

Abstract
Objectives: Sphingosine kinase (SphK), which is regulated by hypoxia, catalyses phosphorylation of sphingosine to produce sphingosine‐1‐phosphate, which stimulates invasiveness of gliomas. However, whether SphK is involved in proliferation of glioma cells under hypoxic conditions is not clearly understood. In this study, we have investigated the role of SphK in of proliferation glioma cells under hypoxia.
Materials and methods: Effects of small interfering RNA (siRNA) on SphKs, SKI (inhibitor of SphK) and U0126 (inhibitor of ERK) on proliferation of glioma cells under hypoxia were studied using CCK‐8 assay and flow cytometry. Protein expression profiles were evaluated by Western blot analysis.
Results: SKI suppressed proliferation of glioma cells under hypoxia. Similarly, downregulation of SphKs by siRNA inhibited glioma cell proliferation, and the cell cycle was arrested in G2/M phase when SphK1 was inhibited. In addition, inhibition of SphK1 attenuated phosphorylation of ERK in hypoxic conditions. Furthermore, U0126 markedly inhibited cell population growth and arrested cells in G2/M as effectively as SKI. However, silencing SphK2 induced cell cycle arrest in the S phase and it showed little effect on hypoxia‐induced activation of ERK.
Conclusions: SphK1 and SphK2 are involved in proliferation of glioma cells in hypoxic conditions through distinct signalling pathways. SphK1, but not SphK2, promotes cell population expansion in hypoxic conditions by activating ERK.
Introduction
Glioblastoma multiforme (GBM) is the most prevalent and lethal type of CNS tumour. Patients with GBM have a median survival of 10–12 months after standard therapy of surgical resection followed by radiation therapy and chemotherapy with temozolomide (1, 2, 3, 4). Hypoxia‐induced necrosis, and neovascularization are two main indices in pathological diagnosis of GBM. Hypoxic regions are frequently found in GBM, and increased levels of tumour hypoxia have been associated with poor clinical outcomes (5, 6, 7, 8). Sphingosine kinase 1 (SphK1), one of SphK isoforms, is upregulated by hypoxia, and overexpression of SphK1 is correlated with poor prognosis in patients with GBM (9, 10, 11). The two identified mammalian isoforms of sphingosine kinase, SphK1 and SphK2, have different developmental and tissue expression patterns. Previous reports have indicated that overexpression of SphK1 promotes tumourigenesis, whereas SphK2 seems to regulate apoptosis (12, 13, 14). However, precise roles of SphK1 and SphK2 in proliferation of glioma cells under hypoxia remain unclear. Sphingosine‐1‐phosphate (S1P) is derived from phosphorylation of sphingosine, which is catalysed by sphingosine kinase. S1P is a potent lipid mediator that regulates a broad variety of cellular processes, such as cell survival, apoptosis, calcium homeostasis, vascular maturation and angiogenesis. In addition, S1P acts as a second messenger to regulate intracellular signalling (15, 16).
Extracellular signal‐regulated kinase (ERK), a member of a subfamily of MAPKs, is a key molecule that promotes cell proliferation in hypoxia (5, 16); hypoxia increases expression of SphK1 and activity of ERK. However, roles of SphK1 and SphK2 in population expansion of glioma cells, and relationships between sphingosine kinases and ERK activity under hypoxic conditions, remain unclear. In this study we have investigated molecular mechanisms that may be used by sphingosine kinases to regulate proliferation of glioma cells under hypoxia.
Materials and methods
Materials and cell culture
S1P, U73122 and CoCl2 were obtained from Sigma (St Louis, MO, USA), SKI and pertussis toxin (PTX) were obtained from Merck (Darmstadt, Germany). Human glioma cell lines U87MG (ATCC) and U251 (CCTCC) were maintained at 37 °C in a humidified incubator with 5% CO2, in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS; Gibco‐BRL, NY, USA). For hypoxia experiments, cells were incubated in presence or absence of 150 μm CoCl2. Further cells were incubated at different times in atmosphere of 5% CO2 and 92% nitrogen, with an oxygen sensor set at 3% O2.
Cell transfection
siRNA sequences were as follows: SphK1 siRNA‐1: 5′‐CCU AGA GAG UGA GAA GUA UTT‐3′; SphK1 siRNA‐2: 5′‐GCG UCA UGC AUC UGU UCU ATT‐3′; SphK2 siRNA‐1: 5′‐GCT GGG CTG TCC TTC AAC CT‐3′; SphK2 siRNA‐2: 5′‐CAG GAT TGC GCT CGC TTT CAT‐3′; and scrambled siRNA: 5′‐UUC UCC GAA CGU GUC ACG UTT‐3′ as negative control. All siRNAs were synthesized by GenePharma (Shanghai, China). U87MG cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following standard procedures. Cells were incubated for 72 h after transfection before further experiments were performed.
Quantitative RT‐PCR
Total RNA was extracted from cells using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Single‐stranded cDNA was synthesized using a Sensiscript RT kit (Takara, Japan). Quantitative real‐time PCR (RT‐PCR) was performed with Mx3005P instrumentation (Strategene, San Diego, CA, USA) and SYBR Premix Ex Taq kit (Nustar Laboratory). Sequences of SphK1 and SphK2 primers were as follows: SphK1 sense 5′‐CTG GCA GCT TCC TTG AAC CAT‐3′ and antisense 5′‐TGT GCA GAG ACA GCA GGT TCA‐3′; and SphK2 sense 5′‐CCA GTG TTG GAG AG CTG AAG GT‐3′and antisense 5′‐GTC CAT TCA TCT GCT GGT CCT C‐3′. C t values were normalized housekeeping gene β‐actin (sense 5′‐ATT GGC AAT GAG CGG TTC C‐3′; antisense 5′‐GGT AGT TTC GTG GAT GCC ACA‐3′). Cycling conditions were 10 min initial denaturation followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. Specificity of PCR products was controlled using a melting curve analysis.
Western blot analysis
Western blot analysis was used to quantitatively detect specific proteins or phosphorylated forms of proteins, in lysed U87MG cells. Equal amounts of proteins were loaded on 10% SDS–polyacrylamide gel for electrophoresis. Proteins were transferred to PVDF membranes and incubated overnight with primary antibodies at 4 °C as follows: anti‐SphK1 (1:1000; Cell Signaling Technologies, Danvers, MA, USA), anti‐SphK2 (1:1000; Abcam, Cambridge, UK), anti‐ERK1/2 and anti‐phosphorylated ERK1/2 (1:1000; Cell Signaling) antibodies. After incubation for 1 hour with secondary antibodies at room temperature, immunoreactive bands were detected using the ECL‐system (ECL Plus; Amersham, NJ, USA).
CCK‐8 assay
Cell viability was determined using Cell Counting Kit 8 (CCK‐8; Dojindo Laboratories, Kamimashikl‐gun, Kumamoto, Japan) assay. CCK‐8 reagent (10 μl) was added to cultures (100 μl) followed by incubation for 3 h. Absorbance at 450 nm was measured using a microplate reader. Each experiment was conducted in triplicate with separate cell culture preparations. Results are expressed as percentage of control values.
Cell cycle analysis
Approximately 1 × 106 cells were collected and then fixed in cold 70% ethanol for 12 h. Fluorescence‐activated cells were sorted on a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and analysed using Cell Quest Pro software (BD Biosciences, Franklin Lakes, NJ, USA), cell cycle analysis being performed according to manufacturer’s instructions (KeyGen, Nanjing, China). Relative percentage of cells in each phase of the cell cycle was determined.
Statistical analysis
Statistical analyses were performed using (SPSS 15.0, USA), multiple comparisons made using one‐way anova. In addition, Student’s unpaired t‐test was used for statistical analysis. P < 0.05 was considered statistically significant difference. Data are represented as mean ± SD.
Results
Effect of hypoxia on expression of SphK1 and SphK2 in U87MG glioma cells
To establish a model of hypoxia, CoCl2, a well‐known hypoxia mimetic, was used to treat U87MG glioma cells, 150 μm being used for treatment; cells were then harvested and assayed by western blot analysis. As shown in Fig. 1a, protein levels of SphK1 increased during CoCl2 administration starting 1 h after initiation of treatment reaching maximum after 4 h. Notably, expression of SphK2 decreased in response to CoCl2 treatment. The data show that in these cells, SphK1 is upregulated during hypoxic stress, which could occur during rapid tumour cell proliferation.
Figure 1.
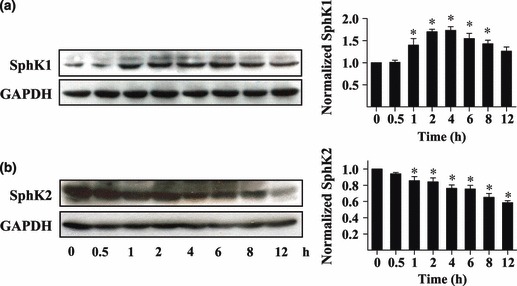
Effects of hypoxia on expression of SphKs in U87MG glioma cells. U87MG cells were incubated for different times in 150 μm CoCl2 and collected for western blot analysis. Protein expression profiles of SphK1 and SphK2 were evaluated. Quantitative analysis of western blot results normalized to those of GADPH are shown. Data shown as mean ± SD (n = 4), *P < 0.05.
Inhibitory effects of SKI on proliferation of glioma cells under hypoxia
Here, we investigated involvement of sphingosine kinases on glioma cell proliferation during hypoxic stress. Both U251 and U87MG glioma cells were pre‐incubated with 20 μm SphK inhibitor (SKI) for 30 min, followed by hypoxia (3% O2 or 150 μm CoCl2). Cell viability was measured over 72 h using the CCK‐8 assay. As shown in Fig. 2a, proliferation of cells treated with SKI under hypoxia (3% O2 incubator) was markedly reduced compared to that of untreated cells (407.5% vs. 696.7% in U251 cells and 497.0% vs. 768.3% in U87MG cells at 72 h). In addition, SKI significantly suppressed proliferation of glioma cells under hypoxic conditions compared to cell proliferation under normal conditions (407.5% vs. 569.4% in U251 cells and 497.0% vs. 686.0% in U87MG cells at 72 h). In parallel experiments, under 150 μm CoCl2‐induced hypoxia, SKI also significantly reduced proliferation of the cells (420.6% vs. 690.0% in U251 cells and 475.6% vs. 722.1% in U87MG cells at 72 h). Compared to normal conditions, inhibitory effects of SKI on proliferation was more effective (420.6% vs. 565.0% in U251 cells and 475.6% vs. 626.0% in U87MG cells at 72 h, Fig. 2b). These results suggested that sphingosine kinase was required for proliferation of our glioma cells in hypoxia.
Figure 2.
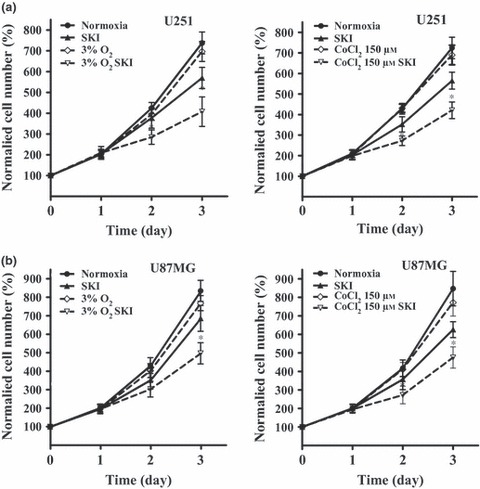
Effects of SKI on proliferation of glioma cells under hypoxia. Growth curve of U251MG cells (a); Growth curve of U87 cells (b). Cells were cultured with vehicle or SKI, and harvested at different intervals for CCK‐8 assay. The growth curve was plotted with normalized cells numbers. Data shown as mean ± SD (n = 6, n = 3). Hypoxia was induced using 3% O2 or 150 μm CoCl2. Cells were pre‐incubated with 20 μm SKI for 30 min before hypoxia treatment. *P < 0.05.
Downregulation of SphK1 or SphK2 inhibited proliferation of U87MG cells under hypoxia
To provide direct evidence that sphingosine kinases are involved in proliferation of glioma cells under hypoxia, SphK1 and SphK2 were specifically knocked down using siRNA technology. U87MG cells were treated with Scr, SphK1, or SphK2 siRNA. As shown in Fig. 3a,b, both sequences were highly specific in downregulation of genes of interest at the mRNA level. Protein expression profiles of SphK1 in SphK1 siRNA‐1‐transfected cells were 34.5% of that found in Scr siRNA‐transfected cells. Numbers of U87MG cells were greatly reduced after downregulation of SphK1 under hypoxic conditions (69.7% of Scr group) and protein expression of SphK2 was also markedly knocked down in SphK2 siRNA‐2‐transfected cells (33.9% vs. 100%). Downregulation of SphK2 inhibited proliferation of U87MG cells (73.7% vs. 100%). These data confirm that both SphK1 and SphK2 are partially responsible for proliferation of glioma cells in hypoxia.
Figure 3.
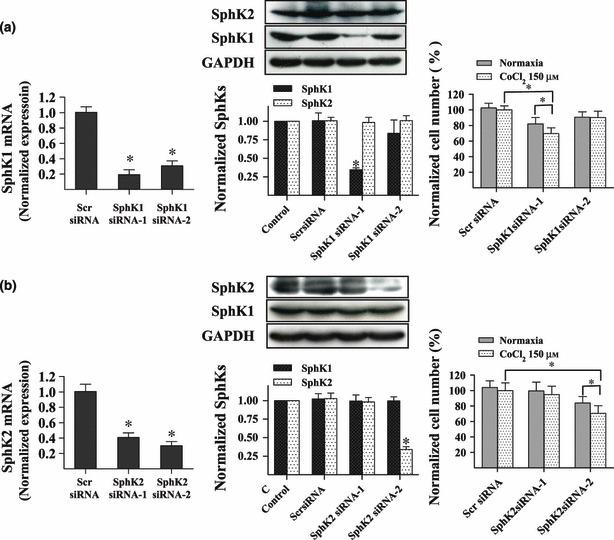
Downregulation of SphK1 and SphK2 suppressed proliferation of U87MG cells under hypoxia. Real‐time PCR and western blot analysis revealed efficiency of siRNA‐mediated suppression of SphK1. Downregulation of SphK1 inhibited U87MG cell proliferation under hypoxia. Cell proliferation was measured using the CCK‐8 assay. Data shown as mean ± SD (n = 8, n = 3) (a). Real‐time PCR and western blot analysis revealed efficiency of siRNA‐mediated suppression of SphK2. Results show that SphK2 was knocked down by SphK2 siRNA‐2. Downregulation of SphK2 inhibited U87MG cell proliferation under hypoxia. Cells were transfected with siRNAs and cell number was counted via CCK‐8 assay. Data shown as the mean ± SD (n = 6, n =3) (b). *P < 0.05.
Downregulation of SphK1 or SphK2 inhibits proliferation of U87MG cells by arresting the cell cycle
Here, we aimed to identify mechanisms by which sphingosine kinases influence proliferation of glioma cells. We examined the cell cycle in U87MG cells after knockdown of sphingosine kinases. As shown in Fig. 4a, flow cytometry assays demonstrated that silencing SphK1 arrested the cell cycle in G2/M phase (Scr siRNA‐transfected cells, 28.10%; SphK1 siRNA‐transfected cells, 11.57%). However, silencing SphK2 arrested the cell cycle in S phase (Scr siRNA‐transfected cells, 26.48%; SphK2 siRNA‐transfected cells, 18.90%, Fig. 4b). These results suggest that SphK1 and SphK2 are, indeed, involved in glioma cell proliferation under hypoxia, but through distinct signal transduction pathways.
Figure 4.
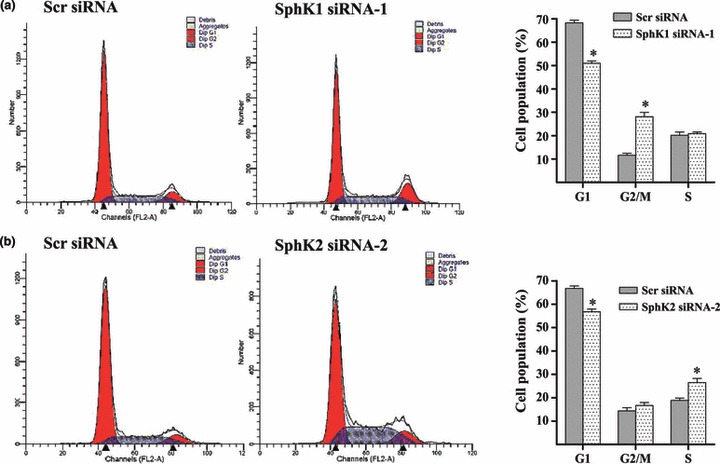
Downregulation of SphK1 or SphK2 caused cell cycle arrest. Downregulation of SphK1 caused cell cycle arrest in G2/M phase. Flow cytometry analysis measured cycle of U87MG cells. Percentage of SphK1 siRNA‐transfected U87MG cells was significantly enhanced in G2/M phase and markedly reduced in G1 phase (a). Downregulation of SphK2 caused cell cycle arrest in S phase. Percentage of SphK2 siRNA‐transfected U87MG cells was significantly enhanced in S phase (b). For hypoxia experiments, cells were treated with 150 μm CoCl2 for 48 h. *P < 0.05.
SphK1, but not SphK2, promotes proliferation of U87MG cells by activating ERK under hypoxia
Activation of ERK has been shown to mediate the proliferative effect in hypoxia (17, 18, 19), so we detected changes in ERK activity during hypoxic conditions to approach weather activation of ERK would be involved in proliferation of U87MG cells. As shown in Fig. 5a, phosphorylation of ERK in U87MG cells reached 1.82‐fold maximum increase after 1 h of CoCl2 treatment; U0126 a MAPK kinase inhibitor and was used to treat them. As shown in Fig. 5c, U0126 significantly inhibited their proliferation under hypoxia (300.3% vs. 405.6% at 48 h, 582.6% vs. 860.7% at 72 h). Proliferation of U0126‐treated cells under hypoxic conditions was markedly reduced compared to U0126‐treated cells under normal conditions. U0126 induced cell cycle arrest in G2/M phase under hypoxia; these results suggesting that ERK is involved in proliferation of U87MG cells.
Figure 5.
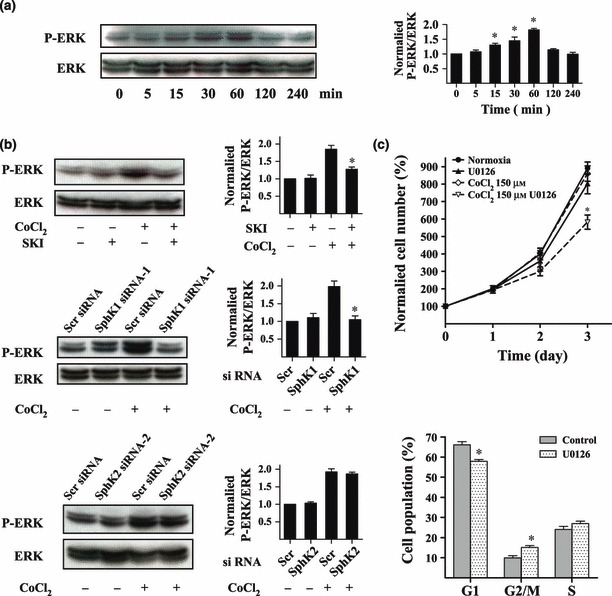
SphK1, but not SphK2, promoted proliferation of U87MG cells by activating ERK under hypoxia. Total ERK and P‐ERK1/2 in U87MG cells were determined using western blot analysis. Cells were treated with 150 μm CoCl2 for different times. Quantitative analysis of western blot results is provided on the right (a). U87MG cells were pre‐treated with 20 μm SKI or transfected with siRNAs. Then cells were incubated with 150 μm CoCl2 for 1 h and collected for western blot analysis to evaluate total ERK and P‐ERK1/2. Quantitative analysis of western blot results is provided on the right (b). Effect of U0126 on proliferation of U87MG cells was measured using CCK‐8 assay. Effect of U0126 on cycle of U87MG cells was detected using flow cytometry. Cells were pre‐incubated with U0126 for 30 min before treatment with 150 μm CoCl2 (c). *P < 0.05.
We investigated whether ERK activity was involved in mechanisms by which sphingosine kinases influenced proliferation of U87MG cells under hypoxia. Our results show that phosphorylation of ERK was significantly attenuated in cells that had either been pre‐treated with SKI (20 μm) or transfected with SphK1 siRNA (Fig. 5b). However, hypoxia‐induced increase in phosphorylation of ERK was not affected in cells transfected with SphK2 siRNA. These results suggest that SphK1‐induced, but not SphK2‐induced, glioma cell proliferation requires ERK activity.
SphK1 enhances proliferation of glioma cells through the GPCR pathway
The glioma cells were pre‐incubated in 0.5 μm S1P for 30 min before treatment with 150 μm CoCl2. S1P significantly increased numbers of U87MG cells that were transfected with SphK1 siRNA‐1, but not SphK2 siRNA‐2 (Fig. 6a). It has been established that S1P can be released into the extracellular milieu or if partitioned into lipid microenvironments has privileged binding to S1P receptors of the G‐protein–coupled receptor type (20). Thus in our studies, U87MG cells were pre‐treated with pertussis toxin (PTX), a specific inhibitor of Gi/o‐coupled receptors, G‐protein‐coupled receptor (GPCR) isoforms, or U73122, an inhibitor of phospholipase C (PLC), to assess possible involvement of GPCR pathway in proliferation of our glioma cells under hypoxia. As shown in Fig. 6b, at 72 h hypoxia, PTX and U73122 markedly reduced proliferation of the cells under hypoxia by 270.6% and 335.6%, respectively. In addition, cells that were treated with PTX or U73122 significantly inhibited cell proliferation in these conditions compared to control cells or PTX‐ or U73122‐treated cells under normal conditions. Furthermore, PTX and U73122 treatments arrested the cell cycle in the G2/M phase (Fig. 6b). PTX and U73122 treatments significantly reduced hypoxia‐induced phosphorylation of ERK (Fig. 6c). These results suggest that under hypoxia, SphK1 enhances proliferation of the cells through the GPCR pathway, to activate ERK.
Figure 6.
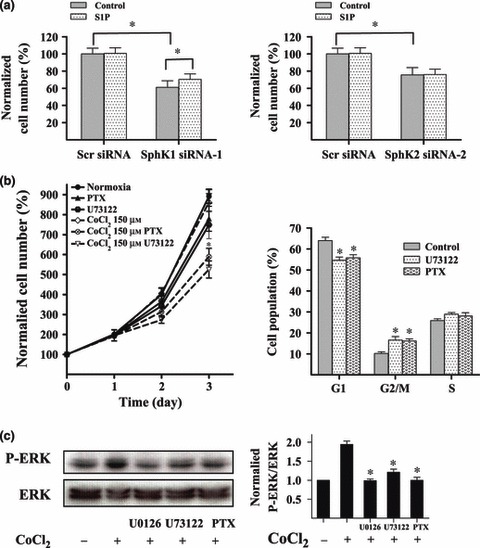
SphK1 enhanced proliferation of glioma cells via the GPCR pathway. U87MG cells were transfected with siRNA and incubated in 0.5 μm S1P for 48 h. Cell number was counted using CCK‐8 assay. Application of 0.5 μm S1P significantly reduced SphK1 siRNA‐1‐mediated inhibition of U87MG cell proliferation under hypoxia for 48 h (a). U87MG cells were cultured with vehicle, PLC inhibitor U73122 (10 μm) or GPCR inhibitor PTX (100 ng/ml), and were harvested at different intervals for CCK‐8 assay. Cell growth curve was plotted with normalized cells numbers. Application of U73122 or PTX inhibited proliferation of the glioma cells under hypoxia. Cells were pre‐incubated with U73122 or PTX for 30 min and administration with 150 μm CoCl2 (b). U73122 and PTX induced cell cycle arrest in G2/M phase under hypoxia. Cells were pre‐incubated with U73122 or PTX for 30 min before the 48 h treatment with 150 μm CoCl2 (c). Total ERK and P‐ERK1/2 were determined using western blot analysis. U73122 (10 μm) or PTX (100 ng/ml) significantly reduced phosphorylation levels of ERK under hypoxia. To induce hypoxia, cells were treated with 150 μm CoCl2 for 1 h (d). *P < 0.05.
Discussion
Glioblastomas are among the most lethal cancers. Recent studies have indicated that sphingosine kinase is overexpressed in a variety of tumours and is linked to cancer development and progression (14, 21). Several studies have implicated a role for SphK1 in progression of oncogenesis and responses to treatment. High levels of SphK1 expression correlate with reduced survival of GBM patients (14). In addition, anti‐cancer treatments have been shown to downregulate SphK1 activity in various cancer cell and animal models, suggesting that SphK1 may act as a ‘sensor’ to anti‐cancer chemotherapy and radiation therapy (22). There is little information concerning SphK2, although some reports suggest that SphK2 may enhance resistance of cancer cells to therapeutic agents.
Hypoxia is a well‐recognized tumour microenvironment linked to poor patient outcome and resistance to therapy (7). Hypoxia increases expression of SphK1, yet the role of SphK1 in survival of glioma cells under hypoxia remains unclear. Therefore, in this study, we proposed a novel function for SphK1 in malignancy to mediate its adaptation to hypoxia. To determine whether sphingosine kinase would affect proliferation of glioma cells under hypoxia, we pre‐incubated U87MG cells with SKI, a sphingosine kinase inhibitor. Proliferation of the cells was strongly suppressed by SKI administration under hypoxia. Furthermore, we downregulated SphK1 and SphK2 using siRNA, is which inhibited cell proliferation under hypoxic conditions. Flow cytometric analysis revealed that SphK1 knockdown induced cell cycle arrest in G2/M phase and that SphK2 knockdown induced cell cycle arrest in S phase. This suggested that SphK1 and SphK2 regulate proliferation of glioma cells under hypoxia using distinct signal transduction pathways; however, tissue levels of oxygen is far less than 20%, and with considerable local and regional variation (23). The true hypoxic levels in tumours are uncertain in vivo; in our studies, we established a model of hypoxia in vitro using 150 μm CoCl2, a well‐known hypoxia mimetic, or 3% oxygen. Thus, under hypoxic conditions, sphingosine kinases regulated proliferation of the glioma cells only in an in vitro model; their true effects in vivo are not yet clear.
Sphingosine kinase generates S1P from its metabolic precursor sphingosine. In addition, S1P is the ligand for a family of specific G‐protein‐coupled receptors that regulate a wide variety of important cellular functions including vascular maturation, angiogenesis, cell population growth and survival, cytoskeletal rearrangements and cell motility (24). Sphingosine kinase, which is responsible for S1P production, is rapidly but transiently stimulated by hypoxia, which reflects its central role in controlling S1P levels. S1P is released from cells to activate cell‐surface S1P receptors to elicit paracrine or autocrine signalling cascades or to act as a second messenger in intracellular spaces (25, 26). Inhibition of sphingosine kinase could impede S1P generation and its downstream effects (27). Thus, sphingosine kinase and its activation play a decisive and necessary role in observed effects ascribed to S1P (20). These findings suggest that hypoxia‐induced effects of sphingosine kinase on proliferation of glioma cells might be mediated by S1P. We therefore analysed glioma cells pre‐incubated with S1P, followed by treatment with CoCl2. Compared to control groups, which lacked pre‐treatment with S1P, survival of U87MG cells transfected with SphK1 siRNA‐1 was partially rescued under hypoxia. However, S1P had no effect on proliferation of U87MG cells transfected with SphK2 siRNA. These results confirm that SphK1 enhanced proliferation of our glioma cells through production of S1P, which is released into the extracellular space. As S1P activates GPCRs, we blocked the GPCR pathway using PTX and U73122. We found that PTX or U73122 treatments significantly reduced proliferation of U87MG cells, providing further evidence for the role of SphK1 through S1P production in glioma cell survival under hypoxia. As S1P is released into the extracellular space it binds to its receptors on plasma membranes. We noted that rescue of cell proliferation by S1P in SphK1 siRNA‐treated cells was not fully effective and hypothesize that there could be further pathways for S1P. In 2010, Alvarez and collaborators found that intracellular S1P could specifically bind to TRAF2 at the amino‐terminal RING domain and stimulate its E3 ligase activity, independently of its cell surface G‐protein‐coupled receptors. Their results highlighted the key role of SphK1 and its product S1P in TNF‐α signalling and the canonical NF‐κB activation pathway, important in anti‐apoptotic processes (28).
Previous reports have shown that hypoxia upregulates ERK phosphorylation (5). ERK is a MAPK subfamily member and is activated by the upstream kinase MEK in response to proliferation stimuli (29). The ERK pathway mediates a number of cell processes, including of proliferation and survival (17). Our results show that phosphorylation of ERK increases during hypoxic stress and that activation of ERK is involved in proliferation of U87MG cells under hypoxia. Sphingosine kinases were inhibited using a chemical agent or siRNA‐mediated knockdown to determine whether sphingosine kinases would affect activation of ERK under hypoxia. Compared to control groups, SKI pre‐treatment or knockdown of SphK1 markedly reduced hypoxia‐induced phosphorylation of ERK in U87MG cells; downregulation of SphK2 had no effect on phosphorylation of ERK under hypoxia. PTX and U73122 also attenuated phosphorylation of ERK in U87MG under hypoxia. These results suggest that the role of SphK1 in proliferation of glioma cells is partly dependent on ERK activation through a GPCR pathway under hypoxia. SphK2 regulates proliferation of glioma cells under hypoxia in an ERK‐independent manner. How SphK2 regulates proliferation of glioma cells remains unclear, and further studies are necessary to answer this question. These findings may help elucidate how signalling pathways cooperate during cell proliferation within hypoxic glioma regions.
In summary, our results show that SphK1 and Sphk2 are involved in proliferation of glioma cells during hypoxia, although they are mediated by distinct signalling pathways. The role of SphK1 in proliferation of the glioma cells required ERK activation through a GPCR pathway; however, function of SphK2 was ERK‐independent. Thus, we propose that relevance of both inhibitory strategies that target SphK1 or SphK2 in cancer might be extended to hypoxia.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 30971193).
References
- 1. Gilbertson RJ (2006) High‐grade glioma: can we teach an old dogma new tricks? Cancer Cell 3, 147–148. [DOI] [PubMed] [Google Scholar]
- 2. Meyer MA (2008) Malignant gliomas in adults. N. Engl. J. Med. 17, 1850; author reply 1850. [DOI] [PubMed] [Google Scholar]
- 3. Bosma I, Vos MJ, Heimans JJ, Taphoorn MJ, Aaronson NK, Postma TJ et al. (2007) The course of neurocognitive functioning in high‐grade glioma patients. Neuro Oncol. 1, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wen PY, Kesari S (2008) Malignant gliomas in adults. N. Engl. J. Med. 5, 492–507. [DOI] [PubMed] [Google Scholar]
- 5. Fang H, Zhang LF, Meng FT, Du X, Zhou JN (2010) Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neurosci. Lett. 3, 173–177. [DOI] [PubMed] [Google Scholar]
- 6. Oliver L, Olivier C, Marhuenda FB, Campone M, Vallette FM (2009) Hypoxia and the malignant glioma microenvironment: regulation and implications for therapy. Curr. Mol. Pharmacol. 3, 263–284. [DOI] [PubMed] [Google Scholar]
- 7. Amberger‐Murphy V (2009) Hypoxia helps glioma to fight therapy. Curr. Cancer Drug Targets 3, 381–390. [DOI] [PubMed] [Google Scholar]
- 8. Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT et al. (2004) Hypoxia is important in the biology and aggression of human glial brain tumors. Clin. Cancer Res. 24, 8177–8184. [DOI] [PubMed] [Google Scholar]
- 9. Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O (2008) Sphingosine kinase 1: a new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 20, 8635–8642. [DOI] [PubMed] [Google Scholar]
- 10. Anelli V, Gault CR, Cheng AB, Obeid LM (2008) Sphingosine kinase 1 is up‐regulated during hypoxia in U87MG glioma cells. Role of hypoxia‐inducible factors 1 and 2. J. Biol. Chem. 6, 3365–3375. [DOI] [PubMed] [Google Scholar]
- 11. Schwalm S, Doll F, Romer I, Bubnova S, Pfeilschifter J, Huwiler A (2008) Sphingosine kinase‐1 is a hypoxia‐regulated gene that stimulates migration of human endothelial cells. Biochem. Biophys. Res. Commun. 4, 1020–1025. [DOI] [PubMed] [Google Scholar]
- 12. Schnitzer SE, Weigert A, Zhou J, Brune B (2009) Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine‐1‐phosphate‐mediated chemoresistance in A549 lung cancer cells. Mol. Cancer Res. 3, 393–401. [DOI] [PubMed] [Google Scholar]
- 13. Vann LR, Payne SG, Edsall LC, Twitty S, Spiegel S, Milstien S (2002) Involvement of sphingosine kinase in TNF‐alpha‐stimulated tetrahydrobiopterin biosynthesis in C6 glioma cells. J. Biol. Chem. 15, 12649–12656. [DOI] [PubMed] [Google Scholar]
- 14. Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW (2005) Sphingosine kinase‐1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 8, 695–705. [DOI] [PubMed] [Google Scholar]
- 15. Payne SG, Milstien S, Spiegel S (2002) Sphingosine‐1‐phosphate: dual messenger functions. FEBS Lett. 1, 54–57. [DOI] [PubMed] [Google Scholar]
- 16. Malchinkhuu E, Sato K, Horiuchi Y, Mogi C, Ohwada S, Ishiuchi S et al. (2005) Role of p38 mitogen‐activated kinase and c‐Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine‐1‐phosphate in glioma cells. Oncogene 44, 6676–6688. [DOI] [PubMed] [Google Scholar]
- 17. Kim JY, Kim YJ, Lee S, Park JH (2009) The critical role of ERK in death resistance and invasiveness of hypoxia‐selected glioblastoma cells. BMC Cancer 27, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Das M, Bouchey DM, Moore MJ, Hopkins DC, Nemenoff RA, Stenmark KR (2001) Hypoxia‐induced proliferative response of vascular adventitial fibroblasts is dependent on g protein‐mediated activation of mitogen‐activated protein kinases. J. Biol. Chem. 19, 15631–15640. [DOI] [PubMed] [Google Scholar]
- 19. Tian Y, Liu Y, Chen X, Zhang H, Shi Q, Zhang J et al. (2010) Tetramethylpyrazine promotes proliferation and differentiation of neural stem cells from rat brain in hypoxic condition via mitogen‐activated protein kinases pathway in vitro. Neurosci. Lett. 1, 26–31. [DOI] [PubMed] [Google Scholar]
- 20. Pyne S, Pyne NJ (2011) Translational aspects of sphingosine 1‐phosphate biology. Trends Mol. Med. 8, 463–472. [DOI] [PubMed] [Google Scholar]
- 21. Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T et al. (2006) Sphingosine kinase 1 is up‐regulated in colon carcinogenesis. FASEB J. 2, 386–388. [DOI] [PubMed] [Google Scholar]
- 22. Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V et al. (2005) Sphingosine kinase‐1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 24, 11667–11675. [DOI] [PubMed] [Google Scholar]
- 23. Csete M (2005) Oxygen in the cultivation of stem cells. Ann. N Y Acad. Sci. 1049, 1–8. [DOI] [PubMed] [Google Scholar]
- 24. Spiegel S, Milstien S (2003) Sphingosine‐1‐phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 5, 397–407. [DOI] [PubMed] [Google Scholar]
- 25. Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK et al. (2009) Regulation of histone acetylation in the nucleus by sphingosine‐1‐phosphate. Science 5945, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S (2006) Role of ABCC1 in export of sphingosine‐1‐phosphate from mast cells. Proc. Natl. Acad. Sci. USA 44, 16394–16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maceyka M, Payne SG, Milstien S, Spiegel S (2002) Sphingosine kinase, sphingosine‐1‐phosphate, and apoptosis. Biochim. Biophys. Acta 2–3, 193–201. [DOI] [PubMed] [Google Scholar]
- 28. Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY et al. (2010) Sphingosine‐1‐phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 7301, 1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K et al. (2001) Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 2, 153–183. [DOI] [PubMed] [Google Scholar]