

Abstract
Abstract. Objectives: A number of stimuli induce cardiac hypertrophy and may lead to cardiomyopathy and heart failure. It is believed that cardiomyocytes withdraw from the cell cycle shortly after birth and become terminally differentiated. However, cell cycle regulatory proteins take part in the development of hypertrophy, and it is important to elucidate the mechanisms of how these proteins are involved in the hypertrophic response in cardiomyocytes. Materials and methods, and Results: In the present study, by immunohistochemistry with a phosphorylation‐specific antibody, we found that cyclin D‐cdk4/6‐phosphorylated retinoblastoma protein (pRb) during hypertrophy and expression of an unphosphorylatable pRb mutant impaired hypertrophic growth in cardiomyocytes. Transcription factor E2F was activated by hypertrophic elicitors but activation was impaired by pharmacological inhibition of cyclin D‐cdk4/6. Inhibition of cyclin E‐cdk2 complex only partly impaired E2F activity and did not prevent hypertrophic growth, but diminished endoreplication during hypertrophy. Conclusions: These results indicate that cyclin D‐cdk4/6‐dependent phosphorylation of pRb and activation of E2F is necessary for hypertrophic growth in cardiomyocytes, whereas cyclin E‐cdk2 kinase is not necessary for hypertrophy but regulates endoreplication in these cells. The data support the notion that hypertrophic growth of cardiomyocytes involves a partial progression through the G1 phase of the cell cycle.
INTRODUCTION
The cell cycle is highly conserved and regulated in all eukaryotic cells, and expression of several cell cycle regulatory proteins is altered in various heart diseases, including cardiomyocyte hypertrophy. Many of the signalling molecules implicated in hypertrophy are proto‐oncogenes, molecules essential for regulating the cell cycle (Reiss et al. 1996; Sadoshima & Izumo 1997; Poolman & Brooks 1998; Tamamori et al. 1998). Cardiomyocytes stop dividing shortly after birth and withdraw from the cell cycle (Poolman & Brooks 1998; Busk & Hinrichsen 2003). Instead, postnatal cardiomyocytes grow by hypertrophy (Zak 1974; Ueno et al. 1988; Busk & Hinrichsen 2003; Busk et al. 2005). Mammalian cardiomyocytes retain only limited cell cycle activity during adult life (Clubb & Bishop 1984; Rumyantsev 1991b; Li et al. 1996; , Busk & Hinrichsen 2003). However, regeneration of injured adult myocardium has been documented in newt (Oberpriller et al. 1988; Busk & Hinrichsen 2003) and zebrafish (Poss et al. 2002; Busk & Hinrichsen 2003), and in one particular mouse strain (Leferovich et al. 2001).
The hypertrophic response is triggered by various stimuli, like left ventricular wall stress, growth factors, cytokines and hormones (Chien & Grace 1999; Busk & Hinrichsen 2003). The increased myocyte size is accompanied by an increase in RNA and protein content (Chien et al. 1999), along with re‐expression of foetal genes. In vitro, cardiomyocyte hypertrophy can also be induced by various mitogens like serum (Lubic et al. 1994; Busk & Hinrichsen 2003) and angiotensin II (AII; Aceto & Baker 1990; Busk & Hinrichsen 2003). These stimuli activate multiple second‐messenger systems and induce activity of various immediate‐early genes, such as c‐fos and c‐jun (Simpson 1989; Bogoyevitch et al. 1995; Sadoshima & Izumo 1997; Busk & Hinrichsen 2003), genes that are essential components of the mitotic machinery. The same hypertrophic stimuli also induce expression of many cell cycle regulatory proteins in cardiomyocytes (Reiss et al. 1996; Sadoshima et al. 1997; Li et al. 1998; Poolman & Brooks 1998; Tamamori et al. 1998; Nozato et al. 2000; Busk et al. 2002; Busk & Hinrichsen 2003). It is thus becoming clear that hypertrophic and proliferative stimuli share certain intracellular signal pathways.
Progression through the four phases of the cell cycle is tightly regulated by the sequential expression, activation, inactivation and degradation of specific cell cycle regulatory proteins. Many of the ‘decisions’ whether a cell should divide or not are taken in G1 phase, before DNA synthesis in S phase (Bartek & Lukas 2001; Busk & Hinrichsen 2003). As hypertrophy does not require DNA synthesis, events in G1 phase are particularly interesting for cardiac hypertrophy. A key regulator of passage from G1 phase into S phase is the E2F transcription factor (Nevins et al. 1997; Dyson 1998; Busk & Hinrichsen 2003). E2F is essential for cell cycle progression and regulation by activating necessary genes. E2F is regulated by the Rb family of proteins that bind to E2F and prevent E2F from activating transcription. E2F is released from this inhibitory complex by phosphorylation of the Rb protein (Nevins et al. 1997; Dyson 1998; Busk & Hinrichsen 2003). The molecules responsible for this phosphorylation are the cyclin‐dependent kinase (cdk) complexes (Li & Brooks 1999; Busk & Hinrichsen 2003). In G1 progression, these complexes include cyclins D1–D3 associated with cdk4/6 kinases and cyclin E associated with cdk2 kinase (Reed 1996; Busk & Hinrichsen 2003). The cyclin D‐cdk4/6 kinase complex phosphorylates the Rb protein, thereby making it possible for E2F to activate transcription of various genes, including the cyclin E gene (Ohtani et al. 1995; Busk & Hinrichsen 2003). Cyclin E is the regulatory subunit for cdk2 kinase, and the cyclin E‐cdk2 complex further phosphorylates the Rb protein, thereby releasing E2F from its inhibitory complex with retinoblastoma protein (pRb). This event controls entry into S phase and DNA synthesis (Ohtsubo et al. 1995; Busk & Hinrichsen 2003). The cyclin‐cdk complexes are regulated in a variety of ways, including by cdk inhibitors (Sherr & Roberts 1999; Bartek & Lukas 2001; Busk & Hinrichsen 2003).
Overexpression of E2F‐1 in adult cardiomyocytes drives a significant number of cells from G1 phase into S phase of the cell cycle (Agah et al. 1997; von Harsdorf et al. 1999). Growing evidence indicates that several hypertrophic stimuli up‐regulate the activities of G1 cyclins, such as cyclins D1, D2, D3 and A, and associated cdks (cdk4/6 or cdk2) in cardiac myocytes in vitro and in vivo (Sadoshima & Izumo 1997; Poolman & Brooks 1998; Tamamori et al. 1998; Busk et al. 2002; Busk & Hinrichsen 2003), while expression of cdk inhibitors is down‐regulated (Li & Brooks 1997; Busk & Hinrichsen 2003). These data strongly suggest that G1 cell cycle regulatory proteins are important factors during the hypertrophic response in cardiomyocytes.
In the present work, we have used inhibitor and expression studies to access the role cell cycle proteins have on hypertrophic development in the myocyte cultures. Results show that phosphorylation of pRb by cyclin D‐cdk4/6 is necessary for hypertrophic growth and that this pathway leads to partial activation of the E2F transcription factor. Cyclin E‐cdk2, which also phosphorylates pRb, had little impact on hypertrophy but prevented endoreplication observed in hypertrophic cardiomyocytes.
METHODS
Cardiomyocyte cultures
Protocols used in this investigation conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, reviser 1996).
Newborn, ventricular myocytes were prepared from 1‐ to 5‐day‐old neonatal Wistar rats (University of Copenhagen, Copenhagen, Denmark) as previously described (Busk et al. 2003).
Cells were plated at a density of 5 × 104 cells/cm2 in ventricular myocyte medium (VMM) [Eagle's minimal essential medium supplemented with 100 nm insulin, 656 mg/L creatine, 396 mg/L carnitine, 626 mg/L taurine, 0.5 g/L bovine serum albumin, 0.1% foetal calf serum (Gibco, Paisley, UK), 50 µg/mL streptomycin (Gibco) and 50 U/mL penicillin (Gibco)] in culture dishes, pre‐coated in PBS with 8% foetal calf serum, for 5 h at 37 °C. Cultures were tested for presence of non‐myocytes by staining with a myocyte‐specific anti‐α‐actin antibody (Sigma, St. Louis, MO, USA) and tetramethylrhodamine isothiocyanate (TRITC)‐labelled phalloidin.
Inhibitors
Roscovitine (10 µm) (Calbiochem, San Diego, CA, USA) or differentiation inducing factor 1 (30 µm) (DIF‐1; Affiniti Research, Exeter, UK) was used to inhibit cdk2 kinase and cyclin D, respectively.
Immunohistochemistry
Immunohistochemistry was performed as previously described (Bartkova et al. 2003) with antibodies (Sigma) to sarcomeric α‐actin (clone 5C5), sarcomeric tropomyosin (clone CH1), α‐smooth muscle actin (clone 1A4), von Willebrand factor and serine 795‐phosphorylated pRb (clone RB‐10), on the myocyte cultures fixed with paraformaldehyde. pRb phosphorylation was visualized using Vectastain Elite ABC kit (Vector Laboratories Inc., Burlingame, CA, USA).
Morphology assay
The morphology assay was performed as described previously (Busk et al. 2002). Briefly, cells were plated in pre‐coated chamber slides and after 3 days of incubation they were fixed in 4% paraformaldehyde, and actin was stained with TRITC‐labelled phalloidin. The preparations were viewed by confocal laser microscopy (LSM510, Zeiss, Switzerland) and cell size was analysed using Metamorph software (Zeiss) and expressed in arbitrary units.
Protein synthesis
Myocytes were plated in pre‐coated wells in VMM. Medium was changed to VMM with 30 µg/mL l‐glutamine 2 days after plating. Cells were treated with roscovitine or DIF‐1 on day 5 and 3H‐phenylalanine (25 Ci/mmol; Amersham/Pharmacia, Buckinghamshire, UK) was added to a final concentration of 0.5 µCi/mL in the medium. After 72 h, medium was removed and protein was precipitated with trichloroacetic acid (Higgins 1994). Precipitated protein was solubilized in 1% sodium dodecyl sulfate and incorporated radioactivity was measured by counting in a scintillation analyser using a 3H program (Tri‐carb 2900TR, Packard Bioscience, Meriden, CT, USA).
DNA synthesis
Myocytes were plated in pre‐coated wells in VMM. Medium was changed to VMM with 30 µg/mL l‐glutamine 2 days after plating. Cells were treated with roscovitine or DIF‐1 on day 4 and 3H‐thymidine (25 Ci/mmol; Amersham/Pharmacia) was added to a final concentration of 0.5 µCi/mL in the medium. After 24 h, medium was removed and DNA was precipitated with trichloroacetic acid (Higgins 1994). Precipitated DNA was solubilized in 0.1 m NaOH; 1% sodium dodecyl sulfate and incorporated radioactivity was measured by counting using a scintillation analyser on a 3H program (Tri‐carb 2900TR, Packard Bioscience).
Recombinant adenovirus
Replication‐deficient adenovirus expressing mock from the cytomegalovirus (CMV) promoter was used as described previously (Busk et al. 2002). The plasmid pECEΔcdk‐HA contains the pRb gene (RbΔcdk) that cannot be phosphorylated by cyclin‐dependent kinases due to mutations in the putative phosphorylation sites (Lukas et al. 1997). RbΔcdk was excised from the vector by digestion with Bsu36I, blunt ended with Klenow fragment, and digested with HindIII. The fragment was inserted into pAdTrack‐CMV (Stratagene, La Jolla, CA, USA) and was digested with HindIII and EcoRV. Replication‐deficient adenovirus expressing RbΔcdk from the CMV promoter was made by homologous recombination of pAdTrack‐CMV containing RbΔcdk with pAdEasy‐1, in Escherichia coli BJ5183 (He et al. 1998). A gene encoding mutated cdk2 (DNcdk2), which is dominant negative (van den Heuvel & Harlow 1993), was excised from the vector by digestion with BamHI and inserted into pAdTrack‐CMV cut with BglII. The viral vector was multiplied and packed by transfection into HEK293 cells (He et al. 1998). Titres were determined by infection of HEK293 cells and counting of green fluorescent protein (GFP)‐stained cells as described previously (Busk et al. 2002). Cardiomyocytes were infected by adding adenovirus to a multiplicity of infection of 10. All cells in infected cultures expressed GFP and did therefore also express the gene of interest.
Plasmid construction
Annealed oligonucleotides (TAG, Copenhagen, Denmark) containing the E2F promoter site (Muller et al. 1997) were cloned into the vector pLuc‐MCS (Stratagene) by digesting the vector with HindIII and XhoI. The fragment containing the E2F promoter and the luciferase gene were excised by digesting the plasmid with BamHI, blunt ended with Klenow fragment and digested with SpeI. This fragment was cloned into pAAV‐LacZ (Stratagene), digested with EcoRV and SpeI. The luciferase gene was removed by digesting the plasmid with SacI and EcoRV. Enhanced green fluorescent protein (EGFP) was excised by digesting pEGFP‐N1 (Clontech, Palo Alto, CA, USA) with NotI, blunt ended with Klenow fragment and digested with SacI, and the fragment was inserted instead of the luciferase gene. The fragment containing the E2F promoter site and the EGFP gene was excised from the plasmid by digesting with XmnI and NotI. The fragment was ligated into the Shuttle vector (Stratagene AdEasy Adenoviral Vector System), digested with NotI and followed by treatment with calf intestine alkaline phosphatase.
E2F activity assay
Cardiomyocytes were plated in pre‐coated 6‐well plates in VMM. Using polyethyleneimine (Sigma), transient transfection was carried out with 1 µg E2F plasmid and 1 µg β‐galactosidase plasmid/well as previously described (Busk et al. 2003). Medium was replaced the next day before treating the cells with roscovitine or DIF‐1 and stimulated with serum. After an additional 3 days, the cells were harvested in 150 µL 1× Cell Culture Lysis Reagent (Promega, Madison, WI, USA) and were treated as described previously.
Luciferase and β‐galactosidase assay
Luciferase and β‐galactosidase assays were performed as previously described (Busk et al. 2003).
Endoreplication assay
Cells were plated in pre‐coated chamber slides in VMM. After incubation, they were fixed in 4% paraformaldehyde and actin was stained with TRITC‐labelled phalloidin (Sigma) (Busk et al. 2002) and Sytox Green nucleic acid stain (Molecular Probes, Eugene, OR, USA). Preparations were viewed by confocal laser microscopy (LSM510, Zeiss) and a number of nuclei per cell were counted.
Statistical analysis
Statistical comparison was performed by two‐sided Student's t‐test. Differences were considered significant at P < 0.05.
RESULTS
Two days after isolation of the myocyte culture, the cell preparation was analysed for possible contamination with other cell types. Immunohistochemical analysis showed that nearly all cells were positive for the cardiomyocyte markers sarcomeric α‐actin (Fig. 1; Beltrami et al. 2003) and sarcomeric tropomyosin (von Harsdorf et al. 1999). Cells were also stained with antibodies against α‐smooth muscle actin and von Willebrand factor, in order to detect non‐myocytes. Very few cells were positive for smooth muscle actin but it stains fibroblasts and smooth muscle cells too, and for the endothelial cell marker von Willebrand factor (Beltrami et al. 2003) (data not shown).
Figure 1.
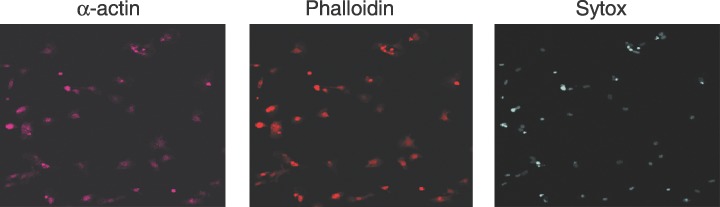
Immunohistochemical analysis of isolated cardiomyocytes. Cardiomyocytes were isolated from heart ventricles of newborn rats as described in the Materials and Methods section. They were plated in 0.1% serum medium and incubated for 2 days before staining with antibodies against sarcomeric α‐actin (actin), non‐specific staining of actin with phalloidin and DNA stained with Sytox Green nucleic acid.
Inhibition of pRb phosphorylation inhibits hypertrophic growth
pRb phosphorylation is a current molecular definition of a restriction point in the cell cycle (Planas‐Silva & Weinberg 1997) and the cyclin D‐cdk4/6 kinase complex plays a key role as growth sensor by phosphorylating pRb in the early stages of G1. To investigate the effect of elimination of pRb phosphorylation on hypertrophic growth, constitutively active pRb, and RbΔcdk, was expressed in cardiomyocytes. They were infected with recombinant adenovirus expressing RbΔcdk protein or a mock virus expressing GFP, and we induced hypertrophy in the myocyte culture by stimulating the cells by addition of 5% serum. Serum is a frequently used inducer of hypertrophy and the size of the cardiomyocytes was subsequently determined as a measure for hypertrophic growth (Fig. 2). This experiment showed that RbΔcdk expression inhibited hypertrophic growth significantly (P < 0.05, Student's t‐test) compared to the control cells.
Figure 2.
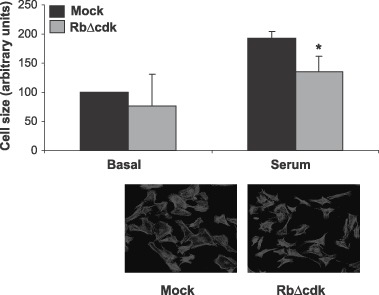
Constitutively active retinoblastoma protein (pRb) inhibited serum induced hypertrophy. Cardiomyocytes were infected with a viral vector expressing mock, or constitutively active pRb (RbΔcdk) (m.o.i. = 10) and were incubated for three days in 5% serum. Size of the cells (arbitrary units) under each condition was determined as described in the Material and Methods section. Error bars denote SD. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments. A minimum of 50 cells was measured.
pRb is phosphorylated by cyclin D‐cdk4/6 during the hypertrophic response
To investigate whether cdk4/6 kinase phosphorylates the pRb, we measured phosphorylation of pRb on serine 795 by immunohistochemistry. Serine 795 is selectively phosphorylated by cyclin D‐dependent kinases (Kitagawa et al. 1996; Grafstrom et al. 1999). Phosphorylation of this site has been shown to reflect activity of cyclin D‐cdk4/6 kinase complex (Bartkova et al. 2003) and appears to be a critical event necessary for inactivation of pRb (Connell‐Crowley et al. 1997). Stimulation of the myocytes lead to a significant (P < 0.05, Student's t‐test) increase in pRb phosphorylation compared to the control cells (Fig. 3a). As expected, this was impaired by DIF‐1, which inhibits cyclin D‐cdk4/6 (Fig. 3b). In contrast, roscovitine did not inhibit pRb phosphorylation on serine 795. Roscovitine is a chemical cdk inhibitor that mainly inhibits cdk2 activity. Roscovitine can inhibit other cdk complexes, but roscovitine IC50 values for cdk4/6 are ≈100 and 1000 times higher than for cdk2, respectively (Penuelas et al. 2003). In summary, these results indicated that hypertrophic growth induces phosphorylation of pRb by activity of cyclin D‐cdk4/6 kinase.
Figure 3.
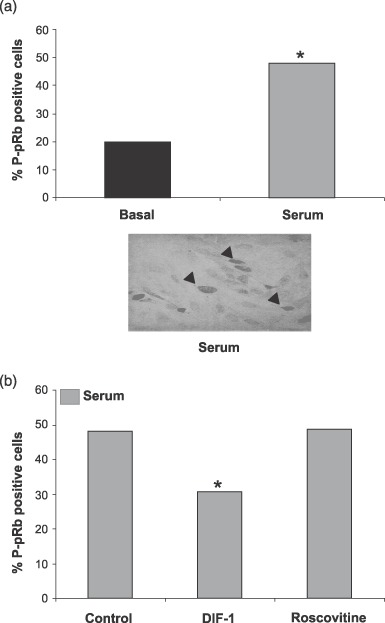
Serum induces retinoblastoma protein (pRb) phosphorylation in neonatal cardiomyocytes. Cells were stimulated with 5% serum (a) and were incubated for 2 days with 30 µm differentiation inducing factor 1 (DIF‐1) or 10 µm roscovitine (b). Fraction of phosphorylated pRb‐positive cells was counted. *indicates P < 0.05, Student's t‐test. Data represent the result of three independent experiments. A minimum of 100 cells was counted.
Cyclin E‐cdk2 kinase complex is not essential for hypertrophic growth
Both cdk4/6 and cdk2 kinase activities are induced during hypertrophic growth in cardiomyocytes (Sadoshima & Izumo 1997; Poolman & Brooks 1998; Tamamori et al. 1998; Busk et al. 2002; Busk & Hinrichsen 2003), and cyclin D‐cdk4/6 is involved in cardiac hypertrophy in vivo and in vitro (Busk et al. 2002). To investigate the role of cyclin E‐cdk2 kinase complex during hypertrophic growth, we induced hypertrophy in the myocyte culture by stimulating the cells with 5% serum and also with 0.5 µm AII (Fig. 4). In addition to serum, AII is a frequently used inducer of hypertrophy. We treated serum‐ or AII‐induced cardiomyocytes with the cdk2 inhibitor roscovitine, and the size of cells was subsequently determined as a measure for hypertrophic growth. Roscovitine treatment had no significant effect on the size of the cells when treating them with serum compared to the control cells (P < 0.05, Student's t‐test) (Fig. 4b). This indicated that cyclin E‐cdk2 complex is not essential for hypertrophic growth of cardiomyocytes. As a control, we also treated the cardiomyocytes with DIF‐1, resulting in expected inhibition of hypertrophic growth in the cardiomyocytes. However, AII treatment did result in a significant reduction in size when treated with roscovitine (Fig. 4d). To test whether this was the result of cdk2 inhibition, we expressed the inhibitory cdk2 mutant DNcdk2 in the cardiomyocytes. Inhibition of the cyclin E‐cdk2 kinase complex with DNcdk2 had no effect on the size of hypertrophic cardiomyocytes (Fig. 5a), but inhibited DNA synthesis (Fig. 5b). This indicated that the discrepancy between roscovitine treatments in serum‐ and AII‐stimulated cells is not due to inhibition of the cyclin E‐cdk2 complex. In summary, inhibition of cyclin E‐cdk2 kinase complex had no effect on the size of the cells. Because roscovitine inhibits entry into S phase, these data support the notion that cardiomyocytes can develop hypertrophy without passing into S phase.
Figure 4.
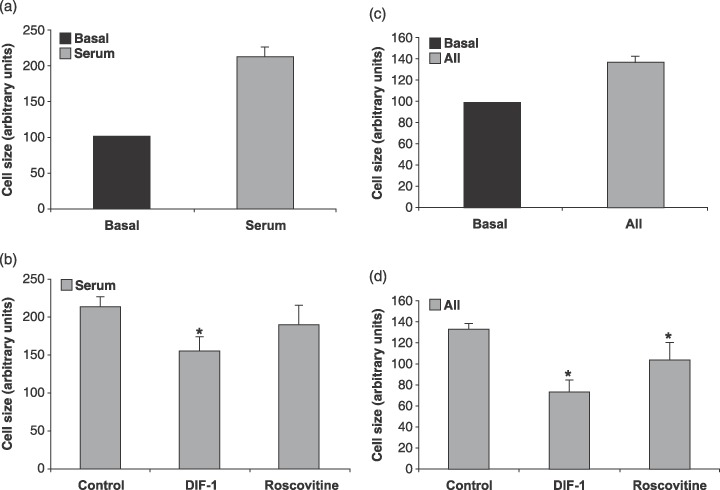
Inhibition of cyclin D impairs hypertrophic growth in neonatal cardiomyocytes. Average cell size in neonatal myocyte cultures after stimulation with 5% serum (a) or 0.5 µm angiotensin II (AII) (c) and were incubated for 3 days with 30 µm differentiation inducing factor 1 (DIF‐1) or 10 µm roscovitine (b) (d). Size of the cells (arbitrary units) under each condition was determined as described in the Material and Methods section. Error bars denote SD. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments. A minimum of 50 cells was measured.
Figure 5.
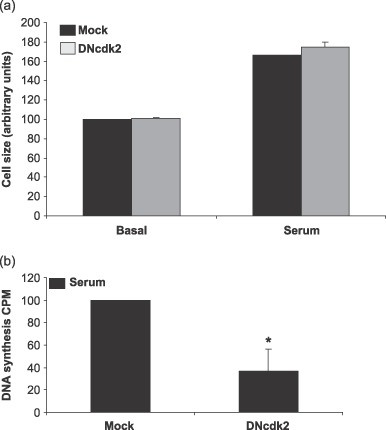
DNcdk2 expression does not impair hypertrophic growth, but inhibits DNA synthesis in neonatal cardiomyocytes. Average cell size in neonatal myocyte cultures infected with dominant‐negative CDK2 (DNcdk2) or adenoviral vectors expressing mock (m.o.i. = 10) and incubated for 3 days in basal serum or with 5% serum (a). Size of the cells (arbitrary units) under each condition was determined as described in the Material and Methods section. Error bars denote SD. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments. A minimum of 50 cells was measured. (b) Cells were infected as in (a) and 1 day after infection DNA synthesis was measured by 3H‐thymidine incorporation over a 24‐h period in cells treated with 5% serum. Data represent results of three independent experiments.
Detection of induced protein synthesis is used as an indicator of hypertrophy. To verify hypertrophic growth observed, total protein synthesis in the cultures was measured by 3H‐phenylalanine incorporation. Serum‐ and AII‐induced hypertrophy up‐regulated protein synthesis in the cultures compared to the control cells (Fig. 6a–d). This induction of protein synthesis was down‐regulated when inhibiting cyclin D‐cdk4/6 with DIF‐1 (Fig. 6a,c). However, protein synthesis was also dependent on cyclin E‐cdk2, as 3H‐phenylalanine incorporation could be impaired by treating the cells with roscovitine (Fig. 6b,d). This result indicated that protein synthesis was not an optimal marker for hypertrophy when using serum or AII as hypertrophic elicitors in the cultures.
Figure 6.
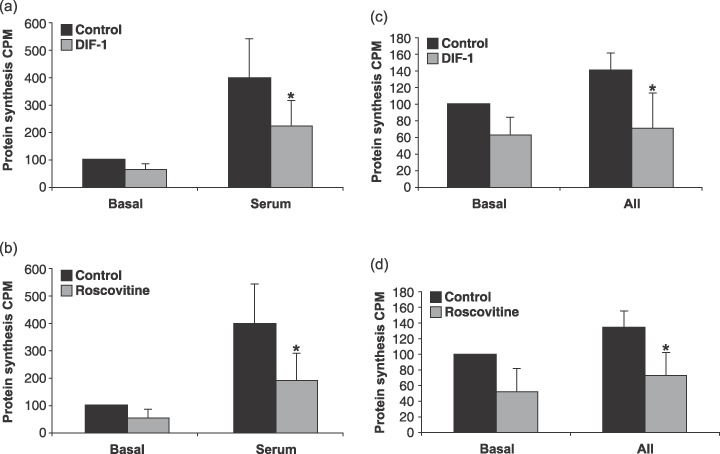
Serum or angiotensin II (AII) induced protein synthesis is inhibited by differentiation inducing factor 1 (DIF‐1) or roscovitine in cardiomyocytes. Cells stimulated with 5% serum (a and b) or 0.5 µm AII (c and d) and incubated for 3 days with 30 µm DIF‐1 or 10 µm roscovitine. Protein synthesis was measured by 3H‐phenylalanin incorporation in the stimulation period of 3 days. Error bars denote SD. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments.
Hypertrophy is almost always associated with increased expression of brain natriuretic peptide and atrial natriuretic peptide in the heart, as well as with increased circulation levels of the two peptides in the blood (reviewed in Tremblay et al. 2002). Therefore, these peptides are often used as markers for the hypertrophic response in cardiomyocytes. As an additional control, we analysed levels of brain natriuretic peptide mRNA and found it to be induced in the cardiomyocyte cultures stimulated with serum or AII (data not shown).
Induced E2F activity during the hypertrophic response
If cyclin D‐cdk4/6‐dependent phosphorylation of pRB and not cyclin E‐cdk2‐dependent phosphorylation of pRB is involved in hypertrophic growth, does this result in altered E2F activity? Because E2F is known to be involved in cell cycle progression in cardiac myocytes (Flink et al. 1998; von Harsdorf et al. 1999), we investigated activity of E2F during the hypertrophic response, using a construct consisting of a minimal promoter controlled by an E2F enhancer element ahead of a luciferase reporter gene. Cardiomyocytes were transiently transfected with the construct and a control plasmid with the CMV promoter ahead of a β‐galactosidase reporter gene. After transfections, the cardiomyocyte cultures were treated with DIF‐1 or roscovitine to inhibit cyclin D and cdk2, respectively. To induce the hypertrophic response, cells were stimulated with serum. Low E2F promoter activity was detected in samples without serum and stimulation of the hypertrophic response induced E2F activity (Fig. 7). DIF‐1 abolished the E2F activity entirely as expected as DIF‐1 leads to inhibition of cyclin D‐cdk4/6, and cyclin E‐cdk2 activity depends on cyclin D‐cdk4/6 (Lundberg & Weinberg 1998). Inhibition of cyclin E‐cdk2 with roscovitine lowered E2F activity significantly (P < 0.05, Student's t‐test), but there was still some activity‐above background. This indicated that phosphorylation by cdk4/6 was sufficient to induce E2F‐regulated transcription in hypertrophic cardiomyocytes. We also analysed expression of E2F1, E2F2 and E2F3 by Western blotting but found no regulation of protein levels in basal cells or cells stimulated with serum and treated with DIF‐1 or roscovitine (data not shown). In summary, these data show that E2F activity was induced during the hypertrophic response of the cardiomyocytes. Transcription factor activity, and not protein expression, seems to be affected by cyclin D‐cdk4/6 and cyclin E‐cdk2 kinase activity.
Figure 7.
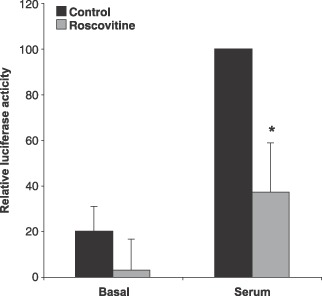
Inhibition of E2F activity in roscovitine treated cardiomyocytes. Cardiomyocytes were polyethyleneimine transfected with an E2F‐luciferase reporter vector and a plasmid expressing β‐galactosidase for 24 h as described in the Material and Methods section. Cells were stimulated with 5% serum and were further incubated for 3 days with 10 µm roscovitin. Error bars denote SD. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments.
Induced E2F activity up‐regulates DNA synthesis
To test whether E2F activity observed during hypertrophic growth induced DNA synthesis in the cardiomyocytes, total DNA synthesis in the cultures was measured by 3H‐thymidine incorporation. Serum‐ or AII‐induced hypertrophy up‐regulated DNA synthesis in the cultures compared to the control cells (Fig. 8a–d). This induction was significantly (P < 0.05, Student's t‐test) down‐regulated by DIF‐1 (Fig. 8a,c). The effect was also dependent on cyclin E‐cdk2, as 3H‐thymidine incorporation could be impaired by treating the cells with roscovitine (Fig. 8b,d). In agreement with the literature, we found that 10 µm roscovitine inhibited DNA synthesis, which depends on cdk2 activity, whereas cdk4/6‐dependent phosphorylation of pRb was not inhibited by this concentration of roscovitine, and these results indicate that 10 µm roscovitine inhibits cdk2 but not cdk4/6 in cardiomyocytes. In addition, DNA synthesis inhibition is observed in basal cardiomyocytes treated with DIF‐1 or roscovitine. Leaving cardiomyocytes to incorporate 3H‐thymidine for an additional 3 days did not absolutely abolish this activity, indicating continuing repair of DNA in the cardiomyocyte culture (data not shown). In summary, even though cardiomyocytes are known to preferentially stay in G1 phase during the hypertrophic response, some myocytes do exceed the S‐phase boundary and start DNA replication. This effect appears to involve both the cyclin D‐cdk4/6 and the cyclin E‐cdk2 kinase complex.
Figure 8.
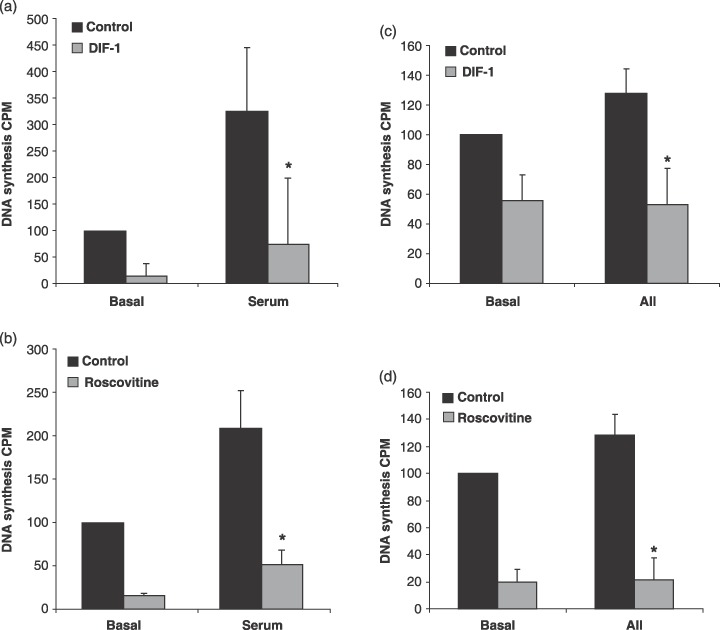
Differentiation inducing factor 1 (DIF‐1) and roscovitine inhibits DNA synthesis in cardiomyocytes. Cells were stimulated with 5% serum (a and b) or 0.5 µm AII (c and d) and 30 µm DIF‐1 or 10 µm roscovitine. After 1 day of incubation, DNA synthesis was measured by 3H‐thymidine incorporation over a 24‐h period. Error bars denote SD. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments.
Endoreplication in hypertrophic cardiomyocytes
Binucleation of cardiomyocytes occurs shortly after birth in mice and rats, and is concurrent with withdrawal from the cell cycle (Brodsky et al. 1980; Rumyantsev 1991a). The molecular and cellular mechanisms responsible for regulating cardiomyocyte binucleation are largely unknown. Both endoreplication and hypertrophic growth involves activation of cell cycle machinery, induced protein synthesis and passage through G1 phase. Because cyclin E‐cdk2 complex is involved in endoreplication in various cell types, we wanted to investigate whether the same were true for cardiomyocytes. Thus, they were serum‐stimulated to undergo hypertrophy with or without roscovitine treatment, and the percentage of binuclear cardiomyocytes was obtained by counting the number of nuclei per cell. Under basal conditions, no considerable endoreplication was observed (Fig. 9). However, serum stimulation resulted in significant induction of binucleation (P < 0.05, Student's t‐test), and this was impaired by cdk2 inhibitor roscovitine, indicating that active cdk2 kinase is necessary for endoreplication. Although cyclin E may bind the cdc2 kinase (Aleem et al. 2005) and this complex could at least in theory be involved in endoreplication and S‐phase transition (Kang & Koh 1997; Flink et al. 1998), roscovitine also inhibits cdc2 kinase (Canduri et al. 2004). This indicates that S‐phase transition and DNA synthesis are inhibited by roscovitine and thus are not necessary for hypertrophic growth in cardiomyocytes.
Figure 9.
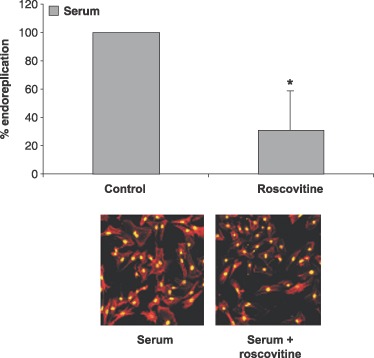
Roscovitine repressed endoreplication in hypertrophic neonatal cardiomyocytes. Cardiomyocytes stimulated with 5% serum were incubated with 10 µm roscovitine for 3 days followed by staining for actin with phalloidin and DNA with Sytox Green nucleic acid. Fraction of cells endoreplicating was counted. *indicates P < 0.05, Student's t‐test. Data represent results of three independent experiments. A minimum of 100 cells was counted.
DISCUSSION
Hypertrophic and mitogenic stimuli share certain intracellular responses in various cell types, and both processes activate the cell cycle (Busk & Hinrichsen 2003). Hypertrophic growth in cardiomyocytes involves exit from G0 and partial progression through the cell cycle. Over the past decade, a large number of studies have revealed the central role of the Rb pathway in regulation of G1‐ to S‐phase transition, and control of the cell cycle, by modulating activity of the E2F transcription factor (reviewed by Stevens & La Thangue 2003). Although regulation of cyclin‐cdk activities is essential to cell size control in eukaryotes (Morgan 1997), and the E2F transcription factor is an important regulator of G1 phase (Johnson et al. 1993; Duronio et al. 1995; Wu et al. 1996), their role in the hypertrophic response in cardiomyocytes is incompletely understood.
pRb phosphorylation by cyclin D‐cdk4/6 kinase complex appears to be a key event necessary for inactivation of pRb in various cells. Despite the fundamental role cyclin D‐cdk4/6 kinase complex plays as growth sensors, to date, only the Rb family and Smad proteins have been found to be substrate for this kinase complex (Matsuura et al. 2004). Cyclin D‐cdk4/6 kinase complex is important for hypertrophic growth and the kinase activity is induced during the hypertrophic response in cardiomyocytes in vivo and in vitro. Inhibition of cyclin D inhibits cyclin D kinase activity and impairs their hypertrophic growth (Busk et al. 2002). This suggests that cyclin D‐dependent kinase activity is necessary for development of hypertrophy in cardiomyocytes. We observed that expression of unphosphorylatable mutant of pRb, RbΔcdk, and inhibition of cyclin D‐cdk4/6 impaired hypertrophic growth. Furthermore, stimuli that activated cyclin D‐cdk4/6 induced phosphorylation of pRb on serine 795 and activity of the E2F transcription factor. This indicates that the pRb pathway is essential for hypertrophic growth of cardiomyocytes. This includes the phosphorylation of pRb by cyclin D‐cdk4/6 kinase complex, followed by induction of E2F activity. Ectopic expression of RbΔcdk could also affect other pRb targets. However, pRb interactions with other proteins besides E2F and histone deacetylases are not that well established and the precise effect is therefore difficult to predict.
We found that inhibition of cyclin E‐cdk2 kinase complex had no effect on hypertrophic growth here. Because cyclin E‐cdk2 kinase complex is a key regulator of transition from G1 to S phase and not involved in hypertrophic growth, the similarities shared by mitogenic stimuli and hypertrophic response are apparently only comparable in passage from G0 to G1 phase, and in the first part of G1.
E2F protein expression is significantly down‐regulated during myocyte development. Highest levels are found in foetal myocytes, but in adult ones E2F protein levels are undetectable (Brooks et al. 1998). In this study, hypertrophic stimuli induced E2F activity and this was either abolished or down‐regulated when inhibiting cyclin D‐cdk4/6 or the cyclin E‐cdk2 kinase complex. This is consistent with the observation that direct inhibition of E2F transcription factor, with a decoy oligonucleotide, inhibits hypertrophic growth of cardiomyocytes (Vara et al. 2003). Induced E2F activity is in agreement with the notion that hypertrophic cardiomyocytes re‐enter the cell cycle from G0 phase, and pass through G1 phase. Earlier studies suggest that complete inactivation of pRb requires phosphorylation by both cyclin D‐cdk4 and cyclin E‐cdk2 (Lundberg & Weinberg 1998). Each kinase complex is activated at different points in G1 phase. Cyclin D‐cdk4 becomes active in mid‐ to late G1 phase, and cyclin E‐cdk2 in late G1 phase is consistent with it contributing to pRb hyperphosphorylation. In this study, we found that only cdk4/6‐dependent phosphorylation of pRb is required for participation of E2F in hypertrophy. This suggests that E2F transcription factor activity released following cyclin D‐cdk4/6 phosphorylation, is activating genes necessary for hypertrophic growth in cardiomyocytes.
Mitogen stimulation leads to activation of cyclin D‐cdk4/6 kinase complex in proliferating cells (Reed 1996), followed by induced E2F transcription factor activity and passage from G1 to S phase. It is possible that mitogen‐induced activation of cyclin D‐cdk4/6 kinase complex in cardiomyocytes (Busk & Hinrichsen 2003) is responsible for previously reported S‐phase passage and DNA synthesis (Yan et al. 1999; Matturri et al. 2002). We also observed DNA synthesis in hypertrophic cardiomyocyte cultures, indicating that cell cycle regulatory proteins, in at least some of the cardiomyocytes, are capable of driving the cells not only to re‐enter the cell cycle, but also to exceed the G1‐ to S‐phase boundary and replicate DNA. Induced DNA synthesis in hypertrophic cardiomyocytes was impaired when either cyclin D or cdk2 was inhibited.
Knockout studies have shown that cyclin E and cdk2 are dispensable for mitosis in mice (Geng et al. 2003; Ortega et al. 2003). However, cyclin E is indispensable for endoreplication (Geng et al. 2003). Endoreplication arises as a consequence of nuclear mitotic division without cytoplasmic separation. Both endoreplication and hypertrophic growth involve induction of the cell cycle and passage through G1 phase. As observed here, some hypertrophic cardiomyocytes are apparently able to pass further through the cell cycle, through G1 phase and undergo endoreplication (Katzberg et al. 1977). This induced endoreplication can be impaired by inhibiting cyclin E‐cdk2 kinase complex. This suggests that cyclin E‐cdk2 kinase complex is necessary for endoreplication in cardiomyocytes. In spite of the molecular similarities between hypertrophic growth and endoreplication, activity of the cdk2 kinase is not a common feature of the two.
Scrutinizing G1 kinase complexes provides valuable information on a possible approach for developing treatment for hypertrophy. In addition, with these data we are one step closer to elucidate the mechanisms necessary for getting cardiomyocytes to undergo mitosis and thereby proliferate. Normal cardiomyocytes have down‐regulated levels of cell cycle regulatory proteins, but maintain the ability to re‐express them during hypertrophy (Li et al. 1998). Because cardiomyocytes do not generally divide, this implies that cell cycle machinery is suppressed under normal conditions. This would in theory mean that by eliminating the suppressive mechanism, cardiomyocytes would be able to divide.
ACKNOWLEDGEMENTS
We thank Jiri Bartek for the pECEΔcdk‐HA plasmid and Jirina Bartkova, both from the Danish Cancer Society, for helping out with the immunohistochemistry. We thank Linda Wulf‐Andersen for advice on technical issues and critical comments, and Katrine Kastberg for technical help. This work was supported by grants from the Danish Heart Association, the Danish Medical Research Council, the Birthe and John Meyer Foundation, Villadsen Family Foundation, Kong Christian X's Foundation, the Illum Foundation and Fru Asta Florida Bolding Foundation.
REFERENCES
- Aceto JF, Baker KM (1990) [Sar1]angiotensin II receptor‐mediated stimulation of protein synthesis in chick heart cells. Am. J. Physiol. 258, H806–H813. [DOI] [PubMed] [Google Scholar]
- Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, Schneider MD (1997) Adenoviral delivery of E2F‐1 directs cell cycle reentry and p53‐independent apoptosis in postmitotic adult myocardium in vivo . J. Clin. Invest. 100, 2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleem E, Kiyokawa H, Kaldis P (2005) Cdc2‐cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 7, 831–836. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2001) Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 490, 117–122. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas C, Sorensen CS, Meyts ER, Skakkebaek NE, Lukas J, Bartek J (2003) Deregulation of the RB pathway in human testicular germ cell tumours. J. Pathol. 200, 149–156. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Marshall CJ, Sugden PH (1995) Hypertrophic agonists stimulate the activities of the protein kinases c‐Raf and A‐Raf in cultured ventricular myocytes. J. Biol. Chem. 270, 26303–26310. [DOI] [PubMed] [Google Scholar]
- Brodsky WY, Arefyeva AM, Uryvaeva IV (1980) Mitotic polyploidization of mouse heart myocytes during the first postnatal week. Cell Tissue Res. 210, 133–144. [DOI] [PubMed] [Google Scholar]
- Brooks G, Poolman RA, Li JM (1998) Arresting developments in the cardiac myocyte cell cycle: role of cyclin‐dependent kinase inhibitors. Cardiovasc. Res. 39, 301–311. [DOI] [PubMed] [Google Scholar]
- Busk PK, Bartkova J, Strom CC, Wulf‐Andersen L, Hinrichsen R, Christoffersen TE, Latella L, Bartek J, Haunso S, Sheikh SP (2002) Involvement of cyclin D activity in left ventricle hypertrophy in vivo and in vitro . Cardiovasc. Res. 56, 64–75. [DOI] [PubMed] [Google Scholar]
- Busk PK, Hinrichsen R (2003) Cyclin D in left ventricle hypertrophy. Cell Cycle 2, 91–95. [PubMed] [Google Scholar]
- Busk PK, Hinrichsen R, Bartkova J, Hansen AH, Christoffersen TE, Bartek J, Haunso S (2005) Cylcin D2 induces proliferation of cardiac myocytes and represses hypertrophy. Exp. Cell Res. 304, 149–161. [DOI] [PubMed] [Google Scholar]
- Busk PK, Wulf‐Andersen L, Strom CC, Enevoldsen M, Thirstrup K, Haunso S, Sheikh SP (2003) Multiprotein bridging factor 1 cooperates with c‐Jun and is necessary for cardiac hypertrophy in vitro . Exp. Cell Res. 286, 102–114. [DOI] [PubMed] [Google Scholar]
- Canduri F, Uchoa HB, De Azevedo WF Jr (2004) Molecular models of cyclin‐dependent kinase 1 complexed with inhibitors. Biochem. Biophys. Res. Commun. 324, 661–666. [DOI] [PubMed] [Google Scholar]
- Chien KR, Grace AA, Hunter JJ (1999) Molecular and Cellular Biology of Cardiac Hypertrophy and Failure. Philadelphia, PA: Saunders. [Google Scholar]
- Clubb FJ Jr, Bishop SP (1984) Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab. Invest. 50, 571–577. [PubMed] [Google Scholar]
- Connell‐Crowley L, Harper JW, Goodrich DW (1997) Cyclin D1/Cdk4 regulates retinoblastoma protein‐mediated cell cycle arrest by site‐specific phosphorylation. Mol. Biol. Cell 8, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH, Xie JE, Brook A, Dyson N (1995) The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 9, 1445–1455. [DOI] [PubMed] [Google Scholar]
- Dyson N (1998) The regulation of E2F by pRB‐family proteins. Genes Dev. 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- Flink IL, Oana S, Maitra N, Bahl JJ, Morkin E (1998) Changes in E2F complexes containing retinoblastoma protein family members and increased cyclin‐dependent kinase inhibitor activities during terminal differentiation of cardiomyocytes. J. Mol. Cell. Cardiol. 30, 563–578. [DOI] [PubMed] [Google Scholar]
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P (2003) Cyclin E ablation in the mouse. Cell 114, 431–443. [DOI] [PubMed] [Google Scholar]
- Grafstrom RH, Pan W, Hoess RH (1999) Defining the substrate specificity of cdk4 kinase‐cyclin D1 complex. Carcinogenesis 20, 193–198. [DOI] [PubMed] [Google Scholar]
- Von Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R (1999) E2F‐1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclin‐dependent kinases in the presence of insulin‐like growth factor I. Circ. Res. 85, 128–136. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, Da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95, 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Heuvel S, Harlow E (1993) Distinct roles for cyclin‐dependent kinases in cell cycle control. Science 262, 2050–2054. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR (1993) Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 365, 349–352. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Koh GY (1997) Differential and dramatic changes of cyclin‐dependent kinase activities in cardiomyocytes during the neonatal period. J. Mol. Cell. Cardiol. 29, 1767–1777. [DOI] [PubMed] [Google Scholar]
- Katzberg AA, Farmer BB, Harris RA (1977) The predominance of binucleation in isolated rat heart myocytes. Am. J. Anat. 149, 489–499. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Jung HK, Suzuki‐Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y (1996) The consensus motif for phosphorylation by cyclin D1‐Cdk4 is different from that for phosphorylation by cyclin A/E‐Cdk2. EMBO J. 15, 7060–7069. [PMC free article] [PubMed] [Google Scholar]
- Leferovich JM, Bedelbaeva K, Samulewicz S, Zhang XM, Zwas D, Lankford EB, Heber‐Katz E (2001) Heart regeneration in adult MRL mice. Proc. Natl. Acad. Sci. USA 98, 9830–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Brooks G (1997) Down‐regulation of cyclin‐dependent kinase inhibitors p21 and p27 in pressure‐overload hypertrophy. Am. J. Physiol. 273, H1358–H1367. [DOI] [PubMed] [Google Scholar]
- Li JM, Brooks G (1999) Cell cycle regulatory molecules (cyclins, cyclin‐dependent kinases and cyclin‐dependent kinase inhibitors) and the cardiovascular system; potential targets for therapy? Eur. Heart J. 20, 406–420. [DOI] [PubMed] [Google Scholar]
- Li JM, Poolman RA, Brooks G (1998) Role of G1 phase cyclins and cyclin‐dependent kinases during cardiomyocyte hypertrophic growth in rats. Am. J. Physiol. 275, H814–H822. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, Gerdes AM (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737–1746. [DOI] [PubMed] [Google Scholar]
- Lubic SP, Giacomini KM, Giacomini JC (1994) Increased 1,4‐dihydropyridine binding sites in serum‐stimulated cardiomyocyte hypertrophy. J. Pharmacol. Exp. Ther. 270, 697–701. [PubMed] [Google Scholar]
- Lukas J, Herzinger T, Hansen K, Moroni MC, Resnitzky D, Helin K, Reed SI, Bartek J (1997) Cyclin E‐induced S phase without activation of the pRb/E2F pathway. Genes Dev. 11, 1479–1492. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, Weinberg RA (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin‐cdk complexes. Mol. Cell. Biol. 18, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F (2004) Cyclin‐dependent kinases regulate the antiproliferative function of Smads. Nature 430, 226–231. [DOI] [PubMed] [Google Scholar]
- Matturri L, Milei J, Grana DR, Lavezzi AM (2002) Characterization of myocardial hypertrophy by DNA content, PCNA expression and apoptotic index. Int. J. Cardiol. 82, 33–39. [DOI] [PubMed] [Google Scholar]
- Morgan DO (1997) Cyclin‐dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell. Dev. Biol. 13, 261–291. [DOI] [PubMed] [Google Scholar]
- Muller H, Moroni MC, Vigo E, Petersen BO, Bartek J, Helin K (1997) Induction of S‐phase entry by E2F transcription factors depends on their nuclear localization. Mol. Cell. Biol. 17, 5508–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR, Leone G, Degregori J, Jakoi L (1997) Role of the Rb/E2F pathway in cell growth control. J. Cell. Physiol. 173, 233–236. [DOI] [PubMed] [Google Scholar]
- Nozato T, Ito H, Tamamori M, Adachi S, Abe S, Marumo F, Hiroe M (2000) G1 cyclins are involved in the mechanism of cardiac myocyte hypertrophy induced by angiotensin II. Jpn Circ. J. 64, 595–601. [DOI] [PubMed] [Google Scholar]
- Oberpriller JO, Oberpriller JC, Arefyeva AM, Mitashov VI, Carlson BM (1988) Nuclear characteristics of cardiac myocytes following the proliferative response to mincing of the myocardium in the adult newt, Notophthalmus viridescens. Cell Tissue Res. 253, 619–624. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Degregori J, Nevins JR (1995) Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 92, 12146–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M (1995) Human cyclin E, a nuclear protein essential for the G1‐to‐S phase transition. Mol. Cell. Biol. 15, 2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M (2003) Cyclin‐dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25–31. [DOI] [PubMed] [Google Scholar]
- Penuelas S, Alemany C, Noe V, Ciudad CJ (2003) The expression of retinoblastoma and Sp1 is increased by low concentrations of cyclin‐dependent kinase inhibitors. Eur. J. Biochem. 270, 4809–4822. [DOI] [PubMed] [Google Scholar]
- Planas‐Silva MD, Weinberg RA (1997) The restriction point and control of cell proliferation. Curr. Opin. Cell Biol. 9, 768–772. [DOI] [PubMed] [Google Scholar]
- Poolman RA, Brooks G (1998) Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J. Mol. Cell. Cardiol. 30, 2121–2135. [DOI] [PubMed] [Google Scholar]
- Poss KD, Wilson LG, Keating MT (2002) Heart regeneration in zebrafish. Science 298, 2188–2190. [DOI] [PubMed] [Google Scholar]
- Reed SI (1996) G1/S regulatory mechanisms from yeast to man. Prog. Cell Cycle Res. 2, 15–27. [DOI] [PubMed] [Google Scholar]
- Reiss K, Cheng W, Giorando A, De Luca A, Li B, Kajstura J, Anversa P (1996) Myocardial infarction is coupled with activation of cyclins and cyclin‐dependent kinases in myocytes. Exp. Cell Res. 225, 44–54. [PubMed] [Google Scholar]
- Rumyantsev PP (1991a) Growth and Hyperplasia in Cardiac Muscle Cells. London: Harwood Academic Publisher. [Google Scholar]
- Rumyantsev PP (1991b) Growth and Hyperplasia of Cardiac Muscle Cells. New York City, NY: Harwood Academic Publishers. [Google Scholar]
- Sadoshima J, Aoki H, Izumo S (1997) Angiotensin II and serum differentially regulate expression of cyclins, activity of cyclin‐dependent kinases, and phosphorylation of retinoblastoma gene product in neonatal cardiac myocytes. Circ. Res. 80, 228–241. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S (1997) The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 59, 551–571. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Simpson PC (1989) Proto‐oncogenes and cardiac hypertrophy. Annu. Rev. Physiol. 51, 189–202. [DOI] [PubMed] [Google Scholar]
- Stevens C, La Thangue NB (2003) E2F and cell cycle control: a double‐edged sword. Arch. Biochem. Biophys. 412, 157–169. [DOI] [PubMed] [Google Scholar]
- Tamamori M, Ito H, Hiroe M, Terada Y, Marumo F, Ikeda MA (1998) Essential roles for G1 cyclin‐dependent kinase activity in development of cardiomyocyte hypertrophy. Am. J. Physiol. 275, H2036–H2040. [DOI] [PubMed] [Google Scholar]
- Tremblay J, Desjardins R, Hum D, Gutkowska J, Hamet P (2002) Biochemistry and physiology of the natriuretic peptide receptor guanylyl cyclases. Mol. Cell. Biochem. 230, 31–47. [PubMed] [Google Scholar]
- Ueno H, Perryman MB, Roberts R, Schneider MD (1988) Differentiation of cardiac myocytes after mitogen withdrawal exhibits three sequential states of the ventricular growth response. J. Cell Biol. 107, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara D, Bicknell KA, Coxon CH, Brooks G (2003) Inhibition of E2F abrogates the development of cardiac myocyte hypertrophy. J. Biol. Chem. 278, 21388–21394. [DOI] [PubMed] [Google Scholar]
- Wu CL, Classon M, Dyson N, Harlow E (1996) Expression of dominant‐negative mutant DP‐1 blocks cell cycle progression in G1. Mol. Cell. Biol. 16, 3698–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SM, Finato N, Di Loreto C, Beltrami CA (1999) Nuclear size of myocardial cells in end‐stage cardiomyopathies. Anal. Quant. Cytol. Histol. 21, 174–180. [PubMed] [Google Scholar]
- Zak R (1974) Development and proliferative capacity of cardiac muscle cells. Circ. Res. 35 (Suppl. II), 17–26. [PubMed] [Google Scholar]