in which malignant cells disseminate from the primary tumour to colonize distant organs. This is a highly inefficient and complex process, which involves initial tumour cell invasion of the microenvironment, migration into the bloodstream, survival during migration and finally extravasation into distant organs. Subsequent steps, including proliferation, induction of angiogenesis and evasion of apoptotic death 3 are crucial for colonization of the secondary site.
Although invasion and migration have been acknowledged as the most lethal attributes of solid tumours, there is limited understanding of the underlying molecular mechanisms of these processes, and development of therapies targeting invasion and metastasis is slow. For these reasons, recent research efforts have been focused on novel therapeutic approaches to address cancer cell metastasis and invasion 4, 5. More specifically, oligonucleotide therapies using small interfering RNAs (siRNAs), short hairpin RNAs (shRNAs), RNA aptamers and ribozymes have received considerable attention because of their potential to allow for specific targeted delivery of anti‐tumour drugs, without significant toxicity or other systemic side effects 6, 7, 8, 9. In this study, we focused on miRNA as a potential therapy for CRC.
miRNAs are small, non‐coding RNAs that regulate gene expression by inducing translational inhibition or direct degradation of target messenger RNAs (mRNAs), through base pairing, to partially complementary sites 10. Moreover, it has been shown that global miRNA expression profiles and expression profiles of specific miRNAs correlate with disease prognosis and clinical outcomes in cancer 11. miR‐497 is one member of the miR‐15/107 group, which is increasingly appreciated as serving key functions in humans. The miR‐15/107 group regulates gene expression involved in cell division, metabolism, stress response and angiogenesis, which are also related to cancer 12, 13, 14.
However, the role of miR‐497 in human malignancy is not yet clear. In this study, we confirmed the relationship between miR‐497, a well‐known tumour suppressor, and one of its targets, vascular endothelial growth factor‐A (VEGF‐A). Also, we provide evidence for inhibition of CRC cell invasion and metastasis by miR‐497 and the role of the VEGF‐A/ERK/MMP‐9 signalling pathway in driving CRC metastasis.
Materials and methods
Reagents
All antibodies (anti‐VEGF‐A, anti‐VEGFR2, anti‐VEGFR3, anti‐MMP‐2, anti‐MMP‐9, anti‐β‐actin, anti‐ERK and anti‐phospho‐ERk) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA); they were used at concentrations recommended by the supplier. Dual luciferase reporter assay system and pGL3‐Basic reporter vector were purchased from Promega Corporation (Madison, WI, USA). miRNA mimics, inhibitor and siRNA were purchased from Biomics Biotechnologies (Nantong, China). All other chemicals were from Sigma‐Aldrich Corporation (St. Louis, MO, USA) unless otherwise stated.
Patient samples
Thirty‐four human colorectal carcinoma samples and corresponding non‐malignant colon samples were collected during surgery at Putuo Hospital, Shanghai University of Traditional Chinese Medicine. Samples were immediately snap‐frozen in liquid nitrogen and stored at −80 °C. Use of human tissues was approved by the Institutional Review Board of our hospital.
Cell culture and plasmid construction
Human colorectal cancer cell lines used in this study were HCT116, HCT8, Ls174, LOVO and HT29. All cells were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) and cultured in Rosewell Park Memorial Institute (RPMI) 1640 medium (HCT116, HCT8, Ls174 and HT29) or F12k medium (LOVO), supplemented with 10% foetal bovine serum (FBS), penicillin (100 U/ml) and streptomycin(l00 mg/ml) (Invitrogen Corporation, Carlsbad, CA, USA) at 37 °C and 5% CO2 in a humidified incubator. Luciferase reporter plasmid driven by a 1772‐bp sequence from the human VEGF‐A promoter was amplified from HCT116 complementary DNA. Primers used were 5′‐CCCAAGCTTCTCACCAGG AAAGACTGAT‐3′ (forward) and 5′‐CATGCCATGGAACTCAAGTCCACAGCAGT‐3′ (reverse). Purified PCR product was cloned into the pGL3‐Basic vector via XhoI and HindIII digestions (Takara Bio Inc., Dalian, China). Transfections were performed using Lipofectamine™ 2000 (Invitrogen Corporation) according to the manufacturer's instructions.
RNA extraction and quantification
Total RNA was isolated from cells or tumours using Trizol (Invitrogen Corporation) according to the manufacturer's instructions. Reverse transcription was performed employng a One Step PrimeScript miRNA cDNA Synthesis Kit (Takara Bio Inc.). Real‐time PCR (RT‐PCR) was performed using SYBR1 Premix Ex TaqTMII (Takara Bio Inc.) and an iCycler thermal cycler (Bio‐Rad, Hercules, CA, USA). Primer for miR‐497 RT‐PCR was 5′‐CAGCAGCACACTGTGGTTTGT‐3′. Quantitative normalization was performed based on expression levels of U6 RNA or of GAPDH for miR or mRNA detection respectively. Relative expression levels between samples were calculated using comparative delta CT (threshold cycle number) method (2−ΔΔc) with a control sample as reference point 15.
Cell viability assays
Cells were seeded in 96‐well plates, 1 × 103 cells/well, in regular growth medium. Cell viability was measured daily for 7 days using the 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐biphenyl tetrazolium bromide (MTT) assay reagent.
Cell motility assay
For cell motility assays, 1 × 105 cells were seeded in 24‐well plates and grown for 24 h. A linear wound was made by scraping a pipette tip across the confluent cell monolayer. Cells were rinsed with PBS and grown in RPMI‐1640 medium supplemented with 10% FBS for an additional 48 h. Cell motility in terms of wound closure was measured by photographing the cell monolayer at three randomly selected fields at the time of wound creation (time 0) and at 48 h later.
Cell migration and invasion assay
Migratory and invasive potentials of cells of the lines were measured using 24‐well Transwell® inserts with a 6.5 mm polycarbonate membrane and 8.0 μm pores (Corning, New York, NY, USA). For Transwell® migration assays, 2.5 × 104 cells were plated in top chambers of wells lined with a non‐coated membrane. For invasion assays, chamber inserts were coated with 200 mg/ml of Matrigel™ (BD Biosciences, Sparks, MD, USA) and dried overnight under sterile conditions, after which cells were plated in top chambers. In both assays, cells were plated in medium containing no serum, and medium supplemented with serum was used as chemoattractant in lower chambers. After incubation at 37 °C for 24 h, top chambers were wiped with cotton wool to remove non‐migratory or non‐invasive cells. Migratory cells on undersides of membranes were fixed in 100% methanol for 10 min, air‐dried, stained using 0.1% crystal violet, and counted when viewed under a microscope (Ti‐E; Nikon, Tokyo, Japan). Each experiment was performed in duplicate.
Western blot analysis
Cell or tissue proteins were extracted and separated using SDS‐PAGE electrophoresis, and western blot analyses were performed according to standard procedures as previously described 16. Western blotting of β‐actin on the same membrane was used as a loading control.
Luciferase reporter assay
Putative wild‐type (WT) and mutated (Mut) targets of miR‐497 in the VEGF‐A 3′‐untranslated (3′‐UTR) region were cloned into pGL3‐promoter vectors. Cells (2 × 104) were co‐transfected with 500 ng of either pGL3‐VEGF‐A 3′‐UTR–WT, pGL3‐VEGF‐A 3ʹ‐UTR–Mut or pGL3‐promoter vector constructs, together with either one of the miR‐497 mimics, the miR‐497 inhibitor or miR‐67 mimics (control). Each sample was transfected with 50 ng of the pRL‐SV40 plasmid expressing Renilla luciferase to monitor transfection efficiency. Luciferase activity assay was performed 24 h after transfection employing the dual luciferase reporter assay system. Relative luciferase activity was normalized with Renilla luciferase activity.
Tumour xenografts
HCT116 cells were infected with lentiviral vector encoding firefly luciferase and miR‐497 or control sequences, under puromycin (Invitrogen Corporation) selection, and called “HCT116‐luc‐miR‐497″ and “HCT116‐luc‐vector”; pooled clones were used for further experiments. Five male athymic nude mice were housed and manipulated according to protocols approved by the Shanghai Medical Experimental Animal Care Commission. For each mouse, 5 × 106 “HCT116‐luc‐vector” and “HCT116‐luc‐miR‐497” sub‐cloned cells in 100 μl PBS were injected subcutaneously into left and right flanks respectively. Every 7 days post‐inoculation, length and width of individual tumours from each mouse were measured with callipers, and volume (mm3) of each tumour was calculated according to the following formula: volume =0.5 × length × width2 17.
In vivo metastasis assay
Male athymic nude mice were randomly divided into two groups (“HCT116‐luc‐control” and “HCT116‐luc‐miR‐497”; five mice per group). Infected cells (1 × 106) were injected intravenously into the tail vein. Tumour growth and metastasis were analysed in situ weekly after injection by luciferase imaging as previously described 18. All mice were euthanized 6 weeks after initial injection and lungs were excised to examine for possible metastases. These tissues were fixed embedded in paraffin wax, sectioned, and stained with haematoxylin and eosin (H&E). Immunohistochemistry was used to detect Ki67 expression in the lungs.
Statistical analysis
Independent Student's t‐tests and analysis of variation (ANOVA) analyses were used to compare differences between groups. Correlation between mRNA expression of miR‐497 and VEGF‐A was measured using Pearson's χ2 test, while statistical significance was determined employing the log rank test. Differences with P‐values of less than 0.05 were considered to be statistically significant. Error bars represent standard error (SE) of the mean, unless otherwise indicated.
Results
Inverse correlation of miR‐497 and VEGF‐A expression in clinical CRC samples
Expression levels of miR‐497 were examined by RT‐PCR in 34 CRC samples and their corresponding non‐malignant colorectal tissue samples. miR‐497 was found to be significantly under‐expressed in 91% (31 of 34) of the CRC cases examined (Fig. 1a,b). To explore the function of miR‐497 in CRC, we further investigated expression of VEGF‐A, a potential target of miR‐497, as predicted by the TargetScan software.
Figure 1.
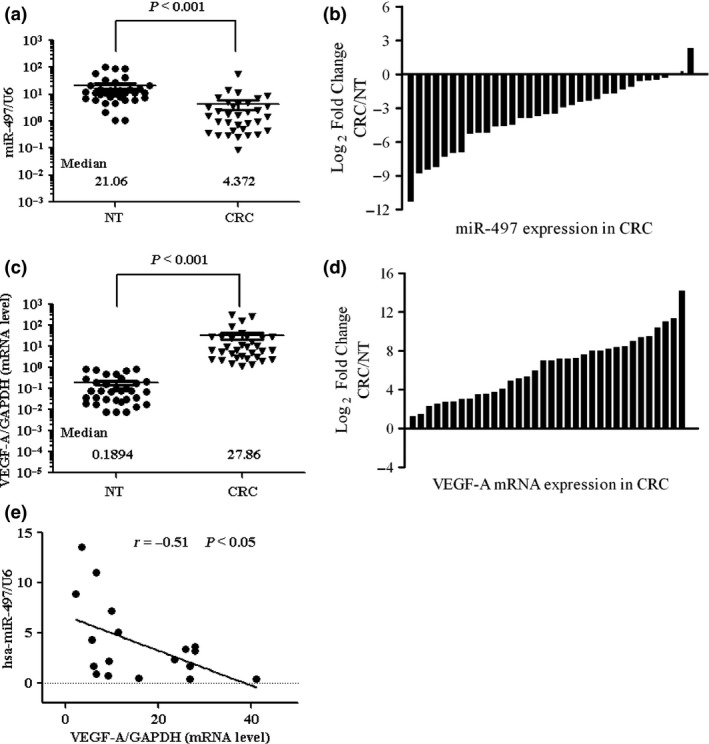
Inverse correlation of miR‐497 and VEGF ‐A expression in clinical CRC samples. miR‐497 and VEGF‐A mRNA expression in 34 pairs of primary CRC samples and the corresponding non‐malignant colorectal tissues (NT) (frozen samples). (a) miR‐497 expression levels were calculated using miR‐497/U6 expression ratio (2−Δct). Median miR‐497 expression levels in normal colorectal tissue (NT) and primary CRC were 21.06 and 4.372 respectively. miR‐497 expression was significantly lower in primary CRC samples relative to the NT tissue expression levels (P < 0.001). (b) miR‐497 expression levels in primary CRC samples relative to their corresponding NT tissue samples. Log2 fold change was considered significant overexpression or down‐regulation. miR‐497 expression was found to be reduced in 91% (31 of 34) of the CRC cases examined. (c) VEGF‐A mRNA expression levels were calculated as the VEGF‐A/GAPDH expression ratio (i.e. 2−Δct). Median VEGF‐A mRNA expression levels in normal colorectal tissue (NT) and primary CRC tissues were 0.1894 and 27.86 respectively. VEGF‐A expression was significantly up‐regulated in primary CRC samples compared to corresponding NT tissue (P < 0.001). (d) VEGF‐A mRNA expression levels in primary CRC samples relative to their corresponding NT tissue samples. Log2 fold change was considered as significant overexpression. VEGF‐A mRNA was found to be up‐regulated in 100% (34 of 34) of the CRC cases examined. (e) A correlation analysis performed using Pearson's χ2 test in 18 pairs of clinical CRC specimens suggests a negative correlation between the expression of miR‐497 and VEGF‐A.
Comparison of VEGF‐A mRNA expression levels between primary CRC tissue and corresponding non‐malignant tissue revealed an increase of 100% (34 of 34) of the CRC cases examined (Fig. 1c,d). Correlation analysis in CRC tissues revealed negative correlation between miR‐497 and VEGF‐A expression (Fig. 1e).
Expression of miR‐497 and VEGF‐A in CRC cell lines
Expression levels of miR‐497 and VEGF‐A were examined by RT‐PCR in CRC cell lines HCT8, LOVO, Ls‐174, HCT116 and HT29. Normal human colorectal tissue was used as control. miR‐497 was found to be significantly lower‐expressed in these cell lines (Fig. 2a,b). Moreover, VEGF‐A expression was significantly up‐regulated in all these five CRC cell lines compared to the control tissues (Fig. 2c,d). All these results were similar as in the clinically obtained CRC samples.
Figure 2.
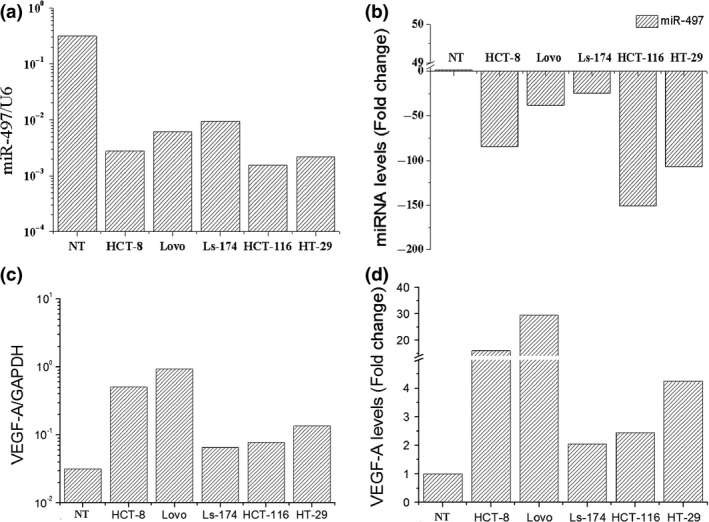
Expression of miR‐497 and VEGF ‐A in CRC cell lines. Expression levels of miR‐497 and VEGF‐A were determined using quantitative RT‐PCR and were normalized against an endogenous control (U6RNA) or GAPDH. Data were analysed using a ΔΔct approach and expressed as either corresponding ratio (2−Δct) or fold change (2−ΔΔc). (a,b) Down‐regulation of miR‐497 was observed in the HCT8, LOVO, Ls‐174, HCT116 and HT29 cell lines. Normal human colorectal tissue was used as a control. (c,d) Expression of VEGF‐A was significantly up‐regulated in all these five CRC cell lines compared to control human colorectal tissues.
VEGF‐A as a direct and functional target of miR‐497
miR‐497 targets such as bcl2 and Cyclin D2 have been identified 19, 20, and we investigated whether other miR‐497 targets would be involved in regulating progression of CRC. Using TargetScan software 21, we identified VEGF‐A to be a likely target of miR‐497 due to presence of a putative miR‐497 target site in its 3ʹUTR (Fig. 3a). We generated a series of luciferase reporter vectors containing full‐length wild‐type/mutant VEGF‐A 3ʹUTR constructs (Fig. 3b). To identify functional targets, target site or its relevant mutant was cloned into an identical reporter vector and used in a luciferase reporter assay. miR‐497 mimics, miR‐497 inhibitors or a non‐functional control miR‐67 were co‐transfected with the above‐mentioned reporter vectors into the CRC cell line HCT116, to assess relative luciferase activity. Our results indicate that miR‐497 targets and full‐length wild‐type VEGF‐A 3ʹUTRs reduced relative luciferase activity only when miR‐497 was present (Fig. 3c). These results demonstrate that VEGF‐A mRNA is a specific target of miR‐497 in HCT116 CRC cells.
Figure 3.
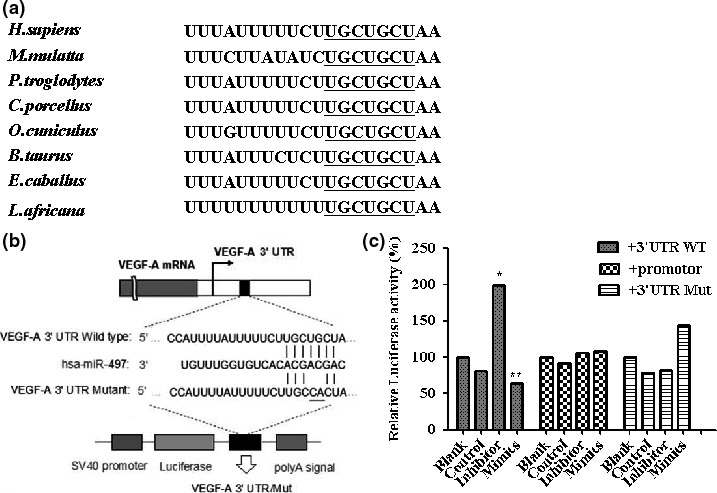
VEGF ‐A is a direct and functional target of miR‐497. (a) Nucleotides 210–216 of the VEGF‐A 3′UTR represent an miR‐497 target site, which is highly conserved across species. Sequence alignment of the miR‐497 binding site within the VEGF‐A 3′UTR of eight species is shown. (b) The wild‐type (WT) and mutant (Mut) forms of the putative miR‐497 target sequences of VEGF‐A 3′UTR. (c) miRNA luciferase reporter assay. WT and Mut miR‐497 target sequences were inserted into luciferase reporter vectors and co‐transfected with miR‐control, miR‐497 mimics or an miR‐497 inhibitor into HCT116 cells. miR‐497 significantly suppressed luciferase activity of WT VEGF‐A 3ʹUTR, normalized against Renilla luciferase activity (*p < 0.05, **p < 0.01).
miR‐497 did not affect cell and tumour growth
We further investigated whether overexpression of miR‐497 could reduce invasiveness and migratory potential of CRC cells. First, we evaluated effects of miR‐497 on proliferation of HCT116 cells in vitro and in vivo (Fig. 4). Results of these experiments indicate that miR‐497 had no significant implication for their proliferation.
Figure 4.
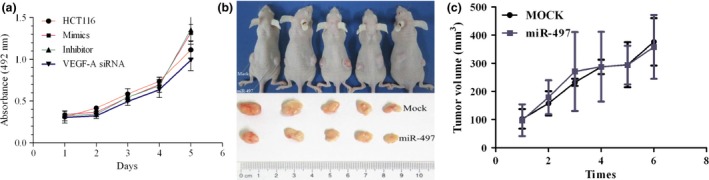
miR‐497 did not affect cell and tumour growth. (a) Viability of cells transfected with miR‐497 mimics, miR‐497 inhibitors or VEGF‐A siRNA. Results are shown as mean ± S.E. of the optical density (OD). Triplicate independent experiments were performed and the results of one representative replicate are shown. (b) Primary tumour growth 4 weeks after subcutaneous injection in the two flanks of nude mice with stably transduced cells “HCT116‐luc‐control” (left) or “HCT116‐luc‐miR‐497” (right). (c) Results are shown as tumour volume.
miR‐497 inhibited invasion and migration of CRC cells
Using a wound healing assay, we found that ectopic expression of miR‐497 reduced cell motility in miR‐497 mimic treatment and VEGF‐A silencing groups, compared to Mock and control groups (Fig. 5a); Figure 5b histogram shows this in detail. Invasive capability was significantly lower in mimic‐treated groups compared to the control group (Fig. 5c, upper panel, 5d), suggesting that miR‐497 seemed to regulate expression of molecules associated with invasion. Simultaneously, migratory potential was also significantly down‐regulated in the mimic‐administered groups compared to the control group (Fig. 5c bottom panel, 5d). Similar results were observed in both invasion and migration assays when cells were transiently transfected with VEGF‐A siRNA (Fig. 5c,d). Collectively, these data suggest that miR‐497, like VEGF‐A, plays a role in silencing inhibition of CRC cell invasion and migration without affecting cell proliferation. These findings indicate that miR‐497 participates in regulation of invasion and migration by directly regulating VEGF‐A expression.
Figure 5.
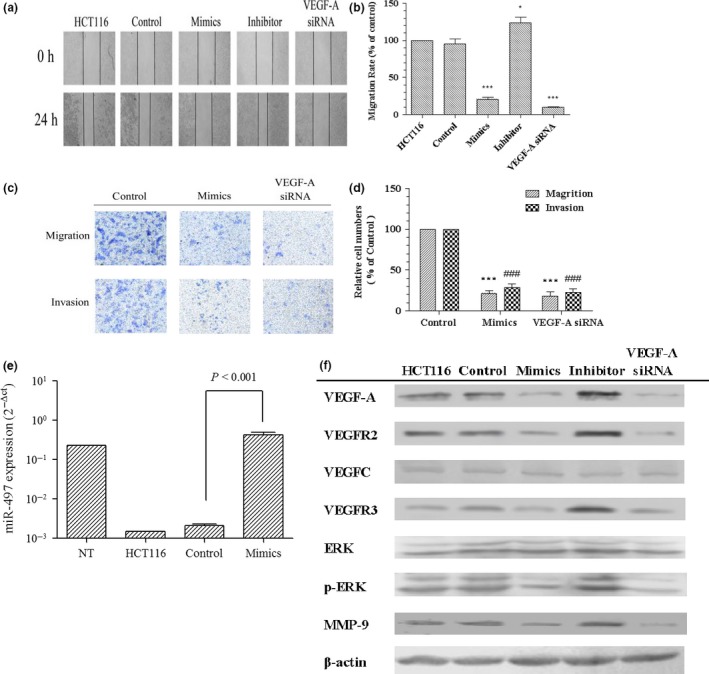
miR‐497 inhibited invasion and migration of CRC cells. (a) Using a wound healing assay, HCT116 cells were transfected with miR‐67 (control), miR‐497 mimics, miR‐497 inhibitor, VEGF‐A siRNA or culture medium (HCT116). Cell motilities were assessed at 0 and 24 h following wound creation. (b) Migration rates are shown by histogram (*p < 0.05, ***p < 0.001). (c,d) Invasion and migration assays show similar and significant inhibition of migration and invasiveness of HCT116 cells following treatment with miR‐497 mimics or VEGF‐A siRNA (***p < 0.001, ### p < 0.001). (e) miR‐497 was up‐regulated after transfection of HCT116 cells with miR‐497. (f) Changes in expression of the VEGF‐A, VEGFR2, VEGFC, VEGFR3, p‐ERK and MMP‐9 genes at the translational level. miR‐497 overexpression significantly inhibits the expression of VEGF‐A, p‐ERK, MMP‐9, VEGFR2 and VEGFR3 proteins, and this inhibition is comparable to that by VEGF‐A siRNA.
These experiments show that synthesis of MMP‐9, VEGFR2 and VEGFR3 decreased by down‐regulation of VEGF‐A. In addition to lower VEGF‐A expression induced by miR‐497 or VEGF‐A siRNA, we found down‐regulation of MMP‐9, VEGFR2 and VEGFR3 expression (Fig. 5e,f). In addition, we discovered changes in protein expression levels of ERK1/2 and p‐ERK1/2 to mirror those of VEGF‐A/MMP‐9 expression.
Overexpression of miR‐497 inhibited invasion and migration of CRC cells in vivo
Figure 6a reveals successively reduced metastatic lesions in lungs of animals in the HCT116‐luc‐miR‐497 group compared to HCT116‐luc‐control group 5 weeks after intravenous injection of HCT116 cells (P < 0.05). Figure 6b shows greater number of tumour foci in the lungs of HCT116‐luc‐control animals than in HCT116‐luc‐miR‐497 mice. In Fig. 6c, H&E staining showed slight alveolar structural damage in HCT116‐luc‐miR‐497 animals and more serious damage in HCT116‐luc‐controls. Ki67, a proliferating cell nuclear antigen that indicates cell proliferation was shown by immunohistochemistry to be expressed in clusters in HCT116‐luc‐control group tissues, and sporadically expressed in HCT116‐luc‐miR‐497 group tissues (Fig. 6d). These results demonstrate inhibition of metastasis and cell proliferation of colorectal cancer in vivo by miR‐497.
Figure 6.
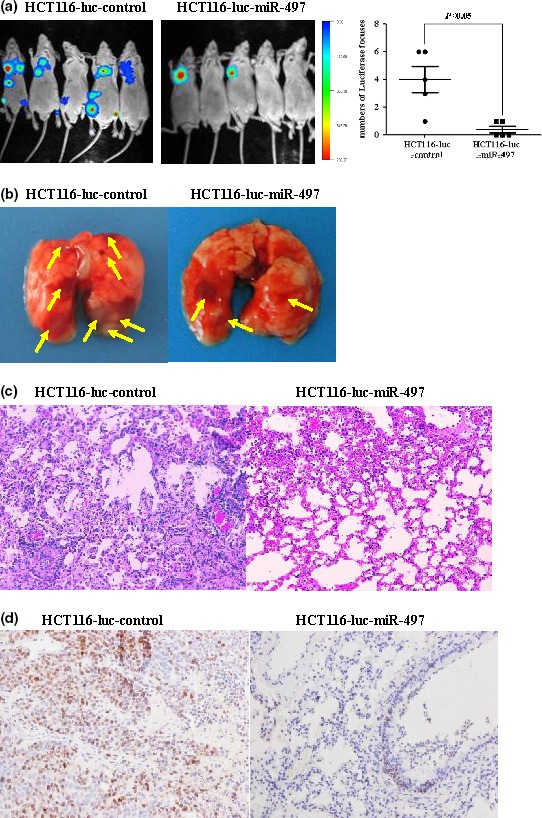
Overexpression of miR‐497 inhibited tumourigenesis and metastasis of CRC cells in vivo. (a) Tumours derived from cells “HCT116‐luc‐control” or “HCT116‐luc‐miR‐497” were injected intravenously via the tail vein. Tumour metastatic lesions in the mice were monitored by a live imaging system detecting the luciferase signal. (b) Representative lung tissues collected from “HCT116‐luc‐control” or “HCT116‐luc‐miR‐497”groups are shown. Tumours were marked with yellow arrows. (c) Representative H&E‐stained sections of the lung tissues collected from “HCT116‐luc‐control” or “HCT116‐luc‐miR‐497”groups are shown. (d) Representative immunohistochemistry sections of the lung tissues collected from “HCT116‐luc‐control” or “HCT116‐luc‐miR‐497” groups are shown.
Discussion
miRNAs serve as master regulators of gene expression in a sequence‐specific fashion. Those under‐expressed in tumours may be tumour suppressors, but can participate in oncogene overexpression 22. miR‐497 has been reported to be under‐expressed in breast cancer (1.57‐fold) 23, cervical squamous cell carcinoma (7.33‐fold) and head and neck squamous cell carcinomas (2.63‐fold) 24, suggesting a possible role for the molecule in tumour carcinogenesis. miRNAs can silence post‐transcription protein expression either by binding to complementary target messenger RNAs (thereby degrading these messengers), or by inhibiting the mRNA from being translated into protein. It is reported miR‐497 has an important role in inhibiting IGF1‐R expression and activation of PI3K/Akt signalling, and in suppressing proliferation, survival and invasive behaviour in human colon cancer cells 14. Usually, it is not a one‐to‐one correspondence relationship between miRNAs and their target genes. An miRNA may correspond to multiple target genes and multiple miRNAs may correspond to the same target genes or different ones within the same family. Here we have presented other targets of miR‐497 and mechanisms.
In this study, miR‐497 expression was 4.8‐fold lower in CRC tissues compared to corresponding non‐malignant tissues. Further investigation revealed a role for miR‐497 in targeting VEGF‐A, a function characteristic of tumour suppressors.
The cytokine VEGF‐A is an angiogenic factor implicated in processes including organ development, wound healing, tissue regeneration, endothelial cell growth and vessel permeability 25. In some solid tumours, overexpression of VEGF‐A is associated with increased angiogenesis, growth and/or metastasis 26, 27, 28. Researchers have also demonstrated that VEGF‐A is not only a promising therapeutic target but also seems to be a good prognostic factor for several cancers 29, 30, 31, 32. Although various intracellular signalling pathways have been proposed to induce the biological activities of VEGF‐A in endothelial cells, signalling events involved in cell migration and invasion in response to VEGF‐A stimulation in CRC have not been fully understood.
In this study, we showed negative correlation between miR‐497 and VEGF‐A expression in CRC samples and the corresponding colorectal tissues. Studies with CRC cell lines showed similar results. In in vitro and in vivo experiments, we found invasion and metastasis of CRC to be inhibited by up‐regulation of miR‐497 and subsequent VEGF‐A targeting. We proceeded to investigate the underlying molecular mechanisms in this process. Previous reports have shown VEGF‐A to be one of the most active factors in secretion of metalloproteinases (MMPs), particularly gelatinases A and B (MMP‐2 and MMP‐9) which hydrolyse collagens IV and V of vessel basal membranes 33. Ability to secrete these enzymes increases metastatic potential of malignant cells and correlates with adverse prognosis in several highly invasive cancers 34. Our findings revealed synthesis of MMP‐9 but not of MMP‐2 to be reduced with down‐regulation of VEGF‐A. Decreasing expression of VEGF‐A by miR‐497 or VEGF‐A siRNA resulted in similar down‐regulation of MMP‐9, while MMP‐2 expression did not change significantly. Furthermore, we found changes in protein expression levels of ERK1/2 and p‐ERK1/2 to mirror the change in expression of VEGF‐A/ MMP‐9, as previously shown 35.
In conclusion, we have shown miR‐497 to function as an inhibitory factor in metastasis of CRC, demonstrating this molecule's potential as a promising and novel therapeutic target. This study also revealed the miR‐497/VEGF‐A/ERK/MMP‐9 pathway to be involved in metastasis of CRC.
Conflict of interest
We declare that there is no conflict of interest regarding this study.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81473482) and Putuo District Committee of Science and Technology, Shanghai China (No. 201102). This research work was also supported by Construct Program of the Key Discipline of State Administration of Traditional Chinese Medicine of People's Republic of China and “apricot grove scholars” training plan of Shanghai University of Traditional Chinese Medicine.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J. Clin. 64, 9–29. [DOI] [PubMed] [Google Scholar]
- 2. Figueredo A, Coombes ME, Mukherjee S (2008) Adjuvant therapy for completely resected stage II colon cancer. Cochrane Database Syst. Rev. 16, CD005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunter KW, Crawford NP, Alsarraj J (2008) Mechanisms of metastasis. Breast Cancer Res. 10(Suppl 1), S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tookman L, Roylance R (2010) New drugs for breast cancer. Br. Med. Bull. 96, 111–129. [DOI] [PubMed] [Google Scholar]
- 5. Hassan MS, Ansari J, Spooner D, Hussain SA (2010) Chemotherapy for breast cancer (Review). Oncol. Rep. 24, 1121–1131. [DOI] [PubMed] [Google Scholar]
- 6. Sibley CR, Seow Y, Wood MJ (2010) Novel RNA‐based strategies for therapeutic gene silencing. Mol. Ther. 18, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ireson CR, Kelland LR (2006) Discovery and development of anticancer aptamers. Mol. Cancer Ther. 5, 2957–2962. [DOI] [PubMed] [Google Scholar]
- 8. Guo P, Coban O, Snead NM, Trebley J, Hoeprich S, Guo S et al (2010) Engineering RNA for targeted siRNA delivery and medical application. Adv. Drug Deliv. Rev. 62, 650–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim SJ, Oh JS, Shin JY, Lee KD, Sung KW, Nam SJ et al (2011) Development of microRNA‐145 for therapeutic application in breast cancer. J. Control Release 155, 427–434. [DOI] [PubMed] [Google Scholar]
- 10. He L, Hannon GJ (2014) MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. [DOI] [PubMed] [Google Scholar]
- 11. Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866. [DOI] [PubMed] [Google Scholar]
- 12. Finnerty JR, Wang WX, Hébert SS, Wilfred BR, Mao G, Nelson PT (2010) The miR‐15/107 group of MicroRNA genes: evolutionary biology, cellular functions, and roles in human diseases. J. Mol. Biol. 24, 491–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T et al (2010) The DLEU2/miR‐15a/16‐1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17, 28–40. [DOI] [PubMed] [Google Scholar]
- 14. Guo ST, Jiang CC, Wang GP, Li YP, Wang CY, Guo XY et al (2013) MicroRNA‐497 targets insulin‐like growth factor 1 receptor and has a tumour suppressive role in human colorectal cancer. Oncogene 32, 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bookout AL, Mangelsdorf DJ (2003) Quantitative real‐time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl. Recept. Signal 1, e012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cao AL, Tang QF, Zhou WC, Qiu YY, Hu SJ, Yin PH (2015) Ras/ERK signalling pathway is involved in Curcumin‐induced cell cycle arrest and apoptosis in human gastric carcinoma AGS cells. J. Asian Nat. Prod. Res. 10, 1–8. [DOI] [PubMed] [Google Scholar]
- 17. Naito S, von Eschenbach AC, Giavazzi R, Fidler IJ (1986) Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 46, 4109–4115. [PubMed] [Google Scholar]
- 18. Qiu YY, Hu Q, Tang QF, Feng W, Hu SJ, Liang B et al (2014) MiR‐497 and bufalin act synergistically to inhibit colorectal cancer metastasis. Tumour Biol. 5, 2599–2606. [DOI] [PubMed] [Google Scholar]
- 19. Yadav S, Pandey A, Shukla A, Talwelkar SS, Kumar A, Pant AB et al (2011) miR‐497 and miR‐302b regulate ethanol‐induced neuronal cell death through BCL2 protein and Cyclin D2. J. Biol. Chem. 286, 37347–37357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J et al (2010) miR‐497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol. Dis. 38, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- 22. Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA (2009) MicroRNAs – the micro steering wheel of tumour metastases. Nat. Rev. Cancer 9, 293–302. [DOI] [PubMed] [Google Scholar]
- 23. Lehmann U, Streichert T, Otto B, Albat C, Hasemeier B, Christgen H et al (2010) Identification of differentially expressed microRNAs in human male breast cancer. BMC Cancer 10, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lajer CB, Garnæs E, Friis‐Hansen L, Norrild B, Therkildsen MH, Glud M et al (2012) The role of miRNAs in human papilloma virus (HPV)‐associated cancers: bridging between HPV‐related head and neck cancer and cervical cancer. Br. J. Cancer 106, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown LF, Detmar M, Claffey K, Nagy JA, Feng D, Dvorak AM et al (1997) Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. EXS 79, 233–269. [DOI] [PubMed] [Google Scholar]
- 26. Folkman J, Klagsbrun M (1987) Vascular physiology. A family of angiogenic peptides. Nature 329, 671–672. [DOI] [PubMed] [Google Scholar]
- 27. Slattery ML, Lundgreen A, Wolff RK (2014) VEGFA, FLT1, KDR and colorectal cancer: assessment of disease risk, tumor molecular phenotype, and survival. Mol. Carcinog. 53, 140–150. [DOI] [PubMed] [Google Scholar]
- 28. Goos JA, de Cuba EM, Coupé VM, Diosdado B, Delis‐Van Diemen PM, Karga C et al (2016) Glucose Transporter 1 (SLC2A1) and Vascular Endothelial Growth Factor A (VEGFA) Predict Survival After Resection of Colorectal Cancer Liver Metastasis. Ann. Surg. 263, 138–145. [DOI] [PubMed] [Google Scholar]
- 29. Pignot G, Bieche I, Vacher S, Güet C, Vieillefond A, Debré B et al (2009) Large‐scale real‐time reverse transcription‐PCR approach of angiogenic pathways in human transitional cell carcinoma of the bladder: identification of VEGFA as a major independent prognostic marker. Eur. Urol. 56, 678–688. [DOI] [PubMed] [Google Scholar]
- 30. Des Guetz G, Uzzan B, Nicolas P, Cucherat M, Morere JF, Benamouzig R et al (2006) Microvessel density and VEGF expression are prognostic factors in colorectal cancer. Meta‐analysis of the literature. Br. J. Cancer 94, 1823–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poon RT, Ho JW, Tong CS, Lau C, Ng IO, Fan ST (2004) Prognostic significance of serum vascular endothelial growth factor and endostatin in patients with hepatocellular carcinoma. Br. J. Surg. 91, 1354–1360. [DOI] [PubMed] [Google Scholar]
- 32. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B et al (2008) Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 68, 6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Munaut C, Noël A, Hougrand O, Foidart JM, Boniver J, Deprez M (2003) Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1‐MMP, MMP‐2 and MMP‐9 in human glioblastomas. Int. J. Cancer 106, 848–855. [DOI] [PubMed] [Google Scholar]
- 34. Foda HD, Zucker S (2001) Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov. Today 6, 478–482. [DOI] [PubMed] [Google Scholar]
- 35. Chakroborty D, Chowdhury UR, Sarkar C, Baral R, Dasgupta PS, Basu S (2008) Dopamine regulates endothelial progenitor cell mobilization from mouse bone marrow in tumor vascularization. J. Clin. Invest. 118, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]