

Abstract
Objectives: Melanoma cells take advantage of impaired ability to undergo programmed cell death in response to different external stimuli and chemotherapeutic drugs; this makes prevention of tumour progression very difficult. The aim of this study was to demonstrate whether 3‐O‐methylfunicone (OMF), a metabolite of Penicillium pinophilum, has the ability to arrest cell population growth and to induce apoptosis in A375P (parental) and A375M (metastasis derivatived) melanoma cell lines.
Materials and methods: Cell proliferation and apoptosis were analysed by flow cytometry, DNA fragmentation, caspase‐3 and caspase‐9 activation, and PARP‐1 cleavage.
Results: We demonstrated that OMF affected cell proliferation in a time‐ and dose‐dependent manner, reaching the best effect at concentration of 80 µg/ml for 24 h. Flow cytometry revealed that OMF caused significant G2 phase arrest, which was associated with marked decrease in cyclin B1/p34cdc2 complex and p21 induction. OMF also induced marked decrease of survivin expression. Reduced levels of apoptosis were evident after silencing p21 expression in both cell lines. Finally, the effect exercised by OMF on hTERT and TEP‐1 gene expression confirmed the ability of this molecule to interfere with replicative ability of cells.
Conclusions: The results reported here seem to suggest that OMF as a promising molecule to include in strategies for treatment of melanoma.
Introduction
Melanoma is the most aggressive form of skin cancer, notoriously resistant to current modalities of cancer therapy, and known to be a tumour with elevated metastatic ability (1). Although today melanoma is diagnosed at its early stages and therefore results in better overall survival, once tumour cells are detected in regional lymph nodes, patients have poor prognosis. Metastatic melanoma cells rarely restrict themselves to single foci, but tend to disseminate to multiple organs, rendering surgical intervention to be of limited use (2). For this reason, it is very important to control the outcome of the disease at its early stages, in order to avoid melanoma cells from spreading. Melanoma cells are resistant to a wide range of anti‐neoplastic treatments, due to their ability to evade cytotoxic action of different insults, such as DNA damage, microtubule destabilization or topoisomerase inhibition (1), showing in contrast, a strong survival feature. Also, in vivo melanoma cells have low levels of spontaneous apoptosis compared to other tumour cell types, and resistance to apoptosis is associated with increased metastasis in animal models of melanoma (3) and increased resistance to chemotherapeutic agents (4). Acquisition of resistance to apoptosis is important in transition from normal melanocyte to melanoma. Apoptosis, or programmed cell death, is critical for epidermal homeostasis, representing a key protective mechanism removing premalignant cells that have acquired mutations (5). Current knowledge concerning apoptotic‐altered mechanisms in melanoma has focused attention of research on molecules that are able to compensate for or bypass cell death defects, to improve the panel of chemotherapeutic drugs actually in use. Thus, development of new chemotherapeutic strategies that can facilitate death of cancer cells is an urgent objective.
3‐O‐methylfunicone (OMF) is a secondary metabolite produced by the soil fungus, Penicillium pinophilum. This compound has been found to be involved in antagonism of P. pinophilum to the plant pathogenic fungus, Rhizoctonia solani (6), and has displayed potent cytostatic properties on human tumour cells (HEp‐2) (7).
Our previous study carried out on tumour cell lines demonstrated the ability of OMF to induce growth arrest and apoptosis and to affect cell motility (8, 9). Strong structural similarity of OMF to more commonly used anticancer drugs, associated to anti‐proliferative and pro‐apoptotic properties demonstrated in HeLa cells, makes this compound a good candidate for arresting melanoma cell proliferation and migration.
The aim of this work was to evaluate the ability of OMF to arrest melanoma cell population growth and to induce apoptosis; these properties prevent tumour progression. We demonstrated that OMF has the ability to affect cell population growth and to induce apoptosis in parental (A375P) and a metastasis derived (A375M) melanoma cell lines of cutaneous origin, which display low and medium metastatic behaviour, respectively.
Materials and methods
OMF preparation
OMF was extracted from liquid cultures of isolate LT4 of P. pinophilum as previously described (10). For biological assays, the compound was dissolved in absolute ethanol at a concentration of 10 mg/ml.
Cell culture and treatments
A375P human primary melanoma cells and A375M human metastatic melanoma cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco Invitrogen, Milan, Italy) supplemented with 10% foetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mm l‐glutamine at 37 °C and 5% CO2. Normal human epidermal melanocytes (NHEM) were cultured in melanocyte growth medium (PromoCell, Heidelberg, Germany) supplemented with Bovine Pituitary Extract (BPE) 0.4%, basic fibroblast growth factor 1 ng/ml, insulin 5 µg/ml, hydrocortisone 0.5 µg/ml, and phorbol‐myristate‐acetate 10 ng/ml at 37 °C and 5% CO2. A preliminary dose–response curve was performed to determine concentration and time by which OMF produced significant effects on cell morphology and population growth inhibition. Aliquots containing 3 × 105 cells were plated in 6 multiwell plates (35 mm diameter) with 2 ml DMEM and treated with increasing OMF concentrations for 12, 24, 48 and 72 h. Control NHEM, A375P and A375M cells were treated or not treated with 16 µl absolute ethanol.
Morphological analysis
Morphological features of A375P and A375M cells treated with OMF were defined by phase‐contrast microscopy (Olympus CDK40, Milan, Italy) at ×20 magnification.
MTT cell proliferation assay
Two thousand OMF‐treated or not cell lines (NHEM, A375P and A375M) were grown in microplates (tissue culture grade, 96 wells, flat bottom), final volume 100 µl DMEM per well, at 37 °C and 5% CO2 for 24 h. Then, 10 µl of MTT labelling reagent (final concentration 0.5 mg/ml) (Roche Diagnostics, Milan, Italy) were added to each well for 4 h at 37 °C and 5% CO2. After 4 h, 100 µl of solubilization solution (10% SDS in 0.01 m HCl) were added to each well and incubated overnight. Spectrophotometrical absorbance was measured using a microplate ELISA reader (Biorad, Milan, Italy) at 600 nm wavelength.
Effect on replicative ability
Three hundred thousand A375P‐ and A375M‐ cell lines were seeded in 6 multiwell plates and treated with 80 µg/ml OMF for 24 h. After this period, medium in each well was changed to fresh medium without OMF; then the cells were incubated in 5% CO2 at 37 °C. Every 2 days cells were trypsinized and counted after trypan blue staining, and aliquots containing 3 × 105 cells were replated in 6‐multiwell plates with 2 ml fresh medium without OMF. Survival ability of cells kept in contact with OMF was calculated with respect to plating efficiency of controls and results obtained were plotted.
Reverse transcription–polymerase chain reaction analysis
Total RNA, isolated by High Pure RNA Isolation Kit (Roche Diagnostics, Milan, Italy) from A375P and A375M cells before and after treatment with 80 µg/ml OMF was transcribed by reverse transcriptase (Expand Reverse Transcriptase, Roche Diagnostics) at 42 °C for 45 min according to the manufacturer's instructions. Two microlitres of complementary DNA were amplified in a reaction mixture containing 10 mm Tris‐HCl (pH 8.3), 1.5 mm MgCl2, 50 mm KCl, 200 µm dNTP, and 2.5 units of Taq DNA polymerase (Roche Diagnostics) in a final volume of 50 µl. For co‐amplification conditions, polymerase chain reaction (PCR) was carried out in the presence of 0.5 µm sense and anti‐sense cyclin B1, p34cdc2, p21, p27, p53, survivin, hTERT, and TEP‐1 primers, and 0.05 µm sense and anti‐sense GAPDH or β‐actin primers. Caspase multiplex reverse transcription–PCR (RT‐PCR) was carried out according to the manufacturer's instructions (Maxim Biotech Inc., San Francisco, CA, USA). Conditions and size of products are reported in Table 1. Reactions were carried out in a DNA thermal cycler (Mastercycler gradient, Eppendorf, Milan, Italy). β‐actin RT‐PCR was performed on mRNA extracted at 24 h to confirm that mRNAs were suitable for RT‐PCR analysis. PCR products were analysed by electrophoresis on 1.8% agarose gel in TBE. Densitometric analysis of ethidium bromide‐stained agarose gel was carried out with NIH Image version 1.6 software. Ratio between the yield of each amplified product and that of co‐amplified internal control allowed a relative estimate of mRNA levels in the sample analysed. Internal control was a housekeeping gene whose PCR product was not overlapping with the gene of interest.
Table 1.
Human primer sense and anti‐sense sequences and expected PCR products (base pairs)
| Gene | Sense and anti‐sense sequences | Conditions | Base pairs |
|---|---|---|---|
| p21 | 5′‐CTGCCCAAGCTCTACCTTCC‐3′ | 30 cycles at 94 °C for 60 s | 123 |
| 5′‐CAGGTCCACATGGTCTTCCT‐3′ | 60 °C for 60 s, 72 °C for 60 s | ||
| p27 | 5′‐ATGTCAAACGTGCGAGTGTCTAAC‐3′ | 30 cycles at 95 °C for 60 s | 596 |
| 5′‐TTACGTTTGACGTCTTCTGAGGCCA‐3′ | 63 °C for 60 s, 72 °C for 60 s | ||
| P53 | 5′‐TTCTTGCATTCTGGGACAGCC‐3′ | 30 cycles at 94 °C for 60 s | 650 |
| 5′‐GCCTCATTCAGCTCTCGGAAC‐3′ | 56 °C for 60 s, 72 °C for 60 s | ||
| Cyclin B1 | 5′‐AAGAGCTTTAAACTTTGGTCTGGG‐3′ | 33 cycles at 94 °C for 30 s | 318 |
| 5′‐CTTTGTAAGTCCTTGATTTACCATG‐3′ | 55 °C for 60 s, 72 °C for 60 s | ||
| p34cdc2 | 5′‐GGAGTATAGGCACCATATTTGC‐3′ | 33 cycles at 95 °C for 60 s | 196 |
| 5′‐GACATGGGATGCTAGGCTTCC‐3′ | 54 °C for 60 s, 72 °C for 60 s | ||
| hTERT | 5′‐CGGAAGAGTGTCTGGAGCAA‐3′ | 35 cycles at 95 °C for 45 s | 163 |
| 5′‐GGATGAAGCGGAGTCTGGA‐3′ | 52 °C for 45 s, 72 °C for 45 s | ||
| TEP‐1 | 5′‐TCAAGCCAAACCTGAATCTGAG‐3′ | 31 cycles at 95 °C for 40 s | 264 |
| 5′‐CCCCGAGTGAATCTTTCTACGC‐3′ | 60 °C for 50 s, 72 °C for 50 s | ||
| Survivin | 5′‐ATGAGATACCATGGGTGCCCCGACG‐3′ | 34 cycles at 94 °C for 45 s | 450 |
| 5′‐TTAAGGATCCCTGCTCGATGGCACG‐3′ | 56 °C for 45 s, 72 °C for 60 s | ||
| GAPDH | 5′‐CGGAGTCAACGGATTTGGTCGTAT‐3′ | 306 | |
| 5′‐AGCCTTCTCCATGGTGGTGAAGAC‐3′ | |||
| β‐actin | 5′‐TGACGGGGTCACCCACACTGTGCCCATCTA‐3′ | 661 | |
| 5′‐CTAGAAGCATTGCGGTGGACGATGGAGGG‐3′ |
Protein extraction and Western blot analysis
A375P and A375M cells were treated with 80 µg/ml OMF for 24 h. Cells were scraped off using 1 ml phosphate‐buffered saline (PBS) and the cell pellet was homogenized with 300 µl ice‐cold buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 1% glycerol, 1% Triton, 1.5 mm MgCl2, 5 mm EGTA) supplemented with 20 mm sodium pyrophosphate, 40 µg/ml aprotinin, 4 mm PMSF, 10 mm sodium orthovanadate, 25 mm NaF. Total extracts were cleared by centrifugation for 30 min at 4 °C at 11 000 g and assayed for protein content using Bradford's method. Fifty micrograms of protein from each cell lysate were separated by 12.5% SDS‐PAGE and transferred to nitrocellulose membranes and filters were stained with 10% Ponceau S solution for 2 min, to verify equal loading and transfer efficiency. Blots were blocked overnight with 5% non‐fat dry milk, and then incubated with p21, p27, p53, caspase‐3 mouse monoclonal antibodies (Santa Cruz, Milan, Italy) and p34cdc2, caspase‐9, PARP‐1 rabbit polyclonal antibodies (Santa Cruz) 1 µg/ml, in TBS (150 mm NaCl, 20 mm Tris‐HCl, pH 8) for 2 h at room temperature. Cyclin B1 antibody conditions were previously described (11, 12). After washing with 0.1% Tween‐20 PBS, the filter was incubated with 1 : 2000 peroxidase‐conjugated anti‐mouse immunoglobulin for 1 h at 22 °C and 1 : 2500 peroxidase‐conjugated anti‐rabbit immunoglobulin for 1 h at 22 °C. Samples were extensively washed and finally analysed using the ECL system (Amersham, Milan, Italy). Protein loading was checked by reprobing membranes with β‐actin or α‐tubulin, to verify that there were no changes in protein level.
Fluorescence‐activated cell sorting analysis
Apoptosis induced by OMF treatment was additionally measured by fluorescence‐activated cell sorting (FACS) analysis. At 48 h of OMF treatment, A375P and A375M cells were trypsinized and fixed in 70% ethanol. Cells were then treated with 10 µg/ml RNase and 50 µg/ml propidium iodide (Sigma, St. Louis, MO, USA) for 2 h. Stained cells were analysed using a FACS Vantage (Becton Dickinson, Milan, Italy) for calculation of relative DNA content.
DNA fragmentation assay
To evaluate possible occurrence of internucleosomal hydrolysis of genomic DNA in OMF‐treated cells, 1 million cells were incubated in standard culture medium at 37 °C for 24 h, in the presence or absence of OMF, then DNA was isolated and 3 µg of total genomic DNA was analysed by agarose gel electrophoresis. At the end of incubation, cells were harvested using a cell scraper, centrifuged at 400 g, washed in PBS, and finally suspended in 100 µl of TNE buffer (10 mm Tris‐HCl (pH 8), 150 mm sodium chloride, 10 mm EDTA). Cell suspensions were lysed with three volumes of lysis buffer (0.2 % SDS, 100 µg/ml RNase in TNE), and the lysate was incubated at 37 °C for 1 h. After incubation, 100 µg/ml proteinase K was added to the lysate and the mixture was incubated for an additional 2.5 h at 56 °C. High molecular weight genomic DNA, extracted from proteinase K‐treated lysates according to a published procedure (13), was analysed by electrophoresis (2 h, 80 V) in ethidium bromide‐containing 1% agarose gel in TBE (0.045 m Tris‐borate, 0.001 m EDTA, pH 8.0).
Small interfering RNA transfection
Cells were grown to 60–70% confluence in 6 multiwell plates and transfected with 100 nmol specific small interfering RNA (siRNA) constructs, for 24 h according to the manufacturer's instructions (HP Validated siRNA, QIAGEN, Milan, Italy). Following silencing, cells were treated with 80 µg/ml OMF for 24 h and efficiency of p21 and p53 silencing verified by RT‐PCR analysis. Efficiency of transfection was assessed using a specific control provided in the kit (data not shown).
Statistical analysis
Each experiment was performed at least five times. Results are expressed as mean ± standard deviation. Analysis of variance (anova) was performed for each type of experiment (for example, MTT proliferation assay, RT‐PCR analysis). anova was supported by Student–Newman–Keuls test correction. P‐value was generally evaluated between 0.01 and 0.05, confirming statistical significance of the results.
Results
Inhibitory effect of OMF on population growth of A375P and A375M cell lines
We have previously shown that OMF can affect cell population growth of HeLa and MCF7 cells and induce apoptosis (8, 9). In the present study, we investigated the effect of OMF on population growth of A375P (parental) and A375M (metastasis derivative), two established melanoma cell lines with differing metastatic properties (14). Cells were treated with 40, 60, 80 and 100 µg/ml of OMF for 12, 24, 48 and 72 h, and cell viability was determined by MTT assay. OMF displayed a strong time‐ and dose‐dependent cell population growth inhibition, with the exception of 40 µg/ml, where only a moderate effect was visible (Fig. 1a,b). In particular, cells treated with 80 µg/ml OMF for 24 h showed typical features of apoptotic death (cell shrinkage, formation of membrane blebs, detachment of blebs from the surface) (Fig. 1d,g), in absence of necrotic phenomena. The lowest concentration (60 µg/ml OMF for 24 h) showed cell morphology modifications (data not shown), but at less extent than 80 µg/ml, whereas with the highest concentration (100 µg/ml OMF for 24 h), cell disruption morphology was noted typical of necrotic cells (Fig. 1e,h). To confirm occurrence of apoptosis in OMF‐treated cells, DNA fragmentation assays were performed. Agarose gel electrophoresis of DNA extracted from A375P and A375M cells treated with 40, 60, 80 and 100 µg/ml of OMF for 24 h revealed typical ladder patterns of internucleosomal fragmentation, only at concentration of 80 µg/ml, whereas the highest (100 µg/ml) showed typical feature of necrotic DNA (Fig. 2). It is worth noting that MTT assay performed on NHEM cell lines treated with 40, 60, 80 and 100 µg/ml of OMF for 12, 24, 48 and 72 h showed the inability of OMF to affect their population growth (Fig. 3), which indicates that the toxic effect was more specific to melanoma cells. By considering the results discussed here, to further investigate the feature of cell population growth inhibition we chose to work with OMF concentration of 80 µg/ml for 24 h.
Figure 1.
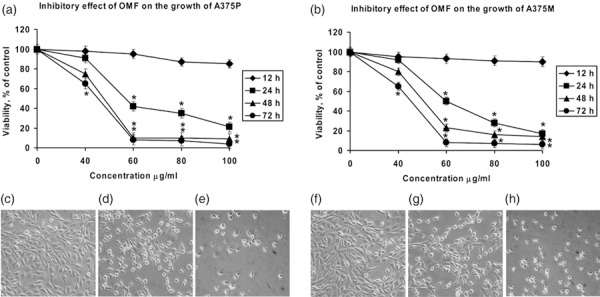
Effect of 3‐O‐methylfunicone (OMF) treatment on cell proliferation (a and b, MTT assay) and on cell morphology of A375P (c–e) and A375M cells (f–h) defined by phase‐contrast microscopy. (a and b) A375P and A375M cells were treated with various concentrations of OMF for 12, 24, 48, and 72 h. Data are presented as means ± standard deviations of the results of five independent experiments. *Significantly different compared to control (P < 0.05). (c) untreated A375P cells (24 h); (d) 80 µg/ml OMF‐treated A375P cells for 24 h; (e) 100 µg/ml OMF‐treated A375P cells for 24 h; (f) untreated A375M cells (24 h); (g) 80 µg/ml OMF‐treated A375M cells for 24 h; and (h) 100 µg/ml OMF‐treated A375M cells for 24 h. Viability was determined by MTT assay (magnification ×20).
Figure 2.
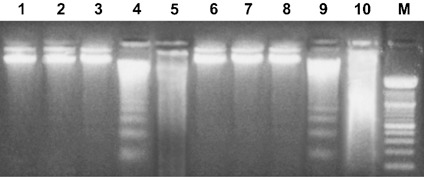
Effect of 3‐O‐methylfunicone (OMF) on DNA fragmentation. Lane 1, untreated A375P cells; lanes 2–5, A375P treated with 40, 60, 80, 100 µg/ml OMF for 24 h, respectively; lane 6, untreated A375M cells; lanes 7–10, A375M treated with 40, 60, 80, 100 µg/ml OMF for 24 h, respectively. M, 100‐bp ladder MW marker (Roche Diagnostics). Data shown are representative of five different experiments.
Figure 3.
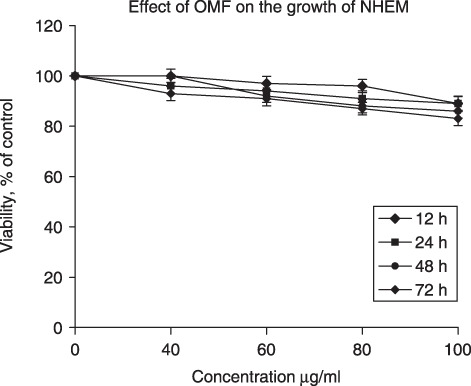
Effect of 3‐O‐methylfunicone (OMF) treatment on normal human epidermal melanocyte (NHEM) cell proliferation (MTT assay). NHEM cells were treated with various concentrations of OMF for 12, 24, 48, and 72 h. Data are presented as mean ± standard deviation of the results of five independent experiments.
OMF induced cell‐cycle arrest
To assess whether OMF‐induced cell population growth inhibition is mediated by alteration in cell‐cycle progression, we evaluated the effect of this compound on cell‐cycle phase distribution. After treatment with 80 µg/ml OMF for 24 h, A375P and A375M cells both arrested in G2 phase of cell cycle progression (Table 2). OMF treatment caused around 20% increase in the sub‐G1 peak in A375P cells, suggestive of apoptotic cell death, compared to OMF‐free culture. On the contrary, treatment of A375M with 80 µg/ml OMF induced the same accumulation of cells in the G2 phase with concomitant decrease in the G1 phase population, but only a slight increase in sub‐G1 peak was evident. These data seem to indicate that A375P parental cell lines were more responsive to OMF treatment, compared to A375M, in terms of apoptosis induction.
Table 2.
Effect of 3‐O‐methylfunicone (OMF) treatment on cell‐cycle distribution and apoptosis. A375P (untreated A375P), A375M (untreated A375M), A375P‐OMF (OMF‐treated A375P), A375M‐OMF (OMF‐treated A375M)
| Groups | Cell cycle | |||
|---|---|---|---|---|
| G0 + G1 | S | G2 + M | Sub‐G1 | |
| Untreated A375P | 60.90 ± 3.56 | 18.73 ± 2.15 | 12.50 ± 0.98 | 0.54 ± 0.27 |
| OMF‐treated A375P | 35.68 ± 1.75 | 16.59 ± 1.86 | 24.91 ± 1.11 | 20.38 ± 2.65 |
| p | A375P vs. A375P‐OMF = 0.001 | A375P vs. siRNA‐OMF1 = 0.00001 | A375P vs. siRNA‐OMF1 = 0.0001 | |
| Untreated A375M | 62.81 ± 3.74 | 16.92 ± 2.85 | 14.88 ± 1.25 | 0.32 ± 0.12 |
| OMF‐treated A375M | 30.04 ± 2.45 | 16.52 ± 2.15 | 36.05 ± 2.13 | 7.86 ± 0.97 |
| p | A375M vs. A375M‐OMF = 0.0001 | A375M vs. A375M‐OMF = 0.00001 | A375M vs. A375M‐OMF = 0.00001 | |
Data expressed as mean ± standard deviation of five experiments. Significant P‐values are reported.
OMF induced alteration in cell cycle‐regulating specific proteins in A375P and A375M cells
Since OMF arrested A375P and A375M cells mainly in the G2 phase of cell cycle, we determined the effect of OMF on cell cycle‐regulating proteins at the G2 boundary. As shown in Fig. 4a and 4c, cyclin B1 and cyclin‐dependent kinase p34cdc2 expression were markedly decreased in OMF‐treated A375P and A375M cells; these results were confirmed by Western blot analysis (Fig. 4b,d). Considering the possibility of different regulation, we also analysed cyclins and CDKs involved in the G1 phase of cell cycle, not observing any prominent change, with respect to untreated A375P or A375M cells (data not shown). Decreased cyclin B1 and p34cdc2 expression in cells exposed to OMF was associated to marked increase of p21 expression, as shown in Fig. 5a,d. We also evaluated possible involvement of p27, another cyclin dependent kinase inhibitor, reported to induce cell‐cycle arrest and apoptosis in melanoma cells (15). As shown in Fig. 5b and 5d, p27 expression was unchanged following OMF treatment in both cell lines. We studied also whether p21 accumulation was dependent on p53 induction. As shown in Fig. 5c and 5d, p53 was not involved in p21 accumulation, since neither RT‐PCR nor Western blotting showed modified levels of expression of p53, in OMF‐treated cells. Consequently, when p53 expression was silenced in A375P and A375M cells, as shown in Fig. 5c, OMF treatment was still able to induce strong up‐regulation of p21 expression (Fig. 5a). All these results pointed out that cell population growth arrest observed in OMF‐treated A375P and A375M cells was associated to p53‐independent activation of p21, excluding the possibility of p53 or p27 involvement in the molecular mechanism.
Figure 4.
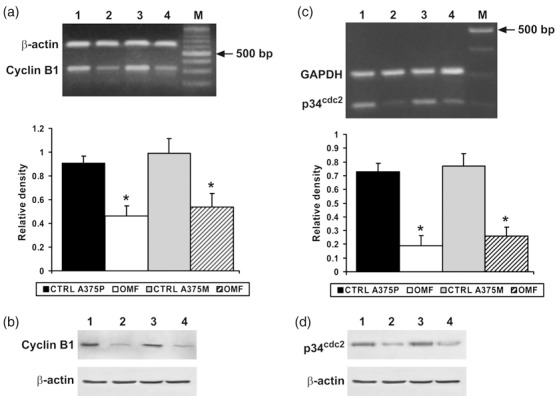
RT‐PCR analysis using specific primers for cyclin B1 and p34cdc2 mRNA expression (a and c) and Western blot analysis (b and d). (a and c) Top: Lane 1, untreated A375P cells mRNA; lane 2, mRNA from A375P treated with 80 µg/ml 3‐O‐methylfunicone (OMF) for 24 h; lane 3, untreated A375M cells mRNA; lane 4, mRNA from A375M treated with 80 µg/ml OMF for 24 h. M, 100‐bp ladder MW marker (Roche Diagnostics). Bottom: Quantitative measurements of the band intensities. (b and d) Lane 1, untreated A375P cells; lane 2, A375P treated with 80 µg/ml OMF for 24 h; lane 3, untreated A375M cells; lane 4, A375M treated with 80 µg/ml OMF for 24 h. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
Figure 5.
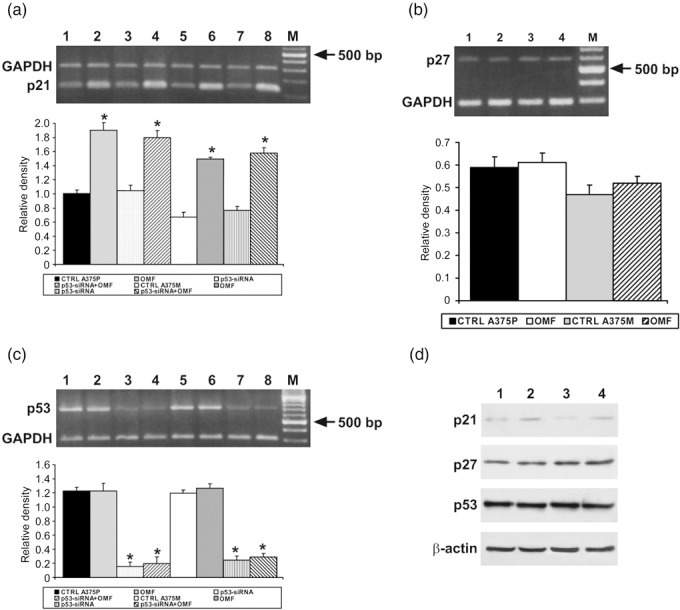
RT‐PCR analysis using specific primers for p21, p27 and p53 mRNA expression (a–c) and Western blot analysis (d). (a, c) Top: Lane 1, untreated A375P cells mRNA; lane 2, mRNA from A375P treated with 80 µg/ml 3‐O‐methylfunicone (OMF) for 24 h; lane 3, mRNA from p53‐siRNA A375P; lane 4, mRNA from p53‐siRNA A375P treated with 80 µg/ml OMF for 24; lane 5, untreated A375M cells mRNA; lane 6, mRNA from A375M treated with 80 µg/ml OMF for 24 h; lane 7, mRNA from p53‐siRNA A375M; lane 8, mRNA from p53‐siRNA A375M treated with 80 µg/ml OMF for 24 h M, 100‐bp ladder MW marker (Roche Diagnostics). (b) Top: Lane 1, untreated A375P cells mRNA; lane 2, mRNA from A375P treated with 80 µg/ml OMF for 24 h; lane 3, untreated A375M cells mRNA; lane 4, mRNA from A375M treated with 80 µg/ml OMF for 24 h. M, 100‐bp ladder MW marker (Roche Diagnostics). Bottom: Quantitative measurements of band intensities. (d) Lane 1, untreated A375P cells; lane 2, A375P treated with 80 µg/ml OMF for 24 h; lane 3, untreated A375M cells; lane 4, A375M treated with 80 µg/ml OMF for 24 h. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
A candidate molecule for the interface between apoptosis and cell cycle is survivin, a member of the inhibitor of apoptosis gene family, selectively expressed at G2 + M and localized at mitotic spindle microtubules (16). Thus, we decided to analyse the effect of OMF treatment on survivin gene expression. Figure 6 clearly shows strong inhibition exercised on survivin expression by OMF treatment, confirming the regulatory effect of OMF on A375P and A375M cell population growth.
Figure 6.
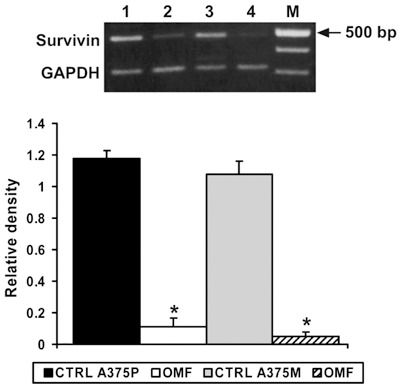
RT‐PCR analysis using specific primers for survivin mRNA expression. Top: Lane 1, untreated A375P cells mRNA; lane 2, mRNA from A375P treated with 80 µg/ml 3‐O‐methylfunicone (OMF) for 24 h; lane 3, untreated A375M cells mRNA; lane 4, mRNA from A375M treated with 80 µg/ml OMF for 24 h. M, 100‐bp ladder MW marker (Roche Diagnostics). Bottom: Quantitative measurements of band intensities. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
Requirement for p21 in OMF‐induced cell population growth inhibition and apoptosis
To evaluate the role of p21 in OMF‐induced apoptosis more directly, we examined the effect on apoptosis of silencing p21 expression and cell cycle distribution in OMF‐treated cells. RT‐PCR analysis confirmed that siRNA construct was able to reduce p21 expression (Fig. 7a). As a consequence, in p21‐silenced, OMF‐treated A375P and A375M cells, reduced levels of apoptosis were also evident, compared to p21‐silenced, OMF‐untreated cells (Table 3). In addition, p21 was only weakly induced in p21‐silenced, OMF‐treated cells, while cyclin B1 and p34 expression were in part restored (Fig. 7b,c, respectively). These results corroborate other findings showing that OMF‐induced accumulation of p21 is at least partly responsible for cell population growth inhibition and apoptosis induction in A375P and A375M cells.
Figure 7.
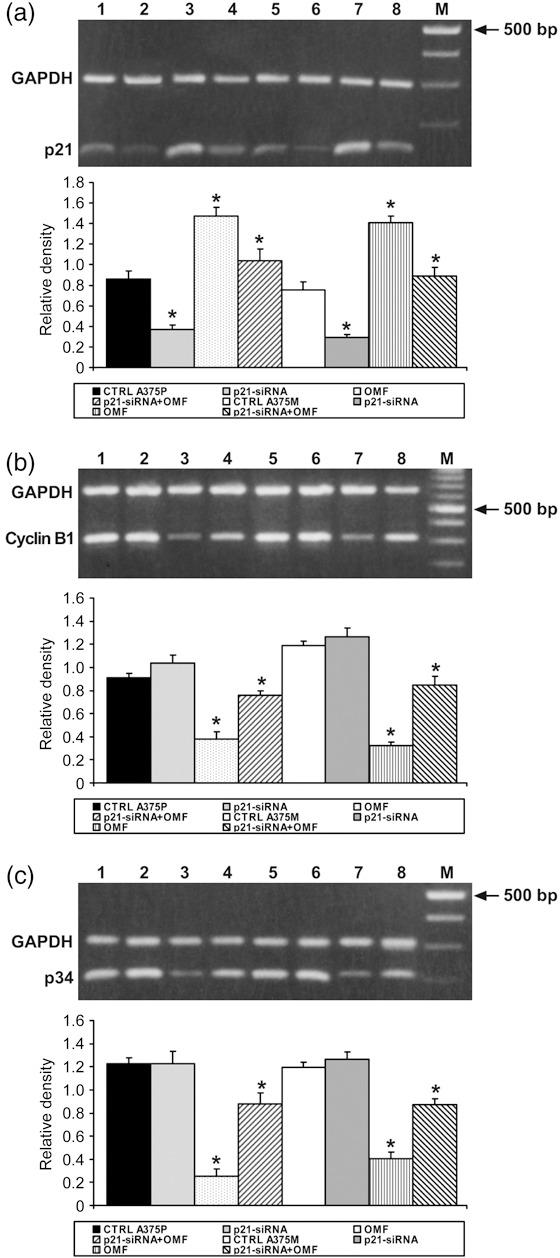
Effect of p21 silencing on cells treated with 3‐O‐methylfunicone (OMF). (a–c) RT‐PCR analysis using specific primers for p21, cyclin B1 and p34 mRNA expression. Top: Lane 1, untreated A375P cells mRNA; lane 2, mRNA from p21‐siRNA A375P; lane 3, mRNA from A375P treated with 80 µg/ml OMF for 24 h; lane 4, mRNA from p21‐siRNA A375P treated with 80 µg/ml OMF for 24 h; lane 5, untreated A375M cells mRNA; lane 2, mRNA from p21‐siRNA A375M; lane 3, mRNA from A375M treated with 80 µg/ml OMF for 24 h; lane 4, mRNA from p21‐siRNA A375M treated with 80 µg/ml OMF for 24 h; M, 100‐bp ladder MW marker (Roche Diagnostics). Bottom: Quantitative measurements of band intensities. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
Table 3.
Effect of p21 siRNA on cell‐cycle distribution and apoptosis of OMF‐treated cells. A375P (untreated A375P), A375M (untreated A375M), siRNA‐OMF1 (p21‐siRNA‐OMF treated A375P), siRNA‐OMF2 (p21‐siRNA‐OMF‐treated A375M)
| Groups | Cell cycle | |||
|---|---|---|---|---|
| G0 + G1 | S | G2 + M | Sub‐G1 | |
| Untreated A375P | 63.85 ± 2.89 | 19.63 ± 2.56 | 11.65 ± 1.23 | 0.45 ± 0.28 |
| p21‐siRNA‐OMF untreated A375P | 59.70 ± 2.33 | 21.42 ± 1.85 | 14.74 ± 3.04 | 1.30 ± 1.34 |
| p21‐siRNA‐OMF treated A375P | 50.65 ± 3.92 | 18.56 ± 1.68 | 17.11 ± 1.11 | 9.46 ± 1.12 |
| p | A375P vs. siRNA‐OMF1 = 0.0046 | A375P vs. siRNA‐OMF1 = 0.002 | A375P vs. siRNA‐OMF1 = 0.00001 | |
| Untreated A375M | 62.95 ± 2.94 | 18.78 ± 3.02 | 17.65 ± 1.25 | 0.62 ± 0.16 |
| p21‐siRNA‐OMF untreated A375M | 57.27 ± 1.07 | 19.56 ± 1.58 | 21.41 ± 1.13 | 1.09 ± 1.02 |
| p21‐siRNA‐OMF treated A375M | 43.05 ± 1.98 | 21.52 ± 2.34 | 25.36 ± 2.13 | 2.34 ± 0.56 |
| p | A375M vs. siRNA‐OMF2 = 0.0003 | A375M vs. siRNA‐OMF2 = 0.002 | A375M vs. siRNA‐OMF2 = 0.003 | |
Data expressed as mean ± standard deviation of five experiments. Significant P‐values are reported.
OMF induces apoptosis by caspase‐3 and caspase‐9 cleavage and PARP‐1 degradation
To further confirm occurrence of apoptosis in OMF‐treated cells, we studied degradation of poly(ADP‐ribose) polymerase‐1 (PARP‐1), an enzyme that influences numerous cellular processes, including DNA repair, transcription regulation, and caspase‐independent cell death (17). It is cleaved during the apoptotic response by caspase‐3 and caspase‐7, thereby inhibiting its activity. Western blot analysis showed cleavage of PARP‐1 in treated A375P and A375M cells (Fig. 8) and activation of the caspase cascade was investigated. As shown in Fig. 9, multiplex RT‐PCR and Western blot analysis showed increased expression and proteolytic cleavage, respectively, of pro‐caspase‐3 and pro‐caspase‐9. In contrast, caspase‐8 was only weakly represented and resulted in no modification, suggesting activation of a caspase cascade typical of the mitochondrial apoptotic pathway.
Figure 8.
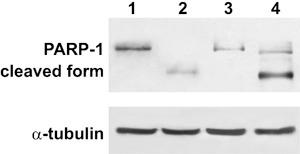
Effect of 3‐O‐methylfunicone (OMF) on PARP‐1 cleavage in A375P and A375M cells. Lane 1, untreated A375P cells; lane 2, A375P treated with 80 µg/ml OMF for 24 h; lane 3, untreated A375M cells; lane 4, A375M treated with 80 µg/ml OMF for 24 h. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
Figure 9.
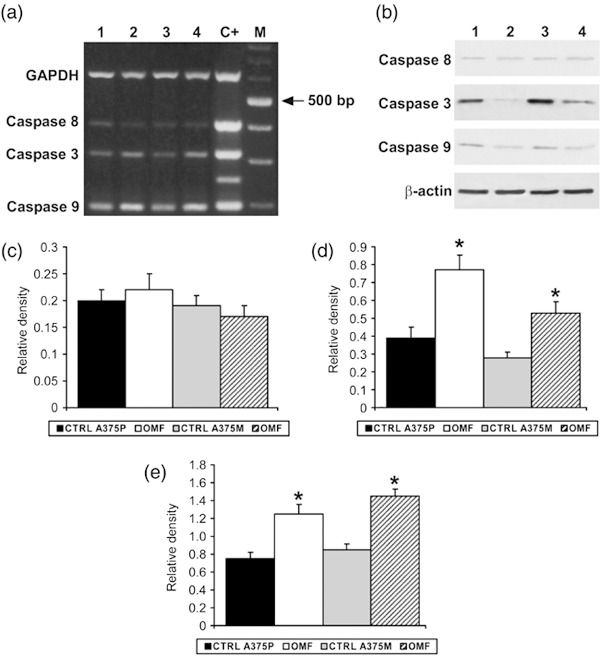
Multiplex RT‐PCR analysis using specific primers for caspase mRNA expression (a) and Western blot analysis (b). Lane 1, untreated A375P cell mRNA; lane 2, mRNA from A375P treated with 80 µg/ml OMF for 24 h; lane 3, untreated A375P cell mRNA; lane 4, mRNA from A375M treated with 80 µg/ml OMF for 24 h; C+, positive control; M, 100‐bp ladder MW marker (Roche Diagnostics). (d,e) Quantitative measurements of the band intensities relative to caspase‐8 (c), caspase‐3 (d) and caspase‐9 (e) RT‐PCR. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
Effect of OMF on cell replication
Since OMF affects cell‐cycle progression, we investigated the effects of the molecule on hTERT, the catalytic subunit of the telomerase enzyme, whose repression leads to cellular death (18). The antiproliferative effect of OMF seem to be exerted on hTERT (Fig. 10a). In fact, it is clearly showed that hTERT is inhibited after treatment with the molecule. The same result was obtained when TEP1, another component of telomerase, was investigated (19), (Fig. 10b). These results are confirmatory of OMF effect on cell proliferation inhibition.
Figure 10.
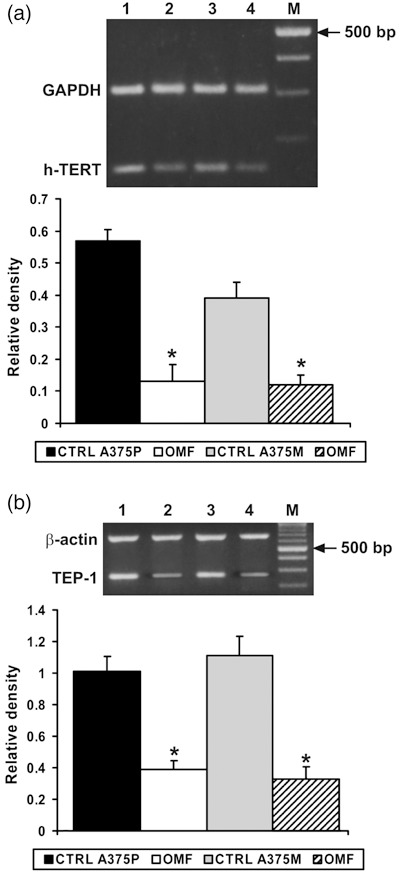
RT‐PCR analysis using specific primers for h‐TERT and TEP‐1 mRNA expression. (a,b) Top: Lane 1, untreated A375P cells mRNA; lane 2, mRNA from A375P treated with 80 µg/ml OMF for 24 h; lane 3, untreated A375M cells mRNA; lane 4, mRNA from A375M treated with 80 µg/ml OMF for 24 h. M, 100‐bp ladder MW marker (Roche Diagnostics). Bottom: Quantitative measurements of band intensities. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
We analysed if OMF has the ability to induce a permanent damage in the cell population, investigating which was the replicative ability of cells treated with 80 µg/ml OMF for 24 h and, successively, grown in OMF‐free medium. A375P and A375M cells were observed until they restored or completely lost their ability to replicate. As showed in Fig. 11 the replicative ability of OMF‐treated cells was significantly decreased in comparison with control cells. This result clearly shows that OMF significantly affects tumour cell replication, inducing an irreversible damage to the progeny.
Figure 11.
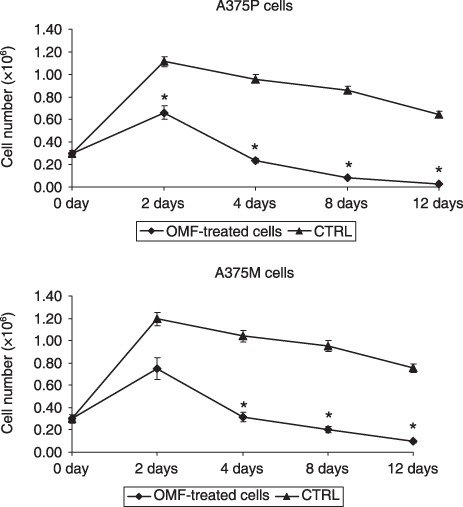
3‐O‐methylfunicone (OMF) affects cell replicative ability. Day 0: OMF‐treated cells for 24 h and replated in OMF‐free medium. Data shown are representative of five different experiments (± standard deviation). *Significantly different compared with control (P < 0.05).
Discussion
One of the earliest events in melanoma progression involves dysregulated proliferation of melanocytes. At this stage, cells lose their ability to maintain cell cycle controls that function in normal unstimulated melanocytes. This loss of control may lead to sustained proliferation, decreased apoptosis, or both. It has also been reported that melanocytes display broad expression of apoptotic inhibitors to maintain their longevity, at the cost of damaged cells with high probability of developing melanoma (5). In contrast, keratinocytes are more prone to undergo apoptosis to ensure rapid turnover and to remove damaged cells efficiently, to meet their functional need in the skin.
Many studies have focused on understanding the cause of such deregulation or have analysed new compounds that are able to induce cell population growth inhibition and apoptosis. OMF has already been characterized as an antiproliferative compound on different tumour cell lines, sharing also anti‐apoptotic and anti‐migratory influence, and thus, capable of comparing possible metastatic features of tumour cell lines. In this work, we have demonstrated that OMF inhibits melanoma cell population growth by inducing apoptosis. This effect was strongly evident in A375P cells, which are characterized as having low metastatic properties and where a more consistent percentage of cells is committed to apoptotic cell death. A375M cells were in the same way affected by OMF, but to lesser extent; this is probably attributable to these cells’ higher metastatic property than A375Ps. The best effect, in terms of population growth inhibition, was shown at 80 µg/ml for 24 h in which more than 60% population growth inhibition, in absence of necrotic phenomena, was demonstrated. FACS analysis clearly revealed that OMF arrested proliferation of A375P and A375M at G2 phase of the cell cycle. This result highlighted greater susceptibility of A395P cells to follow an apoptotic pathway compared to A375M cells. The major regulator of G2 + M transition is a complex consisting of catalytic p34Cdc2 subunit and regulatory cyclin B1 subunit that controls a cell's entry into mitosis (20). Recently, a marked decrease in protein expression of cyclin B1 and its activating partner Cdc2, with concomitant induction of p21WAF1/CIP1, has been reported in human melanoma cells (21). Consistent with these reports, OMF decreased cyclin B1 and p34 mRNA and protein expression in both cell lines and increased p21 expression. The gene Waf1/Cip1 encodes p21 protein (22), whose over‐expression in mammalian cells causes population growth arrest in G1 and/or G2 + M through the inhibition of cyclin/Cdk complexes, which are required for cell cycle progression. Production of p21 can be induced by DNA damage and during apoptosis in specific cell types, as a function of wild‐type p53 activation (23). It can also be induced in a p53‐independent manner as an immediate early gene following mitogenic stimulation of population growth‐arrested cells (22). Furthermore, when DNA damage occurs during G2, cells are able to initiate cell cycle arrest in the absence or presence of p53. Entry into mitosis is prevented by maintaining CDK1/cyclin B1 complex in its inhibited form. Since OMF induces typical features of apoptosis in both cell lines as confirmed by FACS analysis, DNA laddering and PARP‐1 degradation, one might be persuaded to think that inhibited cell population growth is the consequence of DNA damage provoked by OMF treatment. Cells of the A375P and A375M lines are committed to apoptosis, being unable to repair the damage, and to avoid inheritance by their progeny. Overall, the increasing effect on p21 could be the main molecular events contributing to G2 arrest, confirmed also by silencing experiments, preventing replication of damaged DNA. Consequently, cyclin B1 and p34 decrease and the apoptotic process is triggered.
Worthy of note also are the results concerning survivin, originally described as an inhibitor of apoptosis proteins with cell‐cycle‐specific function (24). Survivin is a mitotic substrate of p34cdc2‐cyclin B1, and its phosphorylation on Thr34, regulates apoptosis at cell division via interaction with caspase‐9 (16). Human survivin has become of particular interest as it has been identified as a tumour‐associated antigen, highly expressed in various tumours, but undetectable in normal adult tissues (25). The effect exercised by OMF on survivin, caspase‐3 and caspase‐9 confirms the anti‐apoptotic properties of the molecule, which encouraged us to investigate thoroughly its anti‐tumour properties too.
In all normal somatic cells, each cycle of cell division and DNA replication results in the loss of 50–200 terminal nucleotides from each chromosome. This gradually results in instability of the chromosome and cell death. Telomerase gene is a specialized RNA‐directed DNA polymerase that maintains telomere length by synthesizing hexameric telomeric repeats (18, 26), being essential in many cases for telomere stability and cell proliferation, immortalization and transformation. The importance of modulating expression of this gene or its components relies in that hTERT mRNA has been found to be specifically expressed in most tumour lesions associated with telomerase activity, but it is not detectable in nearly all normal tissues (27). In addition, it has been demonstrated that inhibition of telomerase activity in human cancer cells, by use of anti‐sense hTERT, forces them either to undergo apoptosis or differentiation. Effects of OMF on hTERT and TEP‐1 are very interesting. Results here seemed to suggest that OMF inhibits telomerase activity and consequently, shortens telomere length of cancer cell chromosomes, which disrupt their stability. Cancer cells are forced to not replicate nor die; this is an important goal, since in the clinic high mitotic rate of tumour cells is a predictive attribute for short recurrence‐free state and poor overall survival (28). Consistent with these results is the effect of duration of OMF treatment on A375P and A375M cells. As demonstrated by MTT assay, both cell lines were unable to replicate even though OMF was eliminated from culture medium; this indicates the ability of OMF to induce permanent damage to a cell population, which in turn affects its replicative ability.
Results presented here provide new insight into the mechanism of action of OMF, and contribute to extending the list of drugs with therapeutic potential for treatment of melanoma. OMF has the ability to control proliferation of melanoma cells; therefore, it represents a candidate molecule that is able to block melanoma progression. Indeed, a small molecule like OMF should be further studied in order to generate small compounds to be used in future clinical trials. However, it is important to point out that further in vivo studies on animal models should better clarify anti‐tumour effect of OMF and its ability to prevent melanoma cells from spreading to lymph nodes.
Acknowledgement
This study was supported by grants from Legge 5 (2006), Regione Campania.
References
- 1. Soengas MS, Lowe SW (2003) Apoptosis and melanoma chemoresistance. Oncogene 22, 3138–3151. [DOI] [PubMed] [Google Scholar]
- 2. Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DG et al (2001) Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Invest. 19, 3635–3648. [DOI] [PubMed] [Google Scholar]
- 3. Glinsky GV, Glinsky VV, Ivanova AB, Hueser CJ (1997) Apoptosis and metastasis: increased apoptosis resistance of metastatic cancer cells is associated with the profound deficiency of apoptosis execution mechanism. Cancer Lett. 115, 185–193. [DOI] [PubMed] [Google Scholar]
- 4. Johnstone RW, Ruefli AA, Lowe SW (2002) Apoptosis: a link between cancer genetics and chemiotherapy. Cell 108, 153–164. [DOI] [PubMed] [Google Scholar]
- 5. Bowen AR, Hanks AN, Allen SM, Alexander A, Diedrich MJ, Grossman D (2003) Apoptosis regulators and responses in human melanocytic and keratinocytic cells. J. Invest. Dermatol. 120, 48–55. [DOI] [PubMed] [Google Scholar]
- 6. Nicoletti R, De Stefano M, De Stefano S, Trincone A, Marziano F (2004) Identification of fungitoxic metabolites produced by some Penicillium isolates antagonistic to Rhizoctonia solani . Mycopathologia 158, 465–474. [DOI] [PubMed] [Google Scholar]
- 7. Stammati A, Nicoletti R, De Stefano S, Zampaglioni F, Zucco F (2002) Cytostatic properties of a novel compound derived from Penicillium pinophilum: an in vitro study. Altern. Lab. Anim. 30, 69–75. [DOI] [PubMed] [Google Scholar]
- 8. Buommino E, Nicoletti R, Gaeta GM, Orlando M, Ciavatta ML, Baroni A et al (2004) 3‐O‐methylfunicone, a secondary metabolite produced by Penicillium pinophilum, induces growth arrest and apoptosis in HeLa cells. Cell Prolif. 37, 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buommino E, Boccellino M, De Filippis A, Cozza V, Nicoletti R, Ciavatta ML et al (2007) 3‐O‐methylfunicone produced by Penicillium pinophilum affects cell motility of breast cancer cells, downregulating αVβ5 integrin and inhibiting metalloproteinase‐9 secretion. Mol. Carcinog. 46, 930–940. [DOI] [PubMed] [Google Scholar]
- 10. De Stefano S, Nicoletti R, Milone A, Zambardino S (1999) 3‐O‐Methylfunicone, a fungitoxic metabolite produced by the fungus Penicillium pinophilum . Phytochemistry 52, 1399–1401. [Google Scholar]
- 11. Caputi M, Groeger A, Esposito V, Dean C, De Luca A, Pacilio C et al (1999) Prognostic role of cyclin D1 in lung cancer: relationship with proliferating cell nuclear antigen. Am. J. Respir. Cell Mol. Biol. 20, 746–750. [DOI] [PubMed] [Google Scholar]
- 12. Van Wijnen A, Aziz F, Graña X, De Luca A, Desai RK, Jaarsveld K et al (1994) Transcription of histone H4, H3 and H1 cell cycle genes: Promoter factor HINF‐D contains CDC2, cyclin A, and RB‐related protein. Proc. Natl. Acad. Sci. USA 91, 12882–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: a Laboratory Manual, 2nd edn. New York: Cold Spring Harbor Laboratory. [Google Scholar]
- 14. Gehlsen KR, Davis GE, Sriramarao P (1992) Integrin expression in human melanoma cells with differing invasive and metastatic properties. Clin. Exp. Metastasis. 10, 111–120. [DOI] [PubMed] [Google Scholar]
- 15. Choi HJ, Choi YH, Yee SB, Im E, Jung JH, Kim ND (2005) Ircinin induces cell cycle arrest and apoptosis in SK‐MEL‐2 human melanoma cells. Mol. Carcinog. 44, 162–173. [DOI] [PubMed] [Google Scholar]
- 16. O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A et al (2000) Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. USA 97, 13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. West JD, Ji C, Marnett LJ (2005) Modulation of DNA fragmentation factor 40 nuclease activity by poly (ADP‐ribose) polymerase‐1. J. Biol. Chem. 280, 15141–15147. [DOI] [PubMed] [Google Scholar]
- 18. Greider CW (1998) Telomerase activity, cell proliferation, and cancer. Proc. Natl. Acad. Sci. USA 95, 90–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh U, Das N, Bhattacharyya NP (2007) Inhibition of telomerase activity by reduction of poly (ADP‐ribosyl) ation of hTERT and TEP1/TP1 expression in HeLa cells with knocked down poly (ADP‐ribose) polymerase‐1 (PARP‐1) gene. Mutat. Res. 615, 66–74. [DOI] [PubMed] [Google Scholar]
- 20. Yuan J, Yan R, Kramer A, Eckerdt F, Roller M, Kaufmann M et al (2004) Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 23, 5843–5852. [DOI] [PubMed] [Google Scholar]
- 21. Choi HJ, Yee SB, Park SE, Im E, Jung JH, Chung HY et al (2006) Petrotetrayndiol A induces cell cycle arrest and apoptosis in SK‐MEL‐2 human melanoma cells through cytochrome c‐mediated activation of caspases. Cancer Lett. 232, 214–225. [DOI] [PubMed] [Google Scholar]
- 22. Michieli P, Chedid M, Lin D, Pierce HJ, Mercer WE, Givol D (1994) Induction of Waf1/Cip1 by a p53‐independent pathway. Cancer Res. 54, 3391–3395. [PubMed] [Google Scholar]
- 23. El Deiry W, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J et al (1994) WAF1/CIP1 is induced in p53‐mediated G1 arrest and apoptosis. Cancer Res. 54, 1169–1174. [PubMed] [Google Scholar]
- 24. Temme A, Rieger M, Reber F, Lindemann D, Weigle B, Diestelkoetter‐Bachert P et al (2003) Localization, dynamics, and function of survivin revealed by expression of functional survivinDsRed fusion proteins in the living cell. Mol. Biol. Cell 14, 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ambrosini G, Adida C, Altieri DC (1997) A novel antiapoptosis gene survivin, expressed in cancer and lymphoma. Nat. Med. 3, 917–921. [DOI] [PubMed] [Google Scholar]
- 26. Lee SH, Kim JW, Oh SH, Kim YJ, Rho SB, Park K et al (2005) IFN‐γ/IRF‐1‐induced p27kip1 down‐regulates telomerase activity and human telomerase reverse transcriptase expression in human cervical cancer. FEBS Lett. 579, 1027–1033. [DOI] [PubMed] [Google Scholar]
- 27. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL et al (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 28. Karjalainen JM, Eskelinen MJ, Nordling S, Lipponem PK, Alhava EM, Kosma VM (1998) Mitotic rate and S‐phase fraction as prognostic factors in stage1 cutaneous malignant melanoma. Br. J. Cancer 77, 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]