

Abstract
Abstract. Objective: Various studies have shown that bone marrow stem cells can rescue mice from acute renal tubular damage under a conditioning advantage (irradiation or cisplatin treatment) favouring donor cell engraftment and regeneration; however, it is not known whether bone marrow cells (BMCs) can contribute to repair of acute tubular damage in the absence of a selection pressure for the donor cells. The aim of this study was to examine this possibility. Materials and methods: Ten‐week‐old female mice were assigned into control non‐irradiated animals having only vehicle treatment, HgCl2‐treated non‐irradiated mice, HgCl2‐treated non‐irradiated mice infused with male BMCs 1 day after HgCl2, and vehicle‐treated mice with male BMCs. Tritiated thymidine was given 1 h before animal killing. Results: Donor BMCs could not alleviate non‐irradiated mice from acute tubular damage caused by HgCl2, deduced by no reduction in serum urea nitrogen combined with negligible cell engraftment. However, donor BMCs could home to the bone marrow and spleen and display proliferative activity. This is the first report to show that despite no preparative myeloablation of recipients, engrafted donor BMCs can synthesize DNA in the bone marrow and spleen. Conclusions: Exogenous BMCs do not rescue non‐irradiated mice from acute renal tubular damage caused by HgCl2, despite establishment of chimerism and cell proliferation in bone marrow and spleen.
INTRODUCTION
Studies of gender‐mismatch bone marrow transplant (BMT) and organ allografts have demonstrated that adult bone marrow cells (BMCs) can transdifferentiate into a variety of non‐haematological tissues in rodents and humans. These include skeletal muscle, endothelial cells, cardiomyocytes, neuronal cells and astrocytes, hepatocytes, epidermal cells, pneumocytes, renal tubular epithelium and podocytes, and gut epithelium (reviewed by Poulsom et al. 2002; Herzog et al. 2003; Alison et al. 2004; Fang et al. 2004). Furthermore, stem cell‐based therapy strategy is becoming a realistic option to replace or rebuild damaged organs and tissues. Recently, such strategies have been successfully applied using a variety of cell types, for example, mesenchymal stem cells (MSCs), BMCs, or human umbilical cord blood cells, to rescue animals and humans from organ injury (Azizi et al. 1998; Horwitz et al. 1999, 2001; Orlic et al. 2001a; Strauer et al. 2002; Terai et al. 2003; Ende et al. 2004; Vendrame et al. 2004) and reviewed in Fang & Poulsom (2003), including acute tubular damage (Kale et al. 2003; Lin et al. 2003; Morigi et al. 2004).
Acute renal failure (ARF), defined as rapid loss of the ability of the kidneys to excrete waste, concentrate urine and maintain fluid and electrolyte homeostasis, is a common disease and accounts for 2–15% of hospitalized patients, reviewed by Thadhani et al. (1996). Despite major advances in pharmacological therapy, intensive care and renal replacement therapy, the overall mortality rate of patients with ARF is still high, about 50–80% in a recent series, reviewed in Thadhani et al. (1996) and Schrier et al. (2004), and has changed little over the past 30 years. We have already demonstrated that endogenous BMCs can contribute to tubular regeneration after acute injury (Poulsom et al. 2001; Fang et al. 2005); moreover, several studies have demonstrated the potential of stem cell therapy for ARF (Kale et al. 2003; Lin et al. 2003, 2005; Morigi et al. 2004; Duffield et al. 2005). For example, if sublethally irradiated mice are subjected to ischaemia/reperfusion injury and injected with hematopoietic stem cells (HSCs) (2 × 103 or 5 × 103 cells), then the HSCs can contribute to functional regeneration of damaged renal proximal tubules (Kale et al. 2003; Lin et al. 2003), although mortality rate was high due to the irradiation (three out of seven mice died) (Kale et al. 2003). Lethal irradiation shortly before renal tubular damage can damage stem and progenitor cells not only within the bone marrow, but also within the renal parenchyma (Kale et al. 2003). Morigi et al. (2004) reported that sorted MSCs, but not sorted HSCs, could rescue nonirradiated mice from acute renal tubular damage caused by cisplatin; however, cisplatin can inhibit DNA synthesis in cells of renal tubules (Landrito et al. 1994), and can decrease numbers of stem cells in bone marrow and spleen (Dumenil et al. 1982). These three studies had in common a conditioning environment (irradiation or cisplatin) that inhibits the regenerative ability of the recipient's kidney tubular cells and BMCs, presumably promoting donor cells to contribute to renal repair.
The aim of this study was to test if such preconditioning was a prerequisite for subsequent kidney cell engraftment, so we administered whole BMCs to recipients suffering ARF without any prior treatment. Here, we show that donor BMCs do not ameliorate acute tubular damage in non‐irradiated mice caused by HgCl2, although chimerism was established in the bone marrow and spleen.
MATERIALS AND METHODS
Experimental animals and protocols
Procedures for the animal experiments were carried out under British Home Office procedural and ethical guidelines. Ten‐week‐old female recipient mice (FVB/N) were randomly divided into four groups and were treated as follows (n = 20 in each group): group CON, non‐irradiated mice received vehicle, 0.2 mL phosphate‐buffered saline (PBS); group mBM, mice treated as group CON and given male BMCs (2 × 107 cells) by tail vein injection 1 day after vehicle; group HgCl2, non‐irradiated mice received an intraperitoneal injection of HgCl2 (4 mg/kg body weight); group HgCl2 + mBM: non‐irradiated mice were treated with HgCl2 (4 mg/kg body weight) and were infused with male donor BMCs (2 × 107 cells) by tail vein injection 1 day after HgCl2. The day of HgCl2 administration was designated as day 0. Mice were killed either before HgCl2 administration (day 0), or days 3, 7, 28 after HgCl2, with five mice killed by overdose of pentobarbitone (Sagatal, Rhône Mérieux, UK; 200 mg/kg, intraperitoneally), at each time point. Tritiated thymidine (TRK120, Amersham Biosciences, Munich, Germany) at a dose of 1 µCi/g body weight (intraperitoneally) was injected 1 h before the mice were killed. Terminal blood samples (0.7 mL) were taken by cardiac puncture into an EDTA‐tube. Kidney, spleen and femurs and tibias were harvested and fixed in neutral buffered formalin before being embedded in paraffin wax for later histological examination. For bone specimens, decalcification of femurs and tibias via a combination of 8% formic acid (cat. no. 10115; BDH Laboratory Supplies, Poole, UK) and 8% formic formate (cat. no. 29445‐4; Sigma‐Aldrich, Dorset, UK) for 10–12 days was performed before being embedded in paraffin wax.
Bone marrow cell preparation
Mice were killed humanely by CO2 inhalation. BMCs were collected from the femurs and tibias of male FVB/N mice by flushing three times with sterile ice‐chilled PBS (PBS tablet: cat. no. P4417; Sigma, Surrey, UK) via a 23‐gauge needle attached to a 1‐mL syringe, and then were suspended in sterile ice‐chilled PBS. The cell suspension was centrifuged at 500 g for 5 min, and the supernatant was discarded. The cell pellet was re‐suspended in sterile PBS and after sieving through 70‐µm meshes (cat. no. 352350; BD Falcon, San Jose, CA, USA), and BMCs were re‐suspended in sterile PBS at a concentration of 1 × 108 cells/mL (counted using the haemocytometer chamber method) and kept on ice until use.
Serum urea nitrogen measurements
Blood samples were centrifuged at 5000 g for 10 min at 4 °C and the supernatant was stored at –70 °C for later determination of serum urea nitrogen (SUN) using a commercial kit (cat. no. 0542946, R‐Biopharm GmbH, Darmstadt, Germany) following manufacturer's instructions. The level of SUN was expressed in milligrams per 100 mL.
Histology and semiquantification of morphometric analysis
For histological examination of renal tissue, 4 µm sections were stained with periodic acid–Schiff. Tubular injury was assessed at ×400 overall magnification using 20 consecutive and non‐overlapping fields (adjacent to the corticomedullary junction) of periodic acid–Schiff‐stained specimens. Tubular damage, defined as tubular epithelial swelling, loss of brush border, vacuolar degeneration, necrotic tubules, and desquamation, was scored semiquantitatively. Tubular injury was categorized into one of five scores using the following criteria: 0, normal; 1, 2, 3 and 4 based on the report by Nangaku et al. (1998). The mean tubular injury score (TIS) for each mouse represented average score of the 20 fields immediately adjacent to the corticomedullary junction.
Treatment of tissue sections
Four micron thick sections were dewaxed and were incubated with 3% hydrogen peroxide in methanol, then were rehydrated through a descending series of ethanol in water (100%, 95% and 70%). Sections were then incubated for 5 min in 20% acetic acid in methanol, to block endogenous alkaline phosphatase.
Immunohistochemistry
For detection of megalin expression in proximal renal tubular cells, slides were pre‐incubated in normal swine serum (X0901; DAKO, Glostrup, Denmark) at 1/25 dilution for 10 min. Rabbit antimouse megalin (a kind gift from Dr. Dan Biemesderfer, Yale University School of Medicine, New Haven, CT, USA) was applied to sections at a dilution of 1/500 for 45 min at room temperature. The secondary layer was biotinylated swine antirabbit antibody (E0353; DAKO), diluted to 1/500, for 35 min at room temperature. The third layer was streptavidin‐alkaline phosphatase (D0396; DAKO) at 1/50 dilution for 30 min at room temperature. Following washes in PBS, Vector Red Substrate (SK5100; Vector Laboratories, Burlingame, CA, USA) was applied for 8–10 min at room temperature and sections were again washed in PBS prior to the in situ hybridization protocol.
In situ hybridization
Sections were incubated in 1 m sodium thiocyanate (S7757; Sigma) for 10 min at 80 °C to improve access of the probe to DNA, washed in PBS, and then digested in 0.4% w/v pepsin (P6887; Sigma) in 0.1 m HCl for 10 min at 37 °C to further improve access. Protease was quenched in 0.2% glycine (G4392; Sigma) in double concentration PBS and sections were then rinsed in PBS, post‐fixed in 4% paraformaldehyde (P6148; Sigma) in PBS, dehydrated through graded ethanols, and air dried. FITC‐labelled Y chromosome paint (1189‐YMF‐01; Cambio, Cambridge, UK) was used in the supplier's hybridization mix. Probe mixture was added to the sections, sealed under glass with rubber cement, heated to 60 °C for 10 min, and then incubated overnight at 37 °C. The following day, all slides were washed in 50% formamide (284226P; BDH Laboratory Supplies)/2× standard saline citrate (SSC) at 37 °C, then washed with 2× SSC, and incubated in 4× SSC/0.05% Tween‐20 for 10 min at 37 °C. Slides were then treated by one of the two following methods depending on whether direct fluorescence in situ hybridization (FISH) or indirect FISH was to be performed.
For direct FISH, slides were washed and then coverslipped with Vectashield Hard Set mounting medium with 4′,6‐diamidine‐2‐phenylindole dihydrochloride (DAPI) (H‐1500; Vector Laboratories) for subsequent observation and analysis by fluorescence microscopy using a Zeiss Axioplan 2 fluorescence microscope (Carl Zeiss UK Ltd., Hertfordshire, UK) equipped with a triple band‐pass filter. Images were collected with a cooled charge‐coupled device camera (Quantix Corp., Cambridge, MA, USA) and further analysed using SmartCapture 2 software (DigitalScientific Ltd., Cambridge, UK).
For indirect FISH, slides were washed with PBS and incubated with 1/250 dilution peroxidase‐conjugated antifluorescein antibody (1426346; Boehringer Mannheim, Mannheim, Germany) for 60 min at room temperature, before being developed in 3,3′‐diaminobenzidine (D5637; Sigma) plus 0.3% hydrogen peroxide, washed with PBS and subjected to periodic acid–Schiff staining.
Autoradiography for tritiated thymidine
To evaluate DNA synthesis, slides were dehydrated through graded ethanols and air dried before dipping in ILFORD nuclear track emulsion (L4; ILFORD, Basildon, UK) (diluted 3 : 5 with Q water) at 45 °C in a dark room using a safe light (902 filter). When dry, slides were stored in the dark at 4 °C. After a standard exposure of 1 week, slides were developed for 4 min at 20 °C in Kodak D19 (cat. no. 1464593; Eastman Kodak Company, Rochester, NY, USA) and were stopped with 1% acetic acid (10001 CU; BDH Laboratory Supplies) for 30 s, washed in tap water for 30 s, and fixed in 30% w/v sodium thiosulphate (S‐8503; Sigma) for 8 min. They were then rinsed in gently running cold tap water for 60 min to ensure that all traces of sodium thiosulphate were removed. Finally, sections were counterstained lightly with haematoxylin, dehydrated, cleared and mounted in DePeX (36125‐2B; BDH Laboratory Supplies).
Determination of the tritiated thymidine‐labelling index (3H‐LI)
One thousand consecutively observed megalin‐stained tubular cells of kidney sections, and one thousand consecutively observed bone marrow and spleen cells from the sections were scored per mouse using light‐ and dark‐field microscopy (Nikon Eclipse ME600; Nikon Corporation, Kawasaki, Japan). Each nucleus that was Y chromosome‐positive and had more than five overlying autoradiographic silver grains was considered to be a cell of donor BMC origin in DNA synthesis.
Statistical analysis
SUN, TIS and 3H‐LI (%) were analysed by two‐way analysis of variance (anova) for repeated measures (the first factor being treatment group and the second, time period) for comparison between groups. When a significant effect was detected by anova, the Newman–Keuls test was used to establish which differences between means reached statistical significance (P < 0.05). Results are presented as mean ± SEM.
RESULTS
Renal histological examination, TIS and SUN
Figure 1 illustrates examples of renal damage and regeneration in control mice and HgCl2‐treated mice given or not given donor BMCs. Degenerative changes were apparent by day 3 after HgCl2 administration, had partially resolved at day 7, and fully resolved by day 28. Changes in TIS and SUN are shown in Table 1. Both vehicle alone and administered BMCs had no effect on either parameter throughout the experimental period. HgCl2 treatment significantly increased the TIS at day 3; this had declined by day 7, returning to baseline levels at day 28; this pattern coincided with the pattern of changes in SUN. Patterns of changes in TIS and SUN in HgCl2‐treated mice given BMCs were similar to those in HgCl2‐treated mice not given BMCs; there were no significant differences between these two groups throughout the experimental period.
Figure 1.
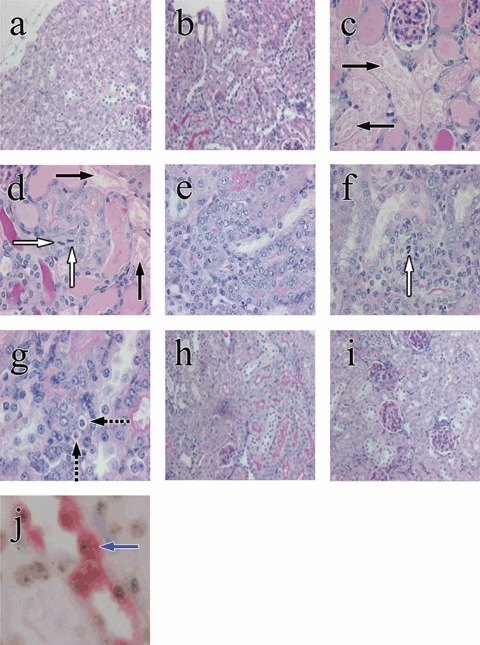
Examples of renal tubular histology in control and HgCl2‐treated mice, given or not given bone marrow cells (BMCs); periodic acid–Schiff stain (original magnifications). Renal tubular cells appeared normal in the control period (a, ×200) and in mice given BMCs throughout the experiment (b, ×200). Three days after HgCl2, renal tubular epithelial damage and tubular cell mitoses were seen in HgCl2‐treated mice given and not given BMCs (c and d, ×400). Black arrows indicate extensive renal tubular damage and necrosis (c, d) and white arrows indicate tubular epithelial mitosis (d). Seven days after HgCl2 damage, regenerative tubules with hypercellularity were seen in HgCl2‐treated mice given (e, ×400) or not given BMCs (f, ×400 and g, ×600). Dashed black arrows indicate apoptotic cells. Four weeks after HgCl2, most damaged tubules have regenerated in HgCl2‐treated mice given or not given BMCs (h and i, ×200). A blue arrow indicates A Y‐positive donor cell (brown dot) with megalin staining (red colour) (j, ×1000).
Table 1.
Effects of administration of HgCl2 alone and in combination with donor BMCs on renal tubular injury score and serum urea nitrogen
| Parameter | n | Pre‐HgCl2 | Post‐HgCl2 and male BMCs | ||
|---|---|---|---|---|---|
| Day 3 | Day 7 | Day 28 | |||
| Tubular injury score | |||||
| Group CON | 5 | 0.12 ± 0.01 | 0.10 ± 0.02 | 0.11 ± 0.02 | 0.11 ± 0.02 |
| Group mBM | 5 | 0.11 ± 0.01 | 0.11 ± 0.01 | 0.12 ± 0.01 | 0.10 ± 0.02 |
| Group HgCl2 | 5 | 0.12 ± 0.01 | 3.66 ± 0.04[Link], [Link], [Link] | 1.37 ± 0.02[Link], [Link], [Link] | 0.10 ± 0.02 |
| Group HgCl2 + mBM | 5 | 0.10 ± 0.02 | 3.72 ± 0.04[Link], [Link], [Link] | 1.4 ± 0.01[Link], [Link], [Link] | 0.11 ± 0.02 |
| Serum urea nitrogen (mg/dL) | |||||
| Group CON | 5 | 20.1 ± 1.9 | 17.8 ± 2.6 | 19.1 ± 2.2 | 17.7 ± 2.5 |
| Group mBM | 5 | 18.2 ± 2.2 | 18.5 ± 2.7 | 18.7 ± 2.3 | 17.1 ± 1.8 |
| Group HgCl2 | 5 | 21.3 ± 2.2 | 69.1 ± 4.2[Link], [Link], [Link] | 44.7 ± 2.0[Link], [Link], [Link] | 18.8 ± 2.1 |
| Group HgCl2 + mBM | 5 | 20.1 ± 2.7 | 70.4 ± 2.6[Link], [Link], [Link] | 48.4 ± 2.9[Link], [Link], [Link] | 18.5 ± 2.0 |
Values are means ± SEM. *P < 0.05 versus the same group at pre‐HgCl2 period;†P < 0.05 versus group CON at the corresponding time point;‡P < 0.05 versus group mBM at the corresponding time point. BMCs, bone marrow cells; CON, control; mBM, male bone marrow.
3H‐LI of megalin‐positive proximal tubular cells
No Y chromosome‐positive megalin‐expressing cells were observed at time zero among the four groups, and only a very small percentage (< 0.1%) of Y chromosome‐positive cells could be found in either the mice given male BMCs alone or the HgCl2‐treated mice given male BMCs (Fig. 1j). Changes in 3H‐LI of megalin‐positive cells are shown in Fig. 2. After HgCl2 treatment, the overall 3H‐LI was significantly higher at day 3 and was still above control values at day 7, but returned to baseline level at day 28. The pattern of changes in HgCl2‐treated mice given BMCs was similar to those given HgCl2 alone. Furthermore, there were no significant differences between 3H‐LI of proximal tubular cells at any time point between these two groups.
Figure 2.
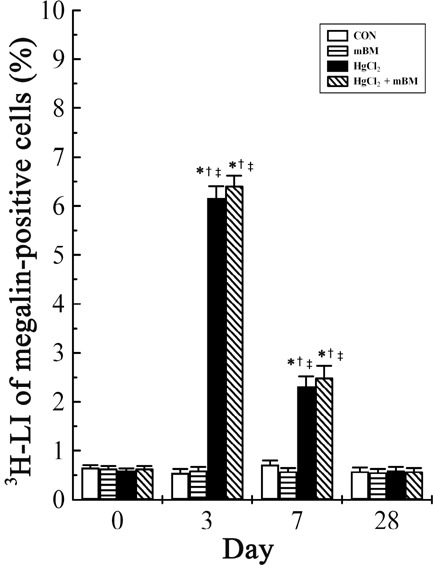
Changes in the total 3H‐LI of megalin‐positive epithelial cells in the kidney of mice treated with HgCl2 with or without bone marrow cells (BMCs). n = 5 mice per treatment time for each group. *P < 0.05 versus the same group at day 0; †P < 0.05 vs. group CON at the corresponding time point; ‡P < 0.05 versus group mBM at the corresponding time point. CON, control; mBM, male bone marrow.
Chimerism and proliferation of donor BMCs in the femoral bone marrow of recipients
Figure 3 depicts changes in the proportion of Y chromosome‐positive cells (Fig. 3a) and changes in 3H‐LI of BMCs (Fig. 3b–d) at day 28 in femoral bone marrow of HgCl2‐treated mice with or without BMCs. Approximately 12% of BMCs were of donor origin in the femoral bone marrow of mice given BMCs alone and in HgCl2‐treated mice also given BMCs, but none were detected in mice not given BMCs (Fig. 3a). Furthermore, the percentage of donor BMCs in the femoral bone marrow was not significantly different between mice given BMCs alone and those HgCl2‐treated mice also given BMCs. At day 28, there was no significant difference in the 3H‐LI of BMCs among groups, ranging from 32.6 ± 1.0% to 33.4 ± 1.2% (Fig. 3b). 3H‐LI of BMCs derived from the host in control and HgCl2‐treated mice was significantly higher than those in mice given BMCs alone and HgCl2‐treated mice also given BMCs (Fig. 3c), presumably because a significant percentage of the 3H‐LI of BMCs was derived from donor BMCs in these latter mice (Fig. 3d). An example of chimerism and proliferation of donor BMCs at day 28 in the femoral bone marrow of HgCl2‐treated mice given BMCs is illustrated in Fig. 4.
Figure 3.
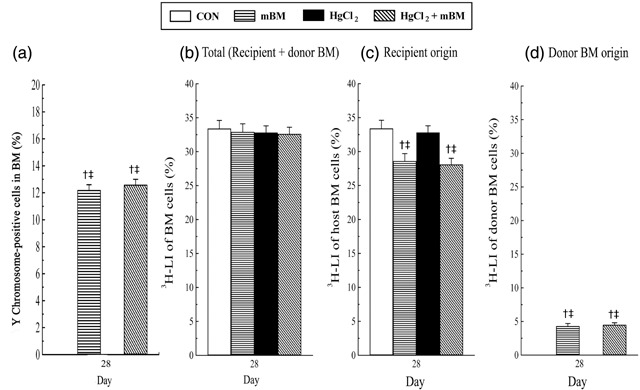
The extent of bone marrow (BM) engraftment was analysed at day 28. The proportion of Y chromosome‐positive cells (a), and changes in the 3H‐LI of bone marrow cells (BMCs) of (b) total (combined recipient and donor BMC origin) (c) recipient origin, and (d) donor BMC origin, at day 28 in the femoral bone marrow of mice treated with or without with HgCl2 and BMCs. n = 5 mice at day 28 per each group. †P < 0.05 versus group CON; ‡P < 0.05 versus group HgCl2. CON, control.
Figure 4.

Examples of chimerism and proliferation of donor bone marrow cells (BMCs) at day 28 in the bone marrow of mice given HgCl2 and infused with donor male BMCs 1 day later: (a) black arrows indicate donor‐derived BMCs (Y chromosome paint seen as brown nuclear signal) under bright field illumination, original magnification ×500; (b) the same field under reflected‐light dark‐field illumination, white arrows point to clusters of silver grains (3H‐thymidine labelling); (c) the images in (a) and (b) were combined to help to show silver grains over cells that are of donor BMC origin. (d), (e) and (f) are 2‐fold magnifications of the boxed area in (a). (g) Engrafted donor BMCs in the femoral bone marrow demonstrated by a FISH method, white arrows point to Y chromosomes (Y chromosome paint seen as green nuclear signal, ×630).
Chimerism and proliferation of donor BMCs in the spleen
Figure 5 illustrates changes in the proportion of Y chromosome‐positive cells (Fig. 5a) and changes in 3H‐LI of spleen cells (Fig. 5b–d) in mice treated with HgCl2 with or without BMCs. No Y chromosome‐positive cells were observed at time zero among the four groups. A gradual and significant increase in Y chromosome‐positive cells was seen in spleens of mice infused with BMCs alone and HgCl2‐treated mice also infused with BMCs during the experimental period, reaching 8.0 ± 0.3% and 8.3 ± 0.3% at day 28, respectively (Fig. 5a). There was no significant difference in overall 3H‐LI of spleen cells among the four groups at all times, being in the region of 16.8 ± 0.6% to 17.6 ± 0.4% (Fig. 5b). However, at day 28, 3H‐LI of spleen cells derived from the host was significantly lower in mice given BMCs alone (15.3 ± 0.5%) and HgCl2‐treated mice also given BMCs (15.2 ± 0.5%), than those in the groups not given BMCs (~18%) (Fig. 5c), again presumably, because a significant percentage of labelled of spleen cells in the mice given BMCs came from the donor BMCs (Fig. 5d). Examples of chimerism and proliferation of donor BMCs in spleens of HgCl2‐treated mice given BMCs are shown in Fig. 6.
Figure 5.

The extent of spleen engraftment at times after treatment with HgCl2 and bone marrow cells (BMCs) was analysed at days 0, 3, 7 and 28. n = 5 mice per treatment time for each group. The proportion of Y chromosome‐positive cells (a), and the 3H‐LI of cells of (b) total (combined recipient and donor BMC origin) (c) recipient origin, and (d) donor BMC origin. †P < 0.05 versus group CON and ‡P < 0.05 versus group HgCl2 at the corresponding time points. CON, control.
Figure 6.
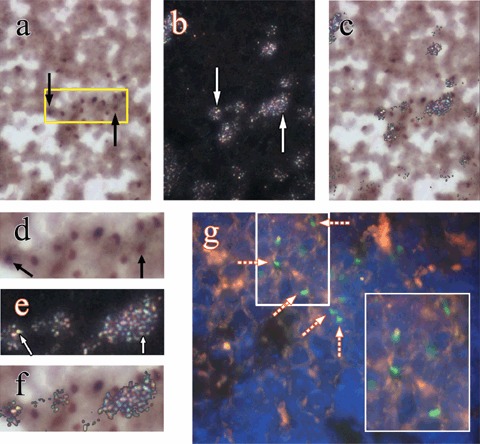
Examples of chimerism and proliferation of donor bone marrow cells (BMCs) at day 28 in the spleen of mice given HgCl2 and infused with donor male BMCs 1 day later: (a) black arrows indicate donor‐derived BMCs (Y chromosome paint seen as brown nuclear signal) under bright field illumination, original magnification ×500; (b) the same field under reflected‐light dark‐field illumination, white arrows point to clusters of silver grains (3H‐thymidine labelling); (c) the images in (a) and (b) were combined to help to show silver grains over cells that are of donor BMC origin. (d), (e) and (f) are 2‐fold magnifications of the boxed area in (a). (g) Engrafted donor BMCs in the spleen demonstrated by a FISH method, broken arrows point to Y chromosomes (Y chromosome paint seen as green nuclear signal, ×630 and ×1000 inset).
DISCUSSION
This study has demonstrated that (1) exogenous donor BMCs do not apparently contribute to repair of the kidney in non‐irradiated mice after acute tubular damage has been caused by HgCl2; (2) without a conditioning regime, injected BMCs can nevertheless home back to the bone marrow and spleen; (3) despite absence of preparative myeloablation, engrafted donor BMCs can synthesize DNA in the bone marrow and spleen, suggesting that these engrafted donor BMCs may play a functional role in homeostasis of the haematopoietic and immunological systems.
Here, administration of donor BMCs into HgCl2‐treated non‐irradiated mice did not ameliorate renal tubular damage as evidenced by the SUN and TIS data, with rare engraftment of BMCs in renal tubules. However, cell therapy with a variety of cell types has been used to aid regeneration in damaged organs (Azizi et al. 1998; Horwitz et al. 1999, 2001; Orlic et al. 2001a; Strauer et al. 2002; Terai et al. 2003; Ende et al. 2004; Vendrame et al. 2004) and has been reviewed in Fang & Poulsom (2003). For example, infusion with BMCs into non‐myeloablated recipients can improve cardiac function in mice with infarcted myocardium (Orlic et al. 2001b) and patients with acute myocardial infarction (Strauer et al. 2002). However, some observations on the value of stem cell therapy to rescue acute organ injury in non‐irradiated recipients are conflicting (Kanazawa & Verma 2003; Terai et al. 2003; Borlongan et al. 2004; Vendrame et al. 2004). For example, one study has shown that after intravenous infusion of GFP(+)–BMCs into non‐irradiated mice with CCl4‐induced liver damage, GFP‐positive hepatocytes could be found 1 day after GFP(+)–BMC infusion that proliferated over the next 4 weeks to account for 25% of all hepatocytes (Terai et al. 2003). In contrast, another study showed that after infusion with either GFP‐ (105 or 107 cells), or lacZ‐marked BMCs (105 or 107 cells) into non‐irradiated mice after CCl4 injury, no donor‐derived cells could be seen at 4 weeks after BMC injection (Kanazawa & Verma 2003). In kidney, the result of stem cells therapy for acute tubular necrosis is conflicting (Kale et al. 2003; Lin et al. 2003, 2005; Morigi et al. 2004; Duffield et al. 2005; Lange et al. 2005; Togel et al. 2005). The discrepancy of stem cell therapy for ARF may be due to (1) existence of a conditioning advantage (cisplatin or irradiation) for donor stem cells, and (2) different cell types and the number of cells injected. For example, considering cisplatin, the study of Morigi et al. (2004) showed a higher level of proliferation (Ki67‐positive nuclei/HPF) at day 29 than at days 4 and 11 after cisplatin although renal function had already recovered to baseline at day 11, implying that regenerative ability of host cells was initially inhibited by cisplatin. In contrast, our study has shown that the regenerative ability of the recipient was not affected, evidenced by the peak level of 3H‐LI consistent with regeneration of renal tubular epithelium (TIS and BUN data). Considering the requirement of lethal irradiation for achieving significant levels of renal tubule engraftment, the consequences are still unclear. Two studies have demonstrated that lethally irradiated mice subjected to ischaemia/reperfusion renal injury and subsequently injected with HSCs, showed that HSCs could contribute to functional regeneration of damaged renal proximal tubules and transdifferentiate into renal tubular cells (Kale et al. 2003; Lin et al. 2003). However, the second study showed that lethally irradiated mice subjected to ischaemia/reperfusion renal injury and subsequently injected with BMCs showed that BMCs did not improve renal function although BMCs consisted of 8.8% of tubular epithelial cells (Lin et al. 2005). Concerning MSCs, several studies have demonstrated that non‐irradiated mice subjected to ischaemia/reperfusion then renal injury and subsequently injected with MSCs, and showed that MSCs could improve renal function without the precondition of lethal irradiation. However, the mechanism of this benefit from MSCs on ischaemia/reperfusion renal injury is unclear, this may be either transdifferentiation (Morigi et al. 2004) or paracrine effects (Duffield et al. 2005; Lange et al. 2005; Togel et al. 2005). Taken together, it seems that lethal irradiation is required for HSCs and BMCs to achieve significant levels of renal tubule engraftment; however, this may not be the same for MSCs to accomplish significant levels of renal tubule engraftment.
In addition, one clinical study supports that autologous bone marrow infusion is beneficial in healing after ischaemic heart disease (Perin et al. 2003). The benefits appear related to preserving or re‐establishing microvessels and limiting the extent and severity of damage (Nishida et al. 2003). Moreover, further information has shown that after intrarenal arterial injection of autologous MSCs, transplanted cells could transdifferentiate into renal tubular cells but did not attenuate the renal injury (Behr et al. 2007). Indeed, if autologous bone marrow should be used, rejection would clearly not be a problem.
Regarding the different cell types and the number of cells injected, the number of HSCs used was a little higher in the study of Lin et al. (2 × 103) and Kale et al. (5 × 103) than that in our investigation, estimated to be around 1–2 × 103 HSCs injected, according to incidence of HSCs in the bone marrow (1 in 5–10 × 105 BMCs) (Spangrude et al. 1988; Morrison et al. 1997). The number of MSCs injected was also higher in the work of Morigi et al. (2 × 105) than that in ours, estimated to be around 20–2000 MSCs, according to incidence of MSCs in bone marrow (1 in 0.1–10 × 105 BMCs) (Barry 2003; Zhao et al. 2004).
It is well known that bone marrow stem cell homing depends on creating spaces or niches for the stem cells to occupy and repopulate, although some animal studies have demonstrated that repeated infusion with a high dose of BMCs (2–8 × 108) into non‐myeloablated recipients can also establish a high level of chimerism in bone marrow (19–88%), spleen (30–100%) and thymus (28–50%) at 5 and 7 weeks after BMC infusion (Blomberg et al. 1998), reviewed in Quesenberry et al. 1999, 2001). However, it is still uncertain whether a single infusion of BMCs into non‐myeloablated recipients can establish chimerism in the bone marrow and spleen (Takada & Takada 1971; Takada et al. 1971; Ramshaw et al. 1995; Nilsson et al. 1997, 1999). Two earlier studies showed that after a single injection, donor BMCs rarely enter the bone marrow in non‐irradiated hosts (Takada et al. 1971; Takada & Takada 1971). In contrast, later studies have demonstrated that a single BMC infusion into non‐myeloablated mice could achieve chimerism in the bone marrow (Ramshaw et al. 1995; Nilsson et al. 1997, 1999). For example, Nilsson et al. reported that a single infusion with BMCs (108 or 1.8 × 108 cells) into non‐myeloablated mice could repopulate bone marrow by up to 30%, analysed 6 weeks after BMC administration by Southern blot analysis (Nilsson et al. 1997, 1999). In our study, where a single infusion of exogenous BMCs was given to recipients without myeloablation, donor BMCs could also home back to haematopoietic areas such as bone marrow, analysed by a different method – direct visualization (in situ hybridization procedure) to detect donor BMCs. This recipient chimerism obviously required infusion with donor BMCs, but was not dependent on HgCl2‐induced damage. It is already known that mixed chimerism can be achieved after HSC transplantation to rescue sufferers of genetic diseases such as haemoglobinopathies and severe combined immunodeficiency disease; however, complication of irradiation to the recipient can not be neglected (Fang & Poulsom 2003). In this present study, we find that even in the absence of irradiation, BMC transplantations can establish chimerism, with obvious therapeutic implications.
It was uncertain whether engrafted donor HSCs are capable of proliferation in the bone marrow and spleen of non‐myeloablated recipients. Earlier work from Hendrikx et al. who transplanted sorted HSCs labelled with PKH26 into non‐myeloablated mice, noted the proliferative activity of engrafted donor HSCs in the bone marrow and spleen (Hendrikx et al. 1996). Later, in contrast, Zhong et al. again using FACS analysis concluded that there was no proliferative activity in engrafted donor HSCs (Zhong et al. 2002). Here, though, not resolving the issue of whether or not injected HSC can proliferate in the bone marrow and spleen of non‐myeloablated recipients, clearly shows that there are cells within whole bone marrow capable of proliferation. However, proliferative ability of engrafted cells within bone marrow does not mean that these cells can participate in some healing process and that they may respond to their host signals. A further investigation is necessary to evaluate whether these engrafted cells would play a role in repair of subsequent renal tubular damage.
In summary, the present study confirmed that donor BMCs seemingly contribute little to the repair of the HgCl2‐damaged kidney in non‐irradiated mice, although chimerism was established in the bone marrow and spleen. These engrafted donor BMCs within spleen and bone marrow had the ability to synthesize DNA, which might account for the rising level of chimerism seen in the spleen. At present, we do not know whether this mixed chimerism of bone marrow established in recipients could play a role in repair of subsequent renal tubular damage.
ACKNOWLEDGEMENTS
This study was supported by Cancer Research UK, and Tzu Chi General Hospital, Hualien, Taiwan. We gratefully acknowledge the support of our colleagues, particularly, Ms. Evangelia Prodromidi and Mr. George Elia.
REFERENCES
- Alison MR, Poulsom R, Otto WR, Vig P, Brittan M, Direkze NC, Lovell M, Fang TC, Preston SL, Wright NA (2004) Recipes for adult stem cell plasticity: fusion cuisine or readymade? J. Clin. Pathol. 57, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi S, Stokes D, Augelli B, Digirolamo C, Prockop D (1998) Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats‐similarities to astrocyte grafts. Proc. Natnl Acad. Sci. USA 95, 3908–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry FP (2003) Biology and clinical applications of mesenchymal stem cells. Birth Defects Res. C Embryo Today 69, 250–256. [DOI] [PubMed] [Google Scholar]
- Behr L, Hekmati M, Fromont G, Borenstein N, Noel LH, Lelievre‐Pegorier M, Laborde K (2007) Intra renal arterial injection of autologous mesenchymal stem cells in an ovine model in the postischemic kidney. Nephron Physiol. 107, 65–76. [DOI] [PubMed] [Google Scholar]
- Blomberg M, Rao S, Reilly J, Tiarks C, Peters S, Kittler E, Quesenberry P (1998) Repetitive bone marrow transplantation in nonmyeloablated recipients. Exp. Hematol. 26, 320–324. [PubMed] [Google Scholar]
- Borlongan CV, Hadman M, Sanberg CD, Sanberg PR (2004) Central nervous system entry of peripherally injected umbilical cord blood cells is not required for neuroprotection in stroke. Stroke 35, 2385–2389. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T, Bonventre JV (2005) Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow‐derived stem cells. J. Clin. Invest. 115, 1743–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumenil D, Droz D, Droz JP, Frindel E (1982) Some effects of chemotherapeutic drugs. III. Short‐ and long‐term effects of cis‐platinum on various hematopoietic compartments and on the kidney of the mouse. Cancer Chemother. Pharmacol. 8, 267–270. [DOI] [PubMed] [Google Scholar]
- Ende N, Chen R, Reddi AS (2004) Transplantation of human umbilical cord blood cells improves glycemia and glomerular hypertrophy in type 2 diabetic mice. Biochem. Biophys. Res. Commun. 321, 168–171. [DOI] [PubMed] [Google Scholar]
- Fang TC, Alison MR, Cook HT, Jeffery R, Wright NA, Poulsom R (2005) Proliferation of bone marrow‐derived cells contributes to regeneration after folic acid‐induced acute tubular injury. J. Am. Soc. Nephrol. 16, 1723–1732. [DOI] [PubMed] [Google Scholar]
- Fang TC, Alison MR, Wright NA, Poulsom R (2004) Adult stem cell plasticity: will engineered tissues be rejected? Int. J. Exp. Pathol. 85, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang TC, Poulsom R (2003) Cell‐based therapies for birth defects: a role for adult stem cell plasticity? Birth Defects Res. C Embryo Today 69, 238–249. [DOI] [PubMed] [Google Scholar]
- Hendrikx PJ, Martens CM, Hagenbeek A, Keij JF, Visser JW (1996) Homing of fluorescently labeled murine hematopoietic stem cells. Exp. Hematol. 24, 129–140. [PubMed] [Google Scholar]
- Herzog EL, Chai L, Krause DS (2003) Plasticity of marrow‐derived stem cells. Blood 102, 3483–3493. [DOI] [PubMed] [Google Scholar]
- Horwitz E, Prockop D, Fitzpatrick L, Koo W, Gordon P, Neel M, Sussman M, Orchard P, Marx J, Pyeritz R, Brenner M (1999) Transplantability and therapeutic effects of bone marrow‐derived mesenchymal cells in children with osteogenesis imperfecta. Nat. Med. 5, 309–313. [DOI] [PubMed] [Google Scholar]
- Horwitz EM, Prockop DJ, Gordon PL, Koo WW, Fitzpatrick LA, Neel MD, McCarville ME, Orchard PJ, Pyeritz RE, Brenner MK (2001) Clinical responses to bone marrow transplantation in children with severe osteogenesis imperfecta. Blood 97, 1227–1231. [DOI] [PubMed] [Google Scholar]
- Kale S, Karihaloo A, Clark PR, Kashgarian M, Krause DS, Cantley LG (2003) Bone marrow stem cells contribute to repair of the ischemically injured renal tubule. J. Clin. Invest. 112, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa Y, Verma IM (2003) Little evidence of bone marrow‐derived hepatocytes in the replacement of injured liver. Proc. Natl. Acad. Sci. USA 100 (Suppl. 1), 11850–11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrito JE, Yoshiga K, Sakurai K, Takada K (1994) Effects of intralesional injection of cisplatin dissolved in urografin and lipiodol on Ehrlich ascites tumor and normal tissues of CD‐1 mice. Cancer Chemother. Pharmacol. 34, 323–330. [DOI] [PubMed] [Google Scholar]
- Lange C, Togel F, Ittrich H, Clayton F, Nolte‐Ernsting C, Zander AR, Westenfelder C (2005) Administered mesenchymal stem cells enhance recovery from ischemia/reperfusion‐induced acute renal failure in rats. Kidney Int. 68, 1613–1617. [DOI] [PubMed] [Google Scholar]
- Lin F, Cordes K, Li L, Hood L, Couser WG, Shankland SJ, Igarashi P (2003) Hematopoietic stem cells contribute to the regeneration of renal tubules after renal ischemia‐reperfusion injury in mice. J. Am. Soc. Nephrol. 14, 1188–1199. [DOI] [PubMed] [Google Scholar]
- Lin F, Moran A, Igarashi P (2005) Intrarenal cells, not bone marrow‐derived cells, are the major source for regeneration in postischemic kidney. J. Clin. Invest. 115, 1756–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi M, Imberti B, Zoja C, Corna D, Tomasoni S, Abbate M, Rottoli D, Angioletti S, Benigni A, Perico N, Alison M, Remuzzi G (2004) Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J. Am. Soc. Nephrol. 15, 1794–1804. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, WandycZ AM, Hemmati HD, Wright DE, Weissman IL (1997) Identification of a lineage of multipotent hematopoietic progenitors. Development 124, 1929–1939. [DOI] [PubMed] [Google Scholar]
- Nangaku M, Alpers CE, Pippin J, Shankland SJ, Kurokawa K, Adler S, Morgan BP, Johnson RJ, Couser WG (1998) CD59 protects glomerular endothelial cells from immune‐mediated thrombotic microangiopathy in rats. J. Am. Soc. Nephrol. 9, 590–597. [DOI] [PubMed] [Google Scholar]
- Nilsson SK, Dooner MS, Tiarks CY, Weier HU, Quesenberry PJ (1997) Potential and distribution of transplanted hematopoietic stem cells in a nonablated mouse model. Blood 89, 4013–4020. [PubMed] [Google Scholar]
- Nilsson S, Dooner M, Weier H, Frenkel B, Lian J, Stein G, Quesenberry P (1999) Cells capable of bone production engraft from whole bone marrow transplants in nonablated mice. J. Exp. Med. 189, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Li TS, Hirata K, Yano M, Matsuzaki M, Hamano K (2003) Improvement of cardiac function by bone marrow cell implantation in a rat hypoperfusion heart model. Ann. Thorac. Surg. 75, 768–773; discussion 773–764. [DOI] [PubMed] [Google Scholar]
- Orlic D, Kajstura J, Chimenti S, Bodine DM, Leri A, Anversa P, Bonner‐Weir S, Montminy M (2001a) Transplanted adult bone marrow cells repair myocardial infarcts in mice. Ann. N. Y. Acad. Sci. 938, 221–229; discussion 229–230. [DOI] [PubMed] [Google Scholar]
- Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal‐Ginard B, Bodine DM, Leri A, Anversa P (2001b) Bone marrow cells regenerate infarcted myocardium. Nature 410, 701–705. [DOI] [PubMed] [Google Scholar]
- Perin EC, Dohmann HF, Borojevic R, Silva SA, Sousa AL, Mesquita CT, Rossi MI, Carvalho AC, Dutra HS, Dohmann HJ, Silva GV, Belem L, Vivacqua R, Rangel FO, Esporcatte R, Geng YJ, Vaughn WK, Assad JA, Mesquita ET, Willerson JT (2003) Transendocardial, autologous bone marrow cell transplantation for severe, chronic ischemic heart failure. Circulation 107, 2294–2302. [DOI] [PubMed] [Google Scholar]
- Poulsom R, Alison MR, Forbes SJ, Wright NA (2002) Adult stem cell plasticity. J. Pathol. 197, 441–456. [DOI] [PubMed] [Google Scholar]
- Poulsom R, Forbes SJ, Hodivala‐Dilke K, Ryan E, Wyles S, Navaratnarasah S, Jeffery R, Hunt T, Alison M, Cook T, Pusey C, Wright NA (2001) Bone marrow contributes to renal parenchymal turnover and regeneration. J. Pathol. 195, 229–235. [DOI] [PubMed] [Google Scholar]
- Quesenberry PJ, Stewart FM, Becker P, D’Hondt L, Frimberger A, Lambert JF, Colvin GA, Miller C, Heyes C, Abedi M, Dooner M, Carlson J, Reilly J, McAuliffe C, Stencel K, Ballen K, Emmons R, Doyle P, Zhong S, Wang H, Habibian H (2001) Stem cell engraftment strategies. Ann. N. Y. Acad. Sci. 938, 54–61; discussion 61–52. [DOI] [PubMed] [Google Scholar]
- Quesenberry PJ, Stewart FM, Zhong S, Habibian H, McAuliffe C, Reilly J, Carlson J, Dooner M, Nilsson S, Peters S, Stein G, Stein J, Emmons R, Benoit B, Bertoncello I, Becker P (1999) Lymphohematopoietic stem cell engraftment. Ann. N. Y. Acad. Sci. 872, 40–45; discussion 45–47. [DOI] [PubMed] [Google Scholar]
- Ramshaw HS, Crittenden RB, Dooner M, Peters SO, Rao SS, Quesenberry PJ (1995) High levels of engraftment with a single infusion of bone marrow cells into normal unprepared mice. Biol. Blood Marrow Transplant. 1, 74–80. [PubMed] [Google Scholar]
- Schrier RW, Wang W, Poole B, Mitra A (2004) Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J. Clin. Invest. 114, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241, 58–62. [DOI] [PubMed] [Google Scholar]
- Strauer BE, Brehm M, Zeus T, Kostering M, Hernandez A, Sorg RV, Kogler G, Wernet P (2002) Repair of infarcted myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans. Circulation 106, 1913–1918. [DOI] [PubMed] [Google Scholar]
- Takada Y, Takada A (1971) Proliferation of donor hematopoietic cells in irradiated and unirradiated host mice. Transplantation 12, 334–338. [DOI] [PubMed] [Google Scholar]
- Takada A, Takada Y, Ambrus JL (1971) Proliferation of donor spleen and bone‐marrow cells in the spleens and bone marrows of unirradiated and irradiated adult mice. Proc. Soc. Exp. Biol. Med. 136, 222–226. [DOI] [PubMed] [Google Scholar]
- Terai S, Sakaida I, Yamamoto N, Omori K, Watanabe T, Ohata S, Katada T, Miyamoto K, Shinoda K, Nishina H, Okita K (2003) An in vivo model for monitoring trans‐differentiation of bone marrow cells into functional hepatocytes. J. Biochem. (Tokyo) 134, 551–558. [DOI] [PubMed] [Google Scholar]
- Thadhani R, Pascual M, Bonventre JV (1996) Acute renal failure. N. Engl. J. Med. 334, 1448–1460. [DOI] [PubMed] [Google Scholar]
- Togel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C (2005) Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation‐independent mechanisms. Am. J. Physiol. Renal Physiol. 289, F31–F42. [DOI] [PubMed] [Google Scholar]
- Vendrame M, Cassady J, Newcomb J, Butler T, Pennypacker KR, Zigova T, Sanberg CD, Sanberg PR, Willing AE (2004) Infusion of human umbilical cord blood cells in a rat model of stroke dose‐dependently rescues behavioral deficits and reduces infarct Volume. Stroke 35, 2390–2395. [DOI] [PubMed] [Google Scholar]
- Zhao RC, Liao L, Han Q (2004) Mechanisms of and perspectives on the mesenchymal stem cell in immunotherapy. J. Lab. Clin. Med. 143, 284–291. [DOI] [PubMed] [Google Scholar]
- Zhong JF, Zhan Y, Anderson WF, Zhao Y (2002) Murine hematopoietic stem cell distribution and proliferation in ablated and nonablated bone marrow transplantation. Blood 100, 3521–3526. [DOI] [PubMed] [Google Scholar]