

Abstract
Abstract. Several methods have been developed for the immortalization of B lymphocytes by Epstein‐Barr virus (EBV). We developed an efficient method which reduces the time from culture initiation to immortalization and cryopreservation. Two infections of EBV to lymphocytes, and the use of phorbol ester‐induced EBV stock significantly improved immortalization efficiency and reduced the time between initiation and immortalization and cryopreservation. The resulting cell bank was used to produce DNA for genetic studies focusing on the genes involved in immune and autistic disorders.
INTRODUCTION
Several methods have been developed for the immortalization of B lymphocytes by Epstein‐Barr virus (EBV) (Werner et al. 1972; Schneider & zur Hausen 1975; Tohda et al. 1978; Neitzel 1986; Pelloquin et al. 1986; Caputo et al. 1991; Wall et al. 1995). Some bypass the separation step and instead use whole blood, either fresh or frozen (Tohda et al. 1978; Ventura et al. 1988), or previously cryopreserved non‐immortalized cells (Louie & King 1991; Pressman & Rotter 1991; Reidy & Wheeler 1992), to simplify the immortalization step and reduce blood volume to that required for establishing a permanent line. Others have used gamma‐irradiated fibroblast type cells as feeder layers to increase the efficiency of culture immortalization (Caputo et al. 1991). To that end, mitogens such as phytohemagglutinin (PHA) (Henderson et al. 1977), pokeweed mitogen (PWM) (Bird et al. 1981), and lipopolysaccharide (LPS) (Henderson et al. 1977) have also been used to stimulate lymphocytes. In addition, cyclosporin A (CSA) has been used to diminish B‐cell regression in proliferation foci by inhibiting suppressor or cytotoxic T‐cell action (Neitzel 1986; Pelloquin et al. 1986; Ventura et al. 1988; Pressman & Rotter 1991).
Although most of above methods are currently useful to immortalize B lymphocytes, the time duration from initiation of culture to immortalization is still the biggest obstacle to be overcome. In addition, the need to bank relatively large numbers of cells as quickly as possible for genome research requires more rapid and reliable methods for immortalization of B lymphocytes. In this study, by modifying the standard method for EBV stock preparation, as well as the infection frequency of EBV, we can greatly reduce the length of time to immortalization and cryopreservation, along with improved efficiency.
MATERIALS AND METHODS
EBV stock preparation
Two different methods were used to prepare virus stock from an EBV‐transformed B95‐8 marmoset cell line. In Method 1, the cells were grown in RPMI 1640 medium, supplemented with 10% fetal bovine serum (FBS). These cultures were maintained for 12–15 days. After centrifugation for 3 min at 850 g, the supernatant was filtered through a 0.22‐µm membrane (Millipore, Bedford, MA, USA) and stored at 4 °C until use. In Method 2, exponentially growing B95‐8 cells (5 × 105/ml) were stimulated with 100 nm tetradecanoyl phorbol acetate (TPA) for 2 h, and then the cells were washed three times with Hanks’ balanced salt solution (HBSS) buffered saline (HBS) to remove PMA in solution. The cells were further incubated for 48 h and the supernatant was prepared as described in Method 1.
Blood collection and lymphocyte separation
Ninety‐seven patients with autism (mean ages 10 ± 4.1) and 105 patients with inflammatory bowel disease (IBD) (mean ages 29 ± 5.5) were selected for this study. On the day of drawing blood samples, we obtained the informed consent from those who participated. For each selected patient, 10 ml of peripheral blood were collected in 10 ml‐Vacutainer tubes containing heparin. All reagents to be used in the lymphocyte isolation were warmed to 37 °C. The blood was divided into 5 ml‐aliquots, underlayered with 4 ml Histopaque 1077, and spun at 850 g for 20 min at room temperature (24 °C). The clear top plasma layer was discarded. The opaque lymphocyte layer, along with the Histopaque, was collected down to the erythrocyte pellet and transferred to another 15‐ml centrifuge tube. The cells were washed twice by repeat centrifugation in RPMI 1640. Finally, the cells were mixed together and re‐suspended in 9 ml RPMI 1640 complete medium. Each 3 ml (∼3 × 106 cells) was used for one of the specific methods (A–C) as described below.
Immortalization of lymphocytes
Three different methods (A–C) were applied for immortalization of B cells. Basically, two methods, A and B, were the same except for the use of EBV supernatant prepared from Methods 1 and 2. Initiation of immortalization was achieved by mixing the cells in 3 ml (∼3 × 106 cells) of RPMI complete medium, 1 ml of active EBV supernatant prepared from Method 1 or 2, and 2 µg/ml CSA (Sandimmun, Sandoz, East Hanover, NJ, USA) in 25‐cm2 tissue culture flask (Corning, New York, NY, USA). Each week, half of the supernatant was replaced by fresh medium containing 1 µg/ml CSA. Although the initial immortalization process was same as the above two methods, Method C allowed a second infection at day 4 after the initial infection. At the day of second infection, the cell aggregates were vigorously shaken and the whole media were changed. After the second infection, every fourth day, total supernatant was replaced by fresh medium containing 1 µg/ml CSA. Each of the lymphoblastoid cell lines cultured by Methods A–C was frozen in 10% dimethylsulfoxide (DMSO) and kept in liquid nitrogen.
Phenotypic analysis of separated lymphocytes
Ten patients with autism and 20 patients with IBD from this study were randomly selected and the ratio of T cells (CD3+) to B cells (CD22+) was compared with normal healthy control groups. These samples were also used to measure DNA synthesis as described below.
Lymphocytes separated from the above method were stained with monoclonal antibodies against CD3 (FITC‐conjugated) and CD22 (phycoerythrin (PE)‐conjugated), and then the percentages of CD3+ and CD22+ cells were determined by flow cytometry on a FACS Calibur (Becton Dickson, San Jose, CA, USA).
Measurement of DNA synthesis
On the day of measurement, cultures (single cells and aggregates) were completely loosened by vigorous agitation (vortex) and triplicate cultures of 200 µl (2 × 105 cells) were seeded in microculture plates (3040; Falcon Labware, Franklin Lakes, NJ, USA). [3H]Thymidine (1 µCi) was added, the cells were incubated for an additional 6 h and then harvested in a cell harvester. The dried filters were counted in a liquid scintillation counter.
RESULTS
Time between infection of EBV and signs of transformation
In general, a small fraction (1–3%) of B95‐8 cells spontaneously enters the viral lytic cycle (Gradoville et al. 2002). However, protein kinase C (PKC) has been assumed to play an essential role in the initiation of the lytic cascade of EBV (Gao et al. 2001; Gradoville et al. 2002). As the phorbol ester tetradecanoyl phorbol acetate (TPA), a PKC agonist, is one of the best understood activators of EBV lytic replication (Baumann et al. 1998), we initially tested whether EBV stock prepared from TPA‐activated B95‐8 cells (Method 2; see Materials and methods) may shorten the time duration between initiation and immortalization and increase the success rate of immortalization.
Signs of transformation (cell aggregates, increased size and rate of growth, etc.) were usually present within 1–3 weeks, as confirmed by the literature (Tohda et al. 1978; Neitzel 1986; Pelloquin et al. 1986; Caputo et al. 1991), when lymphocytes were incubated with EBV stock prepared by Method 1 (see Materials and methods). In contrast, however, the intensity of infection was greatly increased and signs of transformation were seen within 4 days when lymphocytes were incubated with EBV stock prepared by Method 2 (Table 1 and Fig. 1). In addition, the [3H]thymidine uptake experiment revealed higher DNA synthesis in cultures exposed to EBV prepared by Method 2 (Table 1).
Table 1.
DNA synthesis in cultures exposed to EBV prepared by Method 1 versus Method 2
| EBV stock | Number of cell aggregates a | [3H]thymidine uptake (cpm) b | ||
|---|---|---|---|---|
| Day 4 | Day 7 | Day 4 | Day 7 | |
| Method 1 | + | ++ | 2500 ± 309 | 4425 ± 679 |
| Method 2 | +++ | ++++ | 5883 ± 612 | 13 100 ± 1023 |
Intensity of the infection was graded by the number of clusters of EBV‐infected cells. +, small; ++ or +++, intermediate; ++++, large number of clusters.
EBV‐infected cell cultures from 10 patients with autism and 20 patients with IBD were selected and used to analyse DNA synthesis. On the day of measurement, cultures (single cells and aggregates) were completely disturbed by vigorous agitation (vortex) and triplicate cultures of 200 µl (2 × 105 cells) were used for each experiment. DNA synthesis was evaluated by measuring the ability of EBV‐infected cells to uptake [3H]thymidine. Results were expressed means ± SD of all independent experiments.
Figure 1.
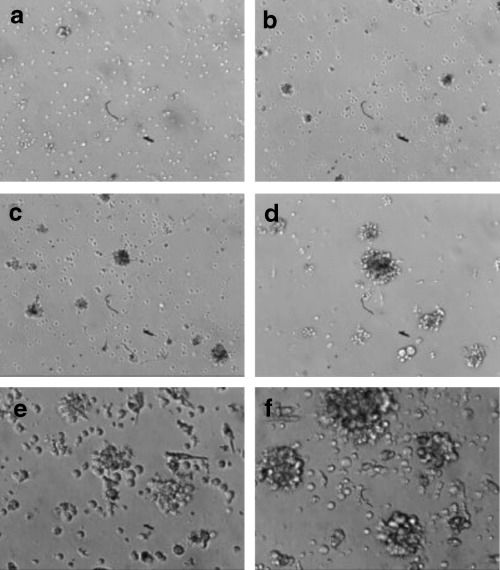
Visualization of signs of immortalization after EBV‐infection. Lymphocytes (∼3 × 106 cells) were incubated with EBV stock prepared by Method 1 (a, b, e) or Method 2 (c, d, f) (see Materials and methods). After 4 (a, c) or 7 (b, d, e, f) days of incubation, the cultures were examined and photographed by inverted microscopy (a–d, × 100; e, f, × 200). Note the cell aggregates of proliferative lymphoblast cells, indicating successful transformation.
This striking difference between the two methods may not be due to the contaminated phorbol ester in EBV stock; TPA alone or TPA in combination with EBV supernatant showed no significant effect on the efficiency of transformation, although it transiently increased cell aggregates (data not shown). Furthermore, three times washing of B95‐8 cells after TPA treatment suggested almost complete removal of TPA in solution. Thus, this result indicates that Method 2 may generate proportionally more, and active, virus particles than Method 1.
Time between initiation and cryopreservation
The number of days after culture initiation was plotted for Methods A and B according to the number of samples cryopreserved (Fig. 2). The average length of time for Method A was 85 ± 24 days, and for Method B was 50 ± 12 days. Method B showed a significant increase in immortalization efficiency compared with Method A. However, during the initial stage, i.e. 1–2 weeks post infection, we observed increasing cell death and complete degeneration of proliferation foci in cultures. This phenomenon was especially more evident in Method B than Method A. Because increased cell death in proliferation foci has been proven to be dependent upon the presence of T cells in the cultures (1978, 1979; Rickinson et al. 1979), CSA, a fungal metabolite, has been used here to prevent the regression of B lymphoblastoid cells. However, we found that the use of CSA was not enough to diminish degeneration of EBV‐activated B cells in Method B. Instead, higher concentrations (> 5 µg/ml) of CSA reduced immortalization efficiency in both methods (data not shown). Because degeneration of B cells in proliferation foci largely depends on the tight junction of B cells with suppressor or cytotoxic T cells, we next tested whether both disturbance of cell aggregates, which may prevent T‐cell‐mediated suppressive or cytotoxic actions by interfering with cell–cell interaction, and re‐infection of EBV, which may increase potential immortalization efficiency, promote the survival and the proliferation of B lymphoblastoid cells (see Method C). As shown in Fig. 2, Method C greatly increased the immortalization efficiency and shortened the time between initiation and cryopreservation. The average length of time for Method C was 36 ± 07 days. Mechanical stress of cultures during second infection had no effect on the viability and proliferation of EBV‐activated B cells as measured by [3H]thymidine uptake test (data not shown).
Figure 2.
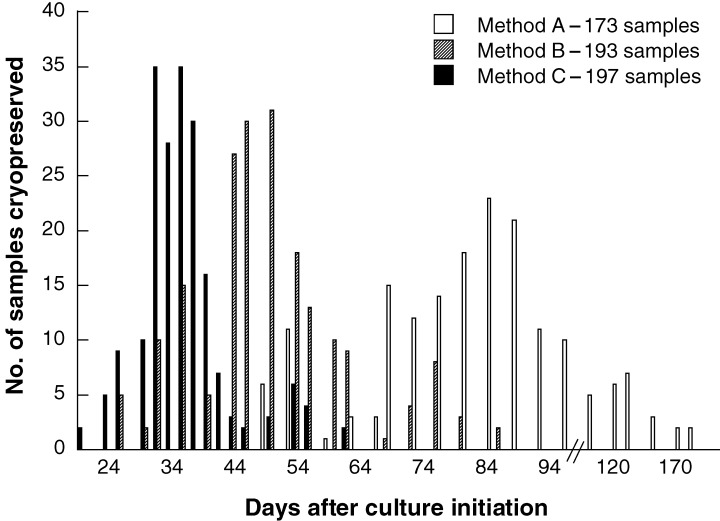
Comparison of Methods A, B, and C for the days from culture initiation to immortalization and cryopreservation. Lymphocytes from patient's blood were immortalized by three different methods as described in MATERIALS and METHODS, and the number of days after culture initiation was plotted according to the number of samples cryopreserved.
Phenotypic surface analysis was carried out to determine the ratio of T cells (CD3+) to B cells (CD22+) in randomly selected blood samples (10 patients with autism and 20 patients with IBD). However, there were no significant alterations of the CD3+/CD22+ ratio for both autism (CD3+ = 66.48 ± 8.23, CD22+ = 11.90 ± 4.19; ratio = 5.59/1) and IBD (CD3+ = 76.09 ± 6.93, CD22+ = 11.04 ± 2.57; ratio = 6.89/1) patients as compared with normal healthy controls (CD3+ = 69.41 ± 7.12, CD22+ = 12.01 ± 3.41; ratio = 5.78/1), indicating that the ratio of T/B cells in both groups may not significantly influence the immortalization efficiency of each method.
Immortalization efficiency
Over a period of 2 years, 198 cell lines were successfully established out of 202. With Method A, of 202 samples processed, 29 samples were unsuccessful due to problems with cell yield or contamination (85.6% success rate). In contrast, only nine and five samples were unsuccessful with Methods B and C, respectively (95.5% with Method B and 98% with Method C).
DISCUSSION
Of the three methods described in this paper, Method C showed the highest rate of culture success, immortalization efficiency, and length of time to cryopreservation. Although differences between the lengths of time to cryopreservation might be due to differences of individual donors, the large numbers of samples and patient sources clearly demonstrated the superiority of Method C.
One of the major reasons why Method A was less successful overall may be due to the length of time required to immortalize cell cultures. In addition, expansion of cultures to large enough numbers for cryopreservation took two or three times longer than for Method B or C. Method B showed increased signs of transformation compared with Method A, as evidenced by cell aggregates, increased size and rate of growth, suggesting that Method 2 (i.e. EBV supernatant prepared from TPA‐activated B95‐8 cells) may generate proportionally more, and active, virus particles than Method 1. However, we could observe that some of the cell aggregates regressed during the initial stage of transformation, i.e. 1–2 weeks post infection, mainly in Method B. Previous reports demonstrated that cell death and degeneration of proliferative foci are largely dependent on T‐cell action (1978, 1979; Rickinson et al. 1979). Thus, in addition to employing CSA, we modified the protocol of Method B, and generated Method C. Interestingly, the disturbance of cell aggregates and re‐infection of EBV greatly improved the survival and the proliferation of B lymphoblastoid cells. We think that vigorous vortexing may decrease the chance of suppressor or cytotoxic T cells meeting EBV‐activated B cells by interfering with specific cell‐to‐cell interaction. In addition, re‐infection of EBV may also increase potential infection efficiency. Mechanical stress itself during second infection did not affect the viability and proliferation of EBV‐activated B cells because DNA synthesis was not decreased even after second infection. With this method, the time between immortalization and cryopreservation was reduced almost 2.6 times as compared with Method A.
In the present data, even though we did not show the data regarding whether re‐infection is necessary or breaking cell aggregates are sufficient for increasing the immortalization efficiency, we strongly suggest that re‐infection is not necessary for all samples, but is important for the improvement of immortalization. For example, as we already observed in Method 2, the intensity of the infection was greatly dependent on the number of active virus particles. The intensity of the first infection can be low due to keeping viral supernatant at 4 °C for more than 4 months or because of slight differences of viral activity from batch‐to‐batch. If this is the case, the second infection can provide a better chance for B cells to meet active virus particles. In fact, we preliminarily tested 22 blood samples (patients with autism) for this purpose, and found that six samples out of 22 showed significant improvement of immortalization (data not shown).
In summary, the procedure described in Method C in this paper is an excellent, routine way of establishing a high number of continuous EBV‐immortalized B cell lines which can be grown to large cell numbers in a relatively short time. This method also can reduce the cost of immortalization. These cellular tools subsequently allow genetic analyses of diseases such as immune and autistic disorders.
ACKNOWLEDGEMENTS
This study was supported by a grant from the Korea Health 21 R & D project, Ministry of Health and Welfare (01‐PJ3‐PG6–01 GN09‐003), a grant from the Korea Science and Engineering Foundation (101‐2002‐000‐00065–0), and, in part, by a Wonkwang University grant 2001.
REFERENCES
- Baumann M, Mischak H, Dammeier S, Kolch W, Gires O, Pich D, Zeidler R, Delecluse HJ, Hammerschmidt W (1998) Activation of the Epstein‐Barr virus transcription factor BZLF1 by 12‐O‐tetradecanoylphorbol‐13‐acetate‐induced phosphorylation. J. Virol. 72, 8105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AG, Britton S, Ernberg I, Nilsson K (1981) Characteristics of Epstein‐Barr virus activation of human B lymphocytes. J. Exp. Med. 154, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo R, Gianotti R, Grimalt R, Monti M, Alessi E (1991) Soft fibroma‐like lesions on the legs of a patient with Kaposi's sarcoma and lymphedema. Am. J. Dermatopathol. 13, 493. [DOI] [PubMed] [Google Scholar]
- Gao X, Ikuta K, Tajima M, Sairenji T (2001) 12‐O‐tetradecanoylphorbol‐13‐acetate induces Epstein‐Barr virus reactivation via NF‐kappaB and AP‐1 as regulated by protein kinase C and mitogen‐activated protein kinase. Virology 286, 91. [DOI] [PubMed] [Google Scholar]
- Gradoville L, Kwa D, El‐Guindy A, Miller G (2002) Protein kinase C‐independent activation of the Epstein‐Barr virus lytic cycle. J. Virol. 76, 5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson E, Miller G, Robinson J, Heston L (1977) Efficiency of transformation of lymphocytes by Epstein‐Barr virus. Virology 76, 152. [DOI] [PubMed] [Google Scholar]
- Louie LG, King MC (1991) A novel approach to establishing permanent lymphoblastoid cell lines: Epstein‐Barr virus transformation of cryopreserved lymphocytes. Am. J. Hum. Genet. 48, 637. [PMC free article] [PubMed] [Google Scholar]
- Moss DJ, Rickinson AB, Pope JH (1978) Long‐term T‐cell‐mediated immunity to Epstein‐Barr virus in man. I. Complete regression of virus‐induced transformation in cultures of seropositive donor leukocytes. Int. J. Cancer 22, 662. [DOI] [PubMed] [Google Scholar]
- Moss DJ, Rickinson AB, Pope JH (1979) Long‐term T‐cell‐mediated immunity to Epstein‐Barr virus in man. III. Activation of cytotoxic T cells in virus‐infected leukocyte cultures. Int. J. Cancer 23, 618. [DOI] [PubMed] [Google Scholar]
- Neitzel H (1986) A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum. Genet. 73, 320. [DOI] [PubMed] [Google Scholar]
- Pelloquin F, Lamelin JP, Lenoir GM (1986) Human B lymphocytes immortalization by Epstein‐Barr virus in the presence of cyclosporin A. In Vitro Cell Dev. Biol. 22, 689. [DOI] [PubMed] [Google Scholar]
- Pressman S, Rotter JI (1991) Epstein‐Barr virus transformation of cryopreserved lymphocytes: prolonged experience with technique. Am. J. Hum. Genet. 49, 467. [PMC free article] [PubMed] [Google Scholar]
- Reidy JA, Wheeler VA (1992) Sample age and Epstein‐Barr virus transformation of cryopreserved lymphocytes. In Vitro Cell Dev. Biol. 28A, 383. [DOI] [PubMed] [Google Scholar]
- Rickinson AB, Moss DJ, Pope JH (1979) Long‐term C‐cell‐mediated immunity to Epstein‐Barr virus in man. II. Components necessary for regression in virus‐infected leukocyte cultures. Int. J. Cancer 23, 610. [DOI] [PubMed] [Google Scholar]
- Schneider U, Zur Hausen H (1975) Epstein‐Barr virus‐induced transformation of human leukocytes after cell fractionation. Int. J. Cancer 15, 59. [DOI] [PubMed] [Google Scholar]
- Tohda H, Oikawa A, Kudo T, Tachibana T (1978) A greatly simplified method of establishing B‐lymphoblastoid cell lines. Cancer Res. 38, 3560. [PubMed] [Google Scholar]
- Ventura M, Gibaud A, Le Pendu J, Hillaire D, Gerard G, Vitrac D, Oriol R (1988) Use of a simple method for the Epstein‐Barr virus transformation of lymphocytes from members of large families of Reunion Island. Hum. Hered. 38, 36. [DOI] [PubMed] [Google Scholar]
- Wall FE, Henkel RD, Stern MP, Jenson HB, Moyer MP (1995) An efficient method for routine Epstein‐Barr virus immortalization of human B lymphocytes. In Vitro Cell Dev. Biol. Anim. 31, 156. [DOI] [PubMed] [Google Scholar]
- Werner J, Henle G, Pinto CA, Haff RF, Henle W (1972) Establishment of continuous lymphoblast cultures from leukocytes of gibbons (Hylobates lar). Int. J. Cancer 10, 557. [DOI] [PubMed] [Google Scholar]