

Abstract
Objectives: Increasing evidence has suggested the close relationship between microRNAs (miRNAs) dysregulation and the carcinogenesis of Ewing's sarcoma (ES), among of which miR‐125b has been reported to be decreased in ES tissues recently. Strikingly, ectopic expression of miR‐125b could suppress cell proliferation of ES cell line A673, suggesting the tumor suppressor role of miR‐125b in ES. However, the other accurate mechanistic functions and relative molecule mechanisms are largely unknown.
Materials and Methods: Herein, we completed a series of experiments to investigate the role of miR‐125b in Ewing's sarcoma. We restored the expression of miR‐125b in ES cell line A673 through transfection with miR‐125b mimics. To further understand the role of miR‐125b in ES, we detected the effects of miR‐125b on the cell proliferation, migration and invasion, cell cycle as well as cell apoptosis.
Results: We found that restored expression of miR‐125b in ES cell line A673 inhibited cell proliferation, migration and invasion, arrested cell cycle progression, and induced cell apoptosis. Moreover, bioinformatic prediction suggested the oncogene, phosphoinositide‐3‐kinase catalytic subunit delta (PIK3CD), was a target gene of miR‐125b in ES cells. Further quantitative RT‐PCR and western blot assays identified over‐expression of miR‐125b suppressed the expression of PIK3CD mRNA and protein. PIK3CD participates in regulating the PI3K signaling pathway, which has been reported to play an important role in the development of ES. Suppression of PIK3CD down‐regulated the expression of phospho‐AKT and phospho‐mTOR proteins and inhibited the biologic progression of A673 cells.
Conclusions: Collectively, these data suggest that miR‐125b functions as a tumor suppressor by targeting the PI3K/Akt/mTOR signaling pathway, and may provide potential therapy strategy for ES patients by targeting miRNA expression.
Introduction
Ewing's sarcoma (ES) is the second most common primary bone and soft malignant tumour in children and adolescents, characterized by small round cells 1. Over the past number of decades, although great effort has been exerted to elucidate underlying mechanisms, and to discover novel diagnostic biomarkers and therapeutic targets for ES patients 2, molecular systems underlying ES carcinogenesis remain largely unknown. Thus, better understanding of the biology of this malignancy is critical to development of prognostic biomarkers and improved novel therapies.
MicroRNAs (miRNAs) compose a large family of non‐coding single‐stranded RNAs 3. By pairing with the 3′‐untranslated region (3′UTR) of specific messenger RNAs (mRNAs), miRNAs can regulate expression of various target mRNAs, at the level of either mRNA degradation or translation. MiRNAs have been reported to be involved in many biological processes including cell proliferation, apoptosis, differentiation, metabolism and more 4. Recently, many reports have indicated that miRNAs play an important role in initiation and progression of ES. Thus, better knowledge of changes in miRNA gene expression during Ewing carcinogenesis may lead to improvements in disease characteristics and therefore provide new avenues for ES diagnostic and treatment regimens.
MiR‐125b is one of the most consistently dysregulated miRNAs. As a homologue of lin‐4, miR‐125b is highly conserved among different species from flies to humans 5. Currently, the role of miR‐125b in progression of ES has been widely explored. Microarray analysis of 11 normal bone tissues and 23 primary ES tumours has indicated that miR‐125b was underexpressed in ES tissues compared to normal bone tissues 6. Moreover, ectopic expression of miR‐125n negatively regulated the insulin‐like growth factor (IGF) signalling pathway and suppressed cell proliferation of A673 cells 7. These data provide a solid foundation for ES utilization of miRNAs in diagnostic and anti‐cancer therapy for the future. However, other accurate mechanistic functions and relative molecular mechanisms remain unclear. In this study, we aimed to investigate cellular consequences of miR‐125b expression in an ES cancer model system, by restoring miR‐125b expression in A673 cells, which have been reported to have low levels of miR‐125b. Restored miR‐125b expression of miRNA mimic transfection‐inhibited cell proliferation, migration and invasion, arrest cell cycle progression, and induced apoptosis of A673 cells. Further bioinformatic prediction has suggested that the tumour suppressor role of miR‐125b in ES cells might be mediated through inhibition of PIK3CD, as ectopic expression of miR‐125b has significantly suppressed expression of PIK3CD and activity of the PI3K/Akt/mTOR signalling pathway. These results suggested that miR‐125b functions as a tumour suppressor miRNA in ES cells and have provided a potential therapeutic strategy for ES patients by targeting miRNA expression.
Materials and methods
Cell culture and transfection
Human ES cell line A673 was obtained from the American Type Culture Collection. They were then maintained in RPMI 1640 medium (PAA, Pasching, Austria) supplemented with 10% foetal bovine serum (PAA), streptomycin (100 μg/ml) and penicillin (100 U/ml). Cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C. MiR‐125b mimic, scramble mimic, siRNA (specifically for PIK3CD) and control siRNA were all purchased from Dharmacon (Austin, TX, USA). Oligonucleotides were transfected into A673 cells to a final concentration of 50 nm by Dhamafect 1 (Dharmacon) according to the manufacturer's instructions. Medium was changed after 6 h transfection.
RNA extraction, reverse transcription and quantitative real‐time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Life Technologies Inc., Darmstadt, Germany) according to the manufacturer's instructions. Expression level of GAPDH was used as internal control for mRNAs, and U6 level was regarded as internal miRNA control. To quantify expression of PIK3CD and GAPDH mRNA, total RNA was reverse transcribed using First‐Strand cDNA Synthesis kit (Invitrogen) according to the manufacturer's protocol. Reverse transcription reaction for PIK3CD and GAPDH was performed using Oligo (dT) (5′‐TTTTTTTTTTTTT‐3′) as primer. To analyse expression of miR‐125b, specific stem‐loop reverse transcription primer was used as follows: 5′‐GTCGTATCCAGTGCAGG GTCCGAGGTATTCGCACTGGTCUCUU‐3′, and reverse transcription reaction primer of U6 was: 5′‐AAAATATGGAACGCTTCACG AATTTG‐3′.
Quantitative real‐time PCR was performed using the Quanti‐TectSYBR Green PCR mixture on an ABI PRISM 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Forward and reverse primers for PIK3CD were 5′‐AACAGCCATAAACGGAAACG‐3′ and 5′‐CGTAGAGGT GTGGTGAGTCA‐3′, and the primers for GAPDH were 5′‐TCAACGACCACTTTGTCAAGC TCA‐3′ and 5′‐GCTGGTGGTCCAGGGGTCTTACT‐3′, respectively. Moreover, forward and reverse primers for miR‐125b were: 5′‐GCUCCCUGAGACCCUAAC‐3′ and 5′‐CAGTGC AGGGTCCGAGGT‐3′, and the PCR primers for U6 were 5′‐CTCGCTTCGGCAGCACATAT ACT‐3′ and 5′‐ACGCTTCACGAATTTGCGTGTC‐3′. Relative expression levels were evaluated using the 2−ΔΔCt method and all experiments were run in triplicate.
Western blot analysis
For western blot assays, cells were harvested in ice‐cold PBS 48 h after transfection, and lysed on ice in cold modified radioimmunoprecipitation buffer supplemented with protease inhibitors. Protein concentration was determined using BCA Protein Assay Kit (Bio‐Rad, Hercules, CA, USA) and equal amounts of protein were analysed by SDS‐PAGE. Gels were electroblotted on to nitrocellulose membranes (Millipore, Darmstadt, Germany). Membranes were blocked for 2 h with 5% non‐fat dry milk in Tris‐buffered saline containing 0.1% Tween‐20, and incubated at 4 °C overnight with primary antibody. Detection was performed using peroxidase‐conjugated secondary antibodies and enhanced chemiluminescence system (Millipore). Primary antibodies used were: PIK3CD, AKT, P‐AKT, mTOR, p‐mTOR and GAPDH (Cell Signaling, Danvers, MA, USA). The experiment was repeated three times.
Cell proliferation analysis
For analysis of cell proliferation, cells were seeded into 24‐well plates at 5 × 103 cells/well and were incubated in 10% Cell Counting Kit‐8 fluids (CCK‐8, Dojindo, Mashikimachi, Japan) diluted in normal culture medium at 37 °C until visual colour conversion occurred. Proliferation level was determined at 0, 24, 48 and 72 h after transfection. Absorbance in each well was measured using a microplate reader set at 450 and 630 nm. All experiments were performed in quadruplicate.
Cell cycle analysis
Cells cycle analysis was performed on the cells 48 h after transfection. Cells were harvested, washed twice in cold PBS, fixed in ice‐cold 70% ethanol, incubated with propidium iodide (PI) and RNase A, then analysed by fluorescence‐activated cell sorting. All experiments were run in triplicate.
Analysis of apoptosis
For analysis of apoptosis, cells were collected and diluted to a concentration of 1 × 106 cells/ml and washed three times in ice‐cold PBS 72 h after transfection. They were incubated in PE annexin‐V and 7AAD according to the PE Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Jose, CA, USA) protocol, then analysed by fluorescence‐activated cell sorting. Cells undergoing early apoptosis bind only to annexin v, and cells binding both are either in late stages of apoptosis or are already dead. The experiment was repeated three times.
Wound‐healing assays
For the wound‐healing assay, cells were grown to near 100% confluence in 24‐well plates, and treated with oligonucleotides. 24 h after transfection, linear scratch wounds were created on the confluent cell monolayers using a 200‐μl pipette tip. To prevent cells from entering the cycle prior to wounding, they were maintained in serum‐free medium. To visualize migrating cells and wound healing, images were captured at 0, 12, 24 and 36 h. A total of ten areas were selected randomly from each well and cells in three wells of each group were quantified. Experiments were independently repeated three times.
Cell invasion assays
For the invasion assay, transwell migration chambers (8 μm) were coated with Matrigel (BD Biosciences, San Jose, CA, USA) and incubated at 37 °C for 1 h allowing it solidify. After 24 h transfection, 4 × 105 cells suspended in serum‐free DMEM were added to the upper chamber, and medium containing 20% foetal bovine serum was added to the lower chamber as chemoattractant. After 24‐h transfection, non‐filtered cells were gently removed with cotton wool swabs. Filtered cells located on the lower side of the chamber were stained with crystal violet, air dried and photographed. Experiments were independently repeated three times.
Vector construction and luciferase assays
The whole 3′‐UTR of PIK3CD gene was amplified from genomic DNA and cloned into pGL‐3‐vector (Promega, San Luis, CA, USA) immediately downstream of the Renilla luciferase gene. Mutation in 3′‐UTR of PIK3CD gene was generated using the QuickChange Site‐Directed Mutagenesis kit (Stratagene, Santa Clara, CA, USA). The whole 3′‐UTR of PIK3CD gene and mutated one have been identified by sequencing, before massive PCR amplification. In the region of 1 × 105 cells per well were seeded into 24‐well plates for 24 h before transfection. Cells were co‐transfected with pGL‐3 firefly luciferase reporter (50 ng), pRL‐TK Renilla luciferase reporter (10 ng) and miR‐125b mimic or scramble mimic (50 nm) using Lipofectamine 2000 (Invitrogen). A luciferase reporter construct containing the miR‐125b consensus target sequence served as positive control (PC) and pRL‐TK vector served as internal control. Cell lysates were prepared using Passive Lysis Buffer (Promega) 48 h after transfection, and firefly and Renilla luciferase activities were measured using Dual‐Luciferase Reporter Assay (Promega). Results were normalized to Renilla luciferase and expressed as relative luciferase activity (Firely LUC/Renilla LUC). Experiments were independently repeated three times.
Statistical analysis
Data were expressed as mean ± SD of at least three independent experiments. Statistical analysis was carried out using spss 15.0 software (SPSS Inc., Chicago, IL, USA). Student's t‐test (two‐tailed) was performed to analyse the data. P‐values <0.05 were considered significant.
Results
Increased miR‐125b inhibited cell proliferation and cell cycle progression
One previous study which has reported reduction in miR‐125b expression in ES tissues and cells, lead us to explore biological significance of it in ES carcinogenesis. To explore relevance of miR‐125b and ES cell growth, miRNA mimic or scramble mimic were transfected into A673 cells. Intracellular level of miR‐125b was in the order of 160‐fold higher in A673 cells upon transfection with miR‐125b mimic, relative to cells transfected with scramble mimic (Fig. 1a). Then, cell proliferation was measured by using CCK‐8 assay. Ectopic expression of miR‐125b suppressed cell proliferation significantly (Fig. 1b). As proliferation is directly linked to cell cycle distribution, the effect of miR‐125b on cell cycle progression was analysed. As expected, percentage of S phase cells was reduced upon transfection with miR‐125b mimic, and subsequently, percentage of G1 phase cells was elevated, suggesting miR‐125b arrested more cells in G1 phase (Fig. 1c). Furthermore, treatment with miR‐125b mimic increased the percentage of early apoptotic cells as judged by PE annexin V staining (Fig. 1d). Taken together, these results indicated that miR‐125b efficiently inhibited cell proliferation and cell cycle progression, and induced apoptosis in vitro, which further validated the tumour suppressor role of miR‐125b in ES.
Figure 1.
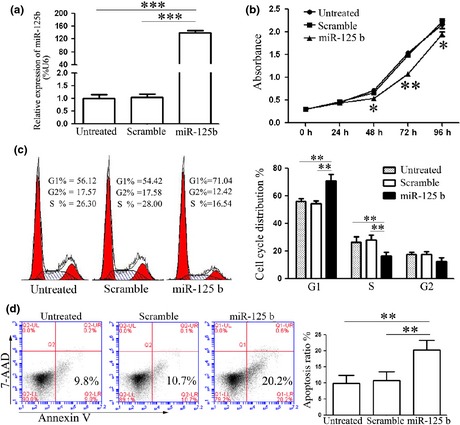
Effects of miR‐125b on Ewing's sarcoma cell proliferation, cell cycle progression and apoptosis. (a) Level of miR‐125b expression detected in A673 cells on transfection with miR‐125b mimics or scramble mimics, by quantitative RT‐PCR. Level of miR‐125b in cells transfected with miR‐125b mimics was up‐regulated. (b) Cell proliferation assay was performed after transfection with miR‐125b mimics or scramble mimics by using CCK‐8. Ectopic expression of miR‐125b suppressed cell proliferation. (c) Cell cycle analysis after transfection was performed by propidium iodide staining. MiR‐125b arrested cell cycle in G1 phase. (d) Apoptosis on transfection with miR‐125b mimics or scramble mimics was detected by PE annexin V staining. Cells undergoing early apoptosis only bound to PE annexin V (bottom right quadrant). MiR‐125b induced apoptosis. Results shown represent one of three experiments performed. Error bars indicate the mean ± SD of three independent experiments. **P < 0.01, ***P < 0.001.
Ectopic expression of miR‐125b inhibited cell migration and invasion
As miR‐125b has been reported to display a close relationship to metastasis in malignancy 8, 9, 10, we proposed that miR‐125b might play an extremely important role in ES migration and invasion. To test this hypothesis, cell migration and invasion assays were performed upon transfection. Re‐expression of miR‐125b significantly reduced cell migration of the A673 cells during closing of an artificial wound created over a confluent monolayer (Fig. 2a). Furthermore, invasive capacity of cells transfected with miR‐125b or scramble mimic was evaluated by Matrigel invasion chamber assays. MiR‐125b dramatically inhibited normally strong invasive capacity of A673 cells as indicated in the transwell invasion assay (Fig. 2b). These data demonstrate that miR‐125b efficiently impaired cell migratory and invasive capacity of ES cells in vitro.
Figure 2.
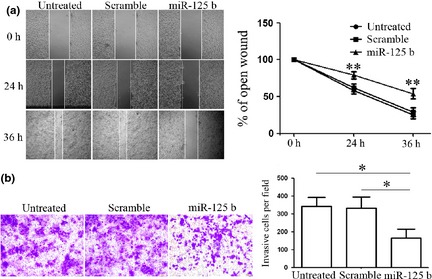
Effects of miR‐125b on Ewing's sarcoma cell migration and invasion. (a) Cell migration after transfection with miR‐125b mimics or scramble mimics was performed by wound‐healing assay and the relative ratio of wound closure per field is shown. Restored expression of miR‐125b reduced cell migration during closing of an artificial wound created over a confluent monolayer. (b) Cell invasion analysis of on transfection with miR‐125b mimics or scramble mimics was performed by transwell assay, and relative ratio of invasive cells per field was determined. MiR‐125b inhibited cell passing through the Matrigel. Results shown represent one of three experiments performed. Error bars indicate the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01.
miR‐125b targeted PIK3CD and suppressed the PI3K/AKT signalling pathway in A673 cells
To explore relative mechanisms involved in tumour progression of ES triggered by miR‐125b, putative target genes of miR‐125b were searched for using prediction programs, TargetScan, PicTar and miRanda. Among them, phosphoinositide‐3‐kinase catalytic subunit delta (PIK3CD) was selected as an ideal candidate as it was a subunit of phosphoinositide‐3‐kinase (PI3K), regulator of the PI3K/AKT signalling pathway, and has been reported to be constitutively activated in ES tissues 11.
Although PIK3CD has been reported to be a target of miR‐125b in cervical cancer cells 12, the interaction between miR‐125b and PIK3CD had not previously been experimentally validated in ES. 3′‐UTR of the PIK3CD gene was cloned into a luciferase reporter construct and mutant 3′UTR lacking the miR‐125b binding site was constructed as a negative control (Fig. 3a). MiR‐125b significantly suppressed luciferase activity of the reporter gene containing wild type 3′UTR, but did not affect luciferase activity of the reporter gene containing mutant 3′UTR (Fig. 3b). Consistent with the reporter assays, we observed strong reduction in mRNA and protein expression of PIK3CD on transfection with miR‐125b mimic (Fig. 3c,d), suggesting that miR‐125b regulates PIK3CD gene expression post‐transcriptionally.
Figure 3.
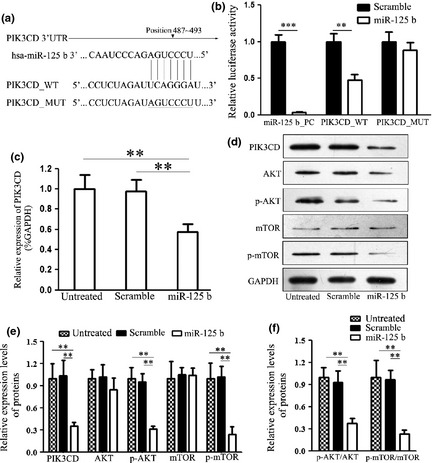
MiR‐125b targeted PIK3CD gene and suppresseed PI3K/AKT/ mTOR signalling pathway in Ewing's sarcoma cells. (a) Schematic representation of PIK3CD 3′UTR showing putative miR‐125b target site. Site‐directed mutagenesis was performed to mutate base pairs in the predicted seed region targeted by miR‐125b in the PIK3CD 3′UTR. Mutant binding site is underlined. (b) Relative luciferase activity of the indicated PIK3CD reporter construct, co‐transfected with miR‐125b mimics or scramble mimics is shown. MiR‐125b down‐regulated luciferase activity controlled by wild PIK3CD 3′UTR (WT), but did not affect luciferase activity controlled by mutant PIK3CD 3′UTR (MUT). Firefly luciferase activity was standardized to Renilla luciferase control. (c) Quantitative RT‐PCR assay was performed to detect expression of PIK3CD on transfection with miR‐125b mimics or scramble mimics. Ectopic expression of miR‐125b down‐regulated the mRNA level of PIK3CD. (d) Western blot analysis of expression of PIK3CD and its relative down‐steam target proteins in cells transfected with miR‐125b mimics or scramble mimics was performed. Ectopic expression of miR‐125b down‐regulated protein level of PIK3CD. All experiments were repeated three times. Error bars indicate the mean ± SD of three independent experiments. **P < 0.01, ***P < 0.001.
To further explore the mechanisms of miR‐125b‐mediated tumour suppression in ES cells, we detected expression of PIK3CD down‐stream genes, AKT, p‐AKT, mTOR and p‐mTOR. Ectopic expression of miR‐125b statistically suppressed expression of p‐AKT, p‐mTOR (Fig. 3d). There was no significant influence on expression of total mTOR. However, expression of total AKT was found to be suppressed at the same time. To discover effects on the AKT/mTOR signalling pathway, we statistically analysed ratios of p‐AKT/AKT and p‐mTOR/mTOR on transfection with miR‐125b. Although expression of total AKT was suppressed, ratio of p‐AKT/AKT significantly decreased; in particular, phosphorylation of AKT was notably suppressed. Moreover, ratio of p‐mTOR/mTOR was significantly down‐regulated with treatment of miR‐125b, suggesting that miR‐125b suppressed activation of the PI3K/AKT/mTOR signalling pathway. Collectively, these findings suggest miR‐125b inhibited expression of PIK3CD directly and suppressed the PI3K/AKT/mTOR signalling pathway.
Suppression of PIK3CD inhibited cell proliferation and cell cycle progression
Because of the oncogenic role of PIK3CD in different cancers, we speculated that suppression of PIK3CD might have an inhibitory effect on cell population growth. We down‐regulated expression of PIK3CD by RNA interference directed against the PIK3CD gene (Fig. 4a). In agreement with reduced expression of target proteins, reduced cell proliferation (Fig. 4b), arrested cell cycle progression (Fig. 4c), induced apoptosis (Fig. 4d), accompanied by impaired cell migration (Fig. 4e) and cell invasion (Fig. 4f), were also observed in A673 cells transfected with si‐PIK3CD. Considered together, these findings suggest that PIK3CD plays an important role in ES, as an oncoprotein. Suppressed cell proliferation and impaired invasion on miR‐125b overexpression were partially due to suppression of PIK3CD.
Figure 4.
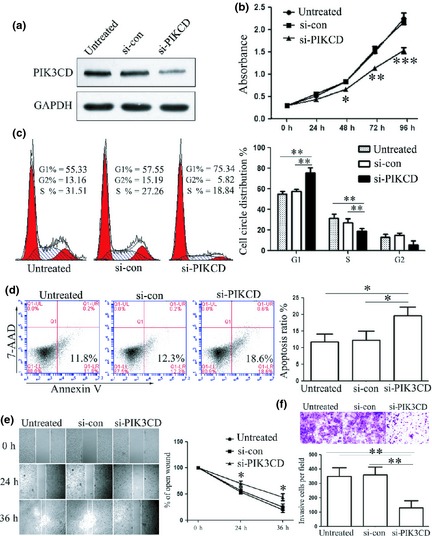
Effects of si‐ PIK 3 CD on the Ewing's sarcoma cell proliferation, cell cycle progression, apoptosis, cell migration and invasion. (a) Level of PIK3CD protein expression was detected on transfection with si‐PIK3CD or si‐control by western blot assay. Si‐PIK3CD suppressed expression of PIK3CD. (b) Cell proliferation assay was performed after transfection with si‐PIK3CD or si‐control by using CCK‐8. Suppression of PIK3CD inhibited proliferation. (c) Cell cycle analysis after transfection was performed by propidium iodide staining. Suppression of PIK3CD arrested cell cycle in G1 phase. (d) Apoptosis after transfection with si‐PIK3CD or si‐control was detected by PE annexin V staining. Si‐PIK3CD induced apoptosis. (e) Cell migration on suppression of PIK3CD was performed by wound‐healing assay and relative ratio of wound closure per field is shown. Down‐regulation of PIK3CD inhibited cell migration. (f) Cell invasion analysis was performed by transwell assay and relative ratio of invasive cells per field is shown. Si‐PIK3CD suppressed invasive capacity. All experiments were repeated three times. Error bars indicate the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
MiRNAs represent approximately 1% of the genome of different species, each of which has hundreds of different conserved or non‐conserved targets, making them key players in various cellular processes 13. Thus, it is extremely important to understand physiological and disease‐associated mechanisms of these small, single‐stranded RNAs. Recently, multiple dysregulated miRNAs have been reported to be closely related to development and progression of ES 7, 14, 15, such as ectopic expression of miR‐30a‐5p inhibiting cell proliferation and invasion of ES cells 15; Repression of miR‐22 promoted EWS/Fli1‐driven carcinogenesis by augmenting IGF pathway activity 7; moreover, restored expression of miR‐145 modulated chemotherapy responsiveness of ES 14. Among these miRNAs, miR‐125b has been reported in several publications to be underexpressed in ES, while the exact functions and relative mechanisms remain unclear. Here, we show that restored expression of miR‐125b in ES cell line A673 resulted in inhibition of cell proliferation, arrested cell cycle progression and increased apoptosis. On the other hand, metastasis is the major cause of mortality for ES patients. Thus, we then explored effects of miR‐125b on cell migration and invasion. Ectopic expression of miR‐125b inhibited migration in wound‐healing assays and suppressed cell invasion in transwell analysis. Our results are in accord with other studies, suggesting the tumour suppressor role of miR‐125b in cancers, and provides a solid foundation for ES utilization of miR125b in anticancer therapy in future.
To further investigate mechanisms of miR‐125b in ES, we predicted putative targets of miR‐125b, using prediction programs. Genes predicted by all three programs were picked out to be candidate targets. Among these genes, PIK3CD was selected out for its key regulation role in the PI3K/AKT signalling pathway. The target role of PIK3CD was identified by luciferase reporter assay, as transfection of miR‐125b caused substantial reduction of luciferase activity by luciferase expression constructs carrying the target PIK3CD fragment. As miRNAs have been reported to regulate gene expression both by translational attenuation and mRNA degradation, we performed both quantitative RT‐PCR and western blot assays to analyse effects of miR‐125b on expression of PIK3CD. We found that ectopic expression of miR‐125b reduced mRNA and protein levels of PIK3CD simultaneously, suggesting that miR‐125b could regulate expression of PIK3CD by targeting its 3′UTR directly. Recently, Anna et al. reported a new function for human miRNA, miR‐328. They found that miR‐328 can act as a decoy by binding to regulatory RNA‐binding protein and preventing it from blocking translation of mRNAs, suggesting a new miRNA regulation method, besides direct binding to their target genes 16. Thus, further research is warranted to elucidate whether miR‐125b‐mediated regulation of PIK3CD is through other indirect mechanisms. As we learn more concerning regulation mechanisms, we gain more opportunities to manipulate them in anti‐cancer therapy.
The PI3K/AKT/mTOR signalling pathway mediates important signalling in cell biological progression such as proliferation and inhibition of apoptosis 17, 18. Activation of PI3K/AKT/mTOR signalling has been shown to improved cell resistance to actinomycin D in ES 19, and inhibition of this signalling pathway in ES cells by PI3K specific inhibitor LY294002 enhanced the level of apoptosis 20. Given that PIK3CD is an important component of the PI3K/AKT/mTOR signalling pathway, frequently activated in human cancers 21 including ES 20, we investigated effects of miR‐125b on expression of PI3K/AKT/mTOR signalling pathway molecules. We found that overexpression of miR‐125b down‐regulated expression of PIK3CD, which in turn suppressed phosphorylation of downstream genes, AKT and mTOR. Interestingly, we found expression of total AKT was simultaneously reduced. Suppression of total AKT by miR‐125b may be mediated through down‐regulation of IL‐6R, which acts as a target gene of miR‐125b 22 upstream of AKT, at the same time 23. Overexpression of miR‐125b may down‐regulate expression of IL‐6R, which in turn mediates suppression of AKT. However, accurate mechanisms need more experimentation for such identification. Also, we analysed p‐AKT/AKT ratio on transfection with miR‐125b. Although expression of AKT was down‐regulated, p‐AKT/AKT ratio was reduced, suggesting that miR‐125b predominately down‐regulated expression of PIK3CD mediated‐suppression of downstream gene phosphorylation. Otherwise, we found no statistically significant difference between expressions of mTOR on transfection. Thus, we analysed ratios of p‐mTOR/mTOR and p‐AKT/AKT on transfection with miR‐125b. Although there was no difference between expression of mTOR, ratio of p‐AKT/AKT, accompanied by ratio of p‐mTOR/mTOR, were significantly reduced. Collectively, restored expression of miR‐125b suppressed activation of the PI3K/AKT/mTOR signalling pathway.
Function of PIK3CD was further identified in this study by observation that PIK3CD knockdown induced retardation of cell proliferation and apoptosis and suppressed cell migration as well as invasion, paralleling tumour suppressive effects induced by miR‐125b restoration. Taken together, down‐regulation of miR‐125b in ES may contribute to increasing expression of PIK3CD at the post‐transcriptional level, which activated the PI3K/AKT/mTOR signalling pathway and in turn facilitated ES biological progression.
In conclusion, our findings demonstrate that miR‐125b acted as a tumour suppressor through suppression of the PI3K/AKT/mTOR signalling pathway by targeting the PIK3CD gene. Re‐introduction of miR‐125b in ES cells down‐regulated PIK3CD, which dampened cell population growth, induced apoptosis and suppressed cell invasion. These results may help us understand the molecular mechanism of ES carcinogenesis, and provide us with a strong rationale to further investigate miR‐125b as a potential biomarker and therapeutic target for ES.
Acknowledgements
Funding: This work was supported by the National Natural Science Foundation of China (81171797) and Anhui Provincial Natural Science Foundation (1308085MH156).
References
- 1. Iwamoto Y (2007) Diagnosis and treatment of Ewing's sarcoma. Jpn. J. Clin. Oncol. 37, 79–89. [DOI] [PubMed] [Google Scholar]
- 2. Borinstein SC, Beeler N, Block JJ, Gorlick R, Grohar P, Jedlicka P et al (2013) A decade in banking Ewing sarcoma: a report from the Children's Oncology Group. Front. Oncol. 3, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- 4. Kong YW, Ferland‐McCollough D, Jackson TJ, Bushell M (2012) microRNAs in cancer management. Lancet Oncol. 13, e249–e258. [DOI] [PubMed] [Google Scholar]
- 5. Olsen PH, Ambros V (1999) The lin‐4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN‐14 protein synthesis after the initiation of translation. Dev. Biol. 216, 671–680. [DOI] [PubMed] [Google Scholar]
- 6. De Vito C, Riggi N, Suva ML, Janiszewska M, Horlbeck J, Baumer K et al (2011) Let‐7a is a direct EWS‐FLI‐1 target implicated in Ewing's sarcoma development. PLoS One 6, e23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McKinsey EL, Parrish JK, Irwin AE, Niemeyer BF, Kern HB, Birks DK et al (2011) A novel oncogenic mechanism in Ewing sarcoma involving IGF pathway targeting by EWS/Fli1‐regulated microRNAs. Oncogene 30, 4910–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Z, Gu X, Fang Y, Xiang J, Chen Z (2012) microRNA expression profiles in human colorectal cancers with brain metastases. Oncol. Lett. 3, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang F, Zhang R, He Y, Zou M, Guo L, Xi T (2012) microRNA‐125b induces metastasis by targeting STARD13 in MCF‐7 and MDA‐MB‐231 breast cancer cells. PLoS One 7, e35435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glud M, Rossing M, Hother C, Holst L, Hastrup N, Nielsen FC et al (2010) Downregulation of miR‐125b in metastatic cutaneous malignant melanoma. Melanoma Res. 20, 479–484. [DOI] [PubMed] [Google Scholar]
- 11. Machado I, Lopez‐Guerrero JA, Navarro S, Alberghini M, Scotlandi K, Picci P et al (2012) Epithelial cell adhesion molecules and epithelial mesenchymal transition (EMT) markers in Ewing's sarcoma family of tumors (ESFTs). Do they offer any prognostic significance? Virchows Arch. 461, 333–337. [DOI] [PubMed] [Google Scholar]
- 12. Cui F, Li X, Zhu X, Huang L, Huang Y, Mao C et al (2012) MiR‐125b inhibits tumor growth and promotes apoptosis of cervical cancer cells by targeting phosphoinositide 3‐kinase catalytic subunit delta. Cell. Physiol. Biochem. 30, 1310–1318. [DOI] [PubMed] [Google Scholar]
- 13. Leonardo TR, Schultheisz HL, Loring JF, Laurent LC. The functions of microRNAs in for review only pluripotency and reprogramming. Nat. Cell Biol. 14, 1114–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ban J, Jug G, Mestdagh P, Schwentner R, Kauer M, Aryee DN et al (2011) Hsa‐mir‐145 is the top EWS‐FLI1‐repressed microRNA involved in a positive feedback loop in Ewing's sarcoma. Oncogene 30, 2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Franzetti GA, Laud‐Duval K, Bellanger D, Stern MH, Sastre‐Garau X, Delattre O (2012) MiR‐30a‐5p connects EWS‐FLI1 and CD99, two major therapeutic targets in Ewing tumor. Oncogene 32, 3915–3921. [DOI] [PubMed] [Google Scholar]
- 16. Anna ME, Jason GH, Paolo N, Christopher G, Joshua JO, Riccardo S et al (2010) MiR‐328 functions as an RNA decoy to modulate hnRNP E2 Regulation of mRNA translation in leukemic blasts. Cell 140, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin‐Sanchez E, Rodriguez‐Pinilla SM, Sanchez‐Beato M, Lombardia L, Dominguez‐Gonzalez B, Romero D et al (2013) Simultaneous inhibition of pan‐phosphatidylinositol‐3‐kinases and MEK as a potential therapeutic strategy in peripheral T‐cell lymphomas. Haematologica 98, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hotfilder M, Sondermann P, Senss A, van Valen F, Jurgens H, Vormoor J (2005) PI3K/AKT is involved in mediating survival signals that rescue Ewing tumour cells from fibroblast growth factor 2‐induced cell death. Br. J. Cancer 92, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamamoto T, Ohno T, Wakahara K, Nagano A, Kawai G, Saitou M et al (2009) Simultaneous inhibition of mitogen‐activated protein kinase and phosphatidylinositol 3‐kinase pathways augment the sensitivity to actinomycin D in Ewing sarcoma. J. Cancer Res. Clin. Oncol. 135, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toretsky JA, Thakar M, Eskenazi AE, Frantz CN (1999) Phosphoinositide 3‐hydroxide kinase blockade enhances apoptosis in the Ewing's sarcoma family of tumors. Cancer Res. 59, 5745–5750. [PubMed] [Google Scholar]
- 21. Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA (2012) Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit. Rev. Oncog. 17, 69–95. [DOI] [PubMed] [Google Scholar]
- 22. Gong J, Zhang JP, Li B, Zeng C, You K, Chen MX et al (2013) MicroRNA‐125b promotes apoptosis by regulating the expression of Mcl‐1, Bcl‐w, and IL‐6R. Oncogene 32, 3071–3079. [DOI] [PubMed] [Google Scholar]
- 23. Chou CH, Lai SL, Chen CN, Lee PH, Peng FC, Kuo ML et al (2013) IL‐6 regulates Mcl‐1L expression through the JAK/PI3K/AKT/CREB signaling pathway in hepatocytes: implication of an anti‐apoptotic role during liver regeneration. PLoS One 8, e66268. [DOI] [PMC free article] [PubMed] [Google Scholar]