

Abstract
Abstract. Objective: Cell immortalization is considered to be a prerequisite status for carcinogenesis. Normal human ovarian surface epithelial (OSE) cells, which are thought to be the origin of most of human ovarian carcinomas, have a very limited lifespan in culture. Establishment of immortalized OSE cell lines has, in the past, required inactivation of pRb and p53 functions. However, this often leads to increased chromosome instability during prolonged culture. Materials and Methods: In this study, we have used a retroviral infection method to overexpress human telomerase reverse transcriptase (hTERT) gene, in primary normal OSE cells, under optimized culture conditions. Results: In vitro and in vivo analysis of hTERT‐immortalized cell lines confirmed their normal epithelial characteristics. Gene expression profiles and functional analysis of p16INK4A, p15INK4B, pRb and p53 confirmed the presence of their intact functions. Our study suggests that inactivation of pRb and p53 is not necessary for OSE immortalization. Furthermore, down‐regulation of p15INK4B in the immortalized cells may indicate a functional role for this protein in them. Conclusion: These immortal OSE cell lines are likely to be an important tool for studying human OSE biology and carcinogenesis.
INTRODUCTION
Telomeres are the repetitive DNA sequences situated at the ends of linear chromosomes. Human telomeres possess more than a thousand copies of the hexanucleotide (T2AG3) at the end of each chromosome. It is now appreciated that the inexorable loss of telomeric repeat DNA at each division imposes a limit proliferative potential of human cells in culture (Harley 1991; Wright & Shay 2000). Mammalian telomerase activity was first discovered in the archetypal immortalized human cell line, HeLa (Morin 1989). Cloning of the catalytic component of human telomerase, human telomerase reverse transcriptase (hTERT), heralded a new era in which it became possible to bypass the proliferative barrier by ecotopic expression of hTERT, and at least for some cell types, to generate stable lines of apparently normal cells (Bodnar et al. 1998; Vaziri & Benchimol 1998).
Immortalization is considered to be a prerequisite for carcinogenesis (Hahn 2002). However, there are multiple barriers to unlimited proliferation such as senescence and crisis (Duncan & Reddel 1997; Bond et al. 1999; Huschtscha & Reddel 1999). Forced expression of telomerase in pre‐crisis cells by transduction with an hTERT construct can allow crisis to be bypassed (Reddel 2000). However, in a wide variety of cell culture models of immortalization, activation of a telomere maintenance mechanism is preceded by loss of functional p53 and pRb pathways, which may create the permissive environment for telomerase activation (Sherr & Depinho 2000; Stewart & Weinberg 2002). In addition to p53, p16INK4A and pRb, some other genes are also involved in senescence and also in immortalization, such as p14ARF, MDM2, CDK4 and D‐cyclin (Hahn 2002). This suggests that senescence can be triggered through multiple pathways and immortalization achieved through cell type‐dependent pathways.
Ovarian cancer remains the most common cause of death from gynaecological cancers in the developed world. Ninety percent of ovarian cancers are carcinomas and are thought to arise from ovarian surface epithelium (OSE) (Wang & Auersperg 2002). Normal OSE cells have a very limited lifespan in culture that limits further studies of their role in carcinogenesis (Li et al. 2004). The first method for the culture of normal human OSE cells in vitro was established in 1984 (Auersperg et al. 1994); since then HPV‐E6E7 and SV40 large T‐immortalized OSE cell lines have been established (Tsao et al. 1995; Davies et al. 2003; Maeda et al. 2005). However, these immortal OSE lines display abrogated cell cycle control and apoptosis machinery, hence undergo spontaneous malignant transformation during prolonged culture (Zalvide et al. 1998; Hurlin et al. 1991; Gregorie et al. 2001; Drayton & Peters 2002). A recent report has indicated that the lifespan of normal OSE cells can be prolonged by overexpressing hTERT alone (Alvero et al. 2004). However, these cells were not characterized for their pRb and p53 functions. Based on the hypothesis that inactivation of pRb and p53 is the requirement for counteracting suboptimal culture conditions, we have previously optimized conditions for primary normal OSE cells (Li et al. 2004). In this study, we have used retroviral infection to overexpress hTERT cDNA in normal OSE cells under the optimal culture conditions. In this way, we successfully generated three immortal cell lines with normal epithelial characteristics and intact pRb and p53 function.
MATERIALS AND METHODS
Generating hTERT immortal OSE lines
Primary normal human OSE cells were cultured in MCDB105/199 (1 : 1)/15% foetal bovine serum/epidermal growth factor (10 ng/mL)/hydrocortisone (0.5 µg/mL)/insulin (5 µg/mL)/bovine pituitary extract (34 µg protein/mL). Early passage cells were infected with a replication‐defective retrovirus containing hTERT cDNA. Briefly, the pBabe‐hTERT‐puro plasmid (Morgenstern & Land 1990) was transiently transfected into the ecotopic retroviral packaging cell line, Phoenix E (Swift et al. 1999), using the calcium phosphate method. The supernatant containing packaged virus was then transferred to an amphotropic packaging cell line, AM12 (Markowitz et al. 1988). Viral supernatant from the AM12 cultures was then used to infect primary normal OSE cells NOSE21R (passage 4). Growth under puromycin (Sigma, St. Louis, MO, USA) selection 2.5 µg/mL continued for 14 days and antibiotic‐resistant clones were picked between 14 and 28 days. Six clones were chosen and were continuously cultured at 5% CO2 in a 37 °C incubator. The growth curves were monitored and telomerase activity was measured by telomeric repeat amplification protocol (TRAP) assay (see below). All cells were maintained in antibiotic‐free medium at no more than 80% confluence. Culture medium was refreshed twice a week. After continuous passaging, clones that proliferated for more than 30 plus population doublings were considered as immortal lines.
Telomerase activity assay
Telomerase activity was measured using the TRAPeze telomerase detection kit (Oncor Inc., Gaithersburg, MD, USA), following the manufacturer's protocol, for the radioactive assay (TRAP assay, telomere repeat amplification protocol). Lysate from a telomerase‐positive cell pellet provided by the kit was used as positive control.
Telomere length assay
Telomere length was determined using the Telo TAGGG telomere length assay kit (Roche Diagnostics Corporation, Indianapolis, IN, USA) following the manufacturer's instructions. Briefly, genomic DNA was isolated from OSE cells and was digested with HinfI and RsaI. After digestion, DNA fragments were separated by gel electrophoresis in a 1% agarose gel, then transferred to a nylon membrane, Southern blotting. Blotted DNA fragments were hybridized to a digoxigenin‐labelled telomere probe. The immobilized probe was visualized by chemiluminescence.
Immunofluoresce cytochemistry
The immortal lines were analysed with immunofluorescence staining using monoclonal antibodies for cytokeratins 7, 8, 14, 16, 18, 19 (a gift from Cancer Research UK monoclonal antibody department), CA125 (Clone OC125, NeoMarkers, Fremont, CA, USA) and E‐cadherin (Clone NCH‐38, NeoMarkers). Comparisons were made between parental normal OSE cells and immortalized OSE cells. The ovarian endometrioid adenocarcinoma cell line TOV112D (Manning et al. 1999; Hendrix et al. 2006) was used as a positive control for CA125 and E‐cadherin staining. Staining procedure followed the protocol described previously (Li et al. 2004).
Serum and growth factor‐dependent growth assay
Firstly, 1 × 105 cells of each immortalized OSE (IOSE) line were plated on a 10‐cm tissue culture dish for 1 day in growth medium. Cultures were then maintained for 14 days in serum‐free and growth factor‐free media and were refreshed twice weekly. Cell morphology changes were monitored and viable cells were counted using Vi‐cell XR cell viability analyser (Beckman Coulter, Miami, FL, USA). The assay was performed in triplicate for each IOSE line and Student's t‐tests were performed to determine statistical significance.
Anchorage‐dependent growth assay
In this assay, 1 × 105 cells were suspended in 2 mL complete medium with 0.3% agar (Nobel Agar, Invitrogen, Paisley, Scotland, UK) and 1 mg/mL bacto‐peptone (Becton Drive Franklin Lakes, NJ, USA), and then were plated on solidified 0.6% agar, in each well of a 6‐well plate. Six replicates were plated for each cell line. The ovarian endometrioid adenocarcinoma cell line TOV112D was plated at the same time as positive control. Cultures were then incubated at 37 °C, 5% CO2 for 28 days. After incubation, plates were stained with 1 mg/mL p‐iodotertazoliumviolet (Sigma) in absolute ethanol, and visible cell clones were counted.
Tumour formation in nude mice
Five million cells were suspended in 0.2 mL PBS and injected into 12 mice intraperitoneally (i.p.) and 3 mice subcutaneously (s.c.). Two i.p. mice were killed at each of the 3‐, 4‐, 5‐, 6‐ and 8‐week time points to monitor tumour formation and two i.p. mice were maintained for long‐term survival. Three s.c. mice were monitored for tumour formation.
Cytogenetic analysis
Molecular cytogenetic analyses were performed using both multiplex fluorescence in situ hybridization (M‐FISH) and chromosomal comparative genomic hybridization (CGH). M‐FISH analysis was performed following the manufacturer's protocol from SpectraVysion Assay (Vysis Inc., Downer Grove, IL, USA). In brief, metaphase chromosomes of three IOSE cell lines were prepared after 0.1 µg/mL colcemid treatment for 5 h. Chromosome spread slides were pre‐treated with RNase and pepsin before being denatured in 70% formamide/2 × Saline Sodium Citrate (SSC) and then were dehydrated in a series of 70%, 85% and 100% ethanol washes. Ten microlitres of SpectraVysion probe were denatured in a 73 °C water bath for 5 min and then were applied on the target area of the chromosome spread slide. Hybridization was performed at 37 °C in a humid incubator overnight. The slides were then washed in 0.4 × SSC/0.3%NP‐40 for 2 min at 72 °C and 2 × SSC/0.1%NP‐40 for 1 min at room temperature. Chromosomes were stained with 4’,6‐diamidino‐2‐phenylindole III. Image capture and analysis were performed using the SpectraVysion Imaging System. Chromosomal CGH analysis was carried out as previously described (Lu et al. 1997). In brief, 1 µg genomic DNA from parental NOSE21R cells and IOSE cells were differentially labelled with tetramethylrhodamine‐5‐dUTP and fluorescein‐12‐dUTP using the nick translation method. Successfully labelled DNA probes were then hybridized on normal metaphase slides made from peripheral blood lymphocytes of healthy individuals. Images were captured using an epifluorescent microscope equipped with a cooled CCD camera controlled by the Macprobe v4.3 CGH capture and analysis software. At least five good quality CGH metaphases were analysed and the average profile was used to detect chromosome copy number changes.
Electron microscopy
Cells were grown on Thermanox® coverslips (Agar Scientific, Stanstead, UK) until confluent and were fixed in 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 M phosphate buffer pH 7.4 (PB). Samples were post‐fix were embedded in Epon resin. Ultrathin sections were contrasted with uranyl acetate and lead citrate and were viewed with using a Jeol 1010 transmission electron microscope with a Gatan 2K × 2K CCD (Jeol Inc., Peabody, MA, USA).
TaqMan low‐density array real‐time RT‐PCR assays
Gene expression comparisons between parental normal OSE cells and immortal cell lines were performed using semiquantitative real‐time RT‐PCR with TaqMan low‐density array (Applied BioSystems, Foster City, CA, USA). In brief, RNA samples from parental normal OSE cells and the immortal cell lines were isolated from 80% confluence cultures using Qiagen RNeasy kit (Qiagen, West Sussex, UK). Two micrograms DNase‐treated RNA from each sample was used for reverse transcription into 100 µL cDNA. For each sample, 40 µL of synthesized cDNA were then mixed with 210 µL TaqMan Universal PCR Master Mix (PE Applied Biosystems, Branchburg, NJ, USA) and 170 µL PCR grade water to form the reaction mix. Four hundred microlitres of this reaction mix was loaded into a low‐density array card containing primers and probes of 96 genes in duplicate. These 96 test genes (Supplementary Table 1) include genes involved in DNA‐damage repair and cell cycle control, common oncogenes, tumour suppressor and metastasis associated genes. Human 18s and glyceraldehyde‐3‐phosphate dehydrogenase were used as internal controls. The real‐time RT‐PCR reaction and laser scanning was performed on an ABI 7900HT genotyper using SDS2.1 software. The ‘relative quantification study’ programme of SDS2.1 software was used for analysing data. Expression level of each gene was calculated as the average of the duplicates. Only genes with reproducible amplification curves of both duplicates were analysed and presented.
Western and in‐cell Western assays
Western blot analysis was used to validate RT‐PCR experiment data for pRb and p53. Polyclonal antihuman p15 antibody (sc‐612) was obtained from Autogen Bioclear UK Ltd. (Wiltshire, UK). Monoclonal antihuman p16 antibody (JS8) was from Abcam Ltd. (Cambridge, UK) and p53 antibody (PAb 1801) was from Merck Bioscience Ltd. (Nottingham, UK). Monoclonal antibody to β‐actin (Sigma) was used as total loading control. pRb and phospho‐pRb antibodies were from Cell Signalling Technology (Beverly, MA, USA), phospho‐p53 monoclonal antibody was from New England Biolabs (Beverly, MA, USA). The ultraviolet (UV)‐induced p53 up‐regulation was analysed using in‐cell Western Li‐Cor Bioscience (Li‐Cor Bioscience, Cambridge, UK) protocol. In brief, four replicates of parental cells and immortal cells were plated into 4‐well plates (NUNC Brand Product, Roskilde, Denmark) 1 day before UV exposure. UV exposure at 6mJ/cm2 dose was applied to cells and then they were then incubated at 37 °C, 5% CO2 incubation for 1, 2, 4 and 8 h before fixation in 3.7% formaldehyde. Monoclonal antihuman p53 antibody and polyclonal anti‐β‐actin antibody (Abcam Ltd.) were hybridized to the cells sequentially; red and green infrared fluorescence‐labelled secondary antibodies (supplied from Li‐Cor Bioscience) were further hybridized following the manufacturer's protocol; fluorescent signal intensities (red for p53 and green for β‐actin) were scanned and measured using an Odyssey Imager scanner. Data were further analysed using Microsoft Excel and SigmaPlot software. Student's t‐test was used for statistical analysis.
Human macrophage co‐culture assay
Human macrophages were derived from peripheral blood mononuclear cells by CD14‐positive selection (Miltenyibiotec, Surrey, UK) and were incubated in Teflon bags (Süd‐Laborbedarf, Gauting, Germany) with AMI V medium (Gibco, Paisley, UK) + 2% human AB serum, until differentiation into macrophages occurred, as assessed by morphological and functional criteria, as described previously (Hagemann et al. 2005); also co‐culture was performed as described previously (Hagemann et al. 2005). In brief, 2 × 105 macrophages/mL RPMI were seeded in transwell inserts (Nunc, Wiesbaden, Germany), the bottom of which consists of a membrane permeable to liquids but not to cells. The transwells were inserted into the upper well of a Boyden chamber. OSE cells were cultured on the bottom of the Boyden chamber as an adherent monolayer. After 48 h incubation, transwell inserts with macrophages were removed and OSE cells in the chamber were either used for direct immunofluorescence staining of p53 with 4’,6‐diamidino‐2‐phenylindole or for preparing whole cell lysates for further Western blot analysis. Ovarian cancer cell line IGROV‐3 was plated simultaneously as control.
RESULTS
hTERT‐immortalized OSE cells showed epithelial characteristics
We used retroviral infection to introduce hTERT cDNA into a primary culture of normal OSE cells and thus generated three immortal lines IOSE‐C21, IOSE‐C10, IOSE‐C9 from six hTERT overexpressing clones. We also performed mock infection of the primary normal OSE cells using empty retroviral vector. Clones that arose from the vector‐only infection underwent senescence after the first or second passage following subcloning. IOSE‐C21, C10 and C9 lines showed relatively slower population growth than the parental normal OSE cells in the first 30 days following subcloning. Thereafter, they grew stably with population‐doubling approximately every 56 h (Fig. 1a). By comparison, the parental normal OSE cells underwent senescence after eight passages. Figure 1a shows the growth curve of IOSE lines over the course of the first 180 days of culture following subcloning. To date, all three IOSE lines have been cultured for more than 190 population doublings with the stable growth rates as shown in Fig. 1a. These immortal lines also have shown serum and growth factor dependence at both early and late passages (Fig. 1b).
Figure 1.
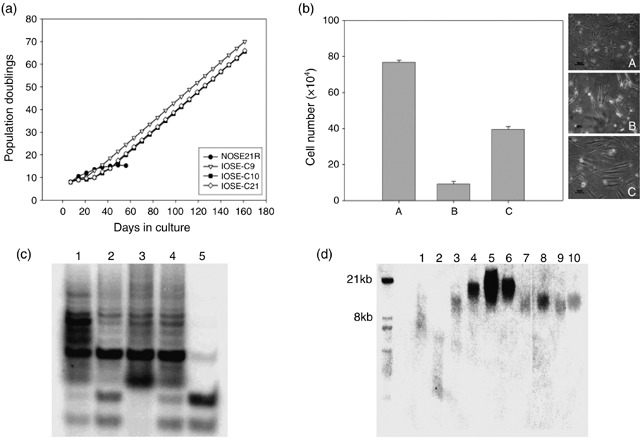
hTERT‐immortalized cell lines show prolonged lifespan with activated telomerase function. (a) Growth curve of hTERT‐immortalized OSE lines (IOSE‐C9, C10, C21) and parental normal OSE cells (NOSE21R) in 180 days of culture. IOSE‐C9, C10 and C21 cells show stable population growth rates 30 days after subcloning. (b) Serum dependence and growth factor dependence analysis. After culturing the hTERT immortal line IOSE‐C21 in complete medium (A), serum‐free medium (B) and growth factor‐free medium (C) for 14 days, cells underwent population growth arrest in serum‐free medium, and slowed their expansion in growth factor‐free medium, but with a change to elongated morphology. Compared to cultures in complete medium, there were significant decreases of live cell populations in both serum and growth factor withdrawn cultures (P < 0.001). (c) TRAP assay show positive telomerase activity in all three immortal lines. 1: Positive control; 2: IOSE‐C21; 3: IOSE‐C9; the missing control band indicates the existence of PCR inhibitor in reaction mix. 4: IOSE‐C10; 5: NOSE21R (parental cells). The lowest two bands of each lane present the PCR endogenous control. Lanes 1–4 show the telomere ladder with 6 base pair (T2AG3) difference between each band, which is a positive indication of telomerase activity. (d) Telomere length assays showed relatively longer telomere length in the three immortal cell lines compared to the parental cells. 1: Telomere ‘high‐level’ control (supplied by the commercial kit); 2: Telomere ‘low‐level’ control (supplied by the commercial kit); 3: NOSE21R (parental cells); 4: IOSE‐C21 cells at passage 9; 5: IOSE‐C21 cells at passage 14; 6: IOSE‐C21 cells at passage 18; 7: IOSE‐C10 cells at passage 4; 8: IOSE‐C10 cells at passage 5; 9: IOSE‐C9 cells at passage 4; 10: IOSE‐C9 cells at passage 5.
Telomeric repeat amplification protocol (TRAP) assays confirmed positive telomerase activity in all three immortalized cell lines (Fig. 1c). There was an increase in telomere lengths in IOSE cells at the early passage (within 30 population doublings; see Fig. 1d), and relatively longer telomere lengths in the IOSEs compared to the parental normal OSE cells. There was no continued increase in telomere length in IOSE cells with subsequent passaging.
Immortalized OSE cell lines maintained epithelial morphology. Transmission electron microscopy displayed a monolayer of epithelial cells with desmosomes at the sites of cell–cell contacts (Fig. 2). We observed that the epithelial morphology was highly dependent on the presence of serum and growth factors, and the cells exhibited an elongated fibroblast appearance when serum and growth factors were withdrawn from the growth medium. Cells of all three lines expressed cytokeratin 18 (Table 1) demonstrated by immunofluorescence cytochemistry assays but not keratins 7, 8 and 14.
Figure 2.
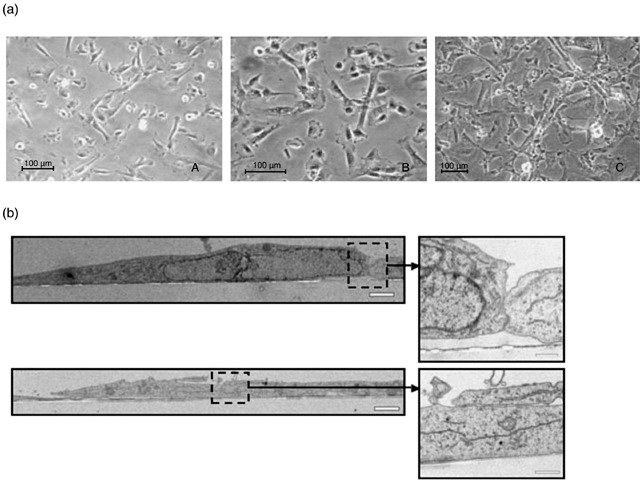
hTERT immortal lines maintain typical epithelial morphology. (a) Light microscopy illuminates that cells of all three immortal lines, IOSE‐C21 (A), IOSE‐C9 (B) and IOSE‐C10 (C), maintain typical epithelial morphology. (b) Transmission electron microscopy (TEM) revealed that the epithelial cells formed a monolayer with desmosome structures visible at sites of cell–cell contact. Inserts are the enlarged images of the areas highlighted by the dashed lines. Scale Bar = 2 µm and 0.5 µm (inserts).
Table 1.
Immunofluorescence staining and karyotyping of hTERT‐immortalized OSE lines
| IOSE‐C21 | IOSE‐C10 | IOSE‐C9 | ||||
|---|---|---|---|---|---|---|
| P9 | P18 | P4 | P18 | P5 | P18 | |
| Cytokeratin 7, 8 | – | – | – | – | – | – |
| Cytokeratin 14 | – | – | – | – | – | – |
| Cytokeratin 18 | + | + | + | + | + | + |
| Cytokeratin 19 | – | – | – | – | – | – |
| CA125 | – | – | – | – | – | – |
| E‐cadherin | – | – | – | – | – | – |
| Karyotyping | 46, xx | 46, xx | 46, xx | 46, xx | 46, xx | 46, xx, del(4)(q26‐q31.3) |
P, passage of cell lines (e.g. P9 means passage 9).
hTERT‐immortalized OSE cells showed no malignant characteristics
We analysed anchorage dependence of all three lines’ cells. None of them showed growth in soft agar at either early (passage 10, 26 cumulative population doublings) or late passage (passage 60, 174 cumulative population doublings). Immunofluorescence cytochemistry analysis of cells from all three lines showed no staining of malignant cell markers such as E‐cadherin and CA125. We did not detect any significant chromosomal abnormalities in IOSE‐C10 and IOSE‐C21 lines using either M‐FISH or metaphase chromosome CGH analysis in early or late passage cells (Table 1, Fig. 3a,b). For IOSE‐C9 cells at passage 18, we found one alteration – an interstitial deletion of chromosome 4q26–q31.3 using the analysis threshold 0.8–1.2 (Fig. 3c). We also performed in vivo tumourigenicity analysis of IOSE‐C21 cells. No tumour growth was observed after 12 months following intraperitoneal and subcutaneous injection of 5 × 106 cells into nude mice (data not shown).
Figure 3.
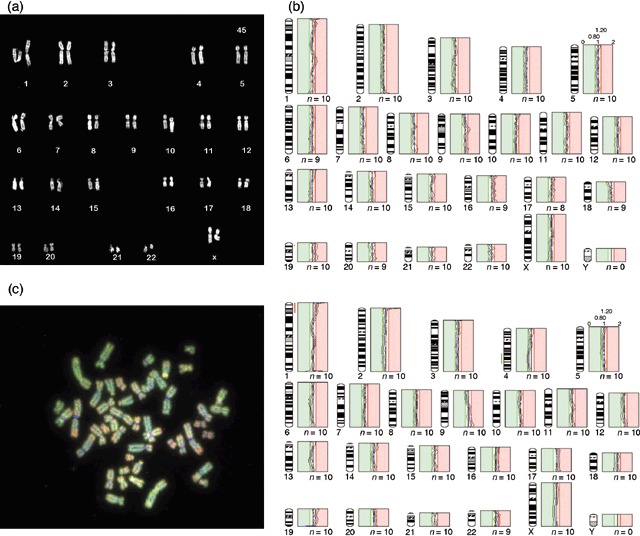
M‐FISH karyotyping and CGH analysis of IOSE cell lines. (a) Representative karyotype of the IOSE‐C21 cell line. (b) Average CGH profile of IOSE‐C10 (passage 18) showed no significant chromosomal alteration at threshold 0.8–1.2 (green: parental NOSE21R cells; red: IOSE cell lines). (c) Representative metaphase CGH hybridization image of IOSE‐C9 passage 18 (left) and average CGH profile of this sample showing an interstitial deletion of chromosome 4. The gain of material at chromosome 1p is a consistent artefact of CGH analysis, and it is not considered as significant alteration.
hTERT‐immortalized OSE cells show increased CSFR1 mRNA, decreased CDH1 and p15INK4B mRNA expression
We performed semiquantitative real‐time RT‐PCR analysis to compare mRNA expression of 94 genes (supplementary Table 1) associated with DNA damage repair, cell cycle control, metastasis associated extracellular matrix formation, known oncogenes and tumour suppressor genes. Gene expression in the hTERT‐immortalized lines (IOSE‐C9, IOSE‐C10 and IOSE‐C21) was compared to the parental OSE cells (NOSE21R). Among the 95 genes investigated, the expression of colony stimulating factor receptor 1 gene (CSFR1) was increased 1000–10 000 fold in all three immortalized cell lines. There were small increases in expression (2–20‐fold) for BRCA1, MLH1, RAD51, RAD54, EGFR, LMNB1, MMP1, 3 and 7, ITGA6 (integrin α6) and ITGAX (integrin αx) (Fig. 4a). We also observed significant decreases in expression for CDH1 (E‐cadherin), VCAM1 and KISS1 genes, but small decreases for CDKN2A, MUC1, CTSF, GADD45A and ITGB7 (integrin β7), again in all three immortalized lines (Fig. 4b). The remainder of the genes examined did not show significant changes in any of the three lines. Interestingly, there was a very low level of mRNA expression for the p15INK4B (CDKN2B) gene in IOSE‐C21 and IOSE‐C10 cells, but hardly any changes in p16INK4A (CDKN2A) gene expression in any of the three immortalized lines (Fig. 4c). Western blot analysis of parental (NOSE21R) and immortalized (IOSE‐C21) cells showed that p15INK4B expression was much lower in immortalized cells grown at no more than 80% confluence. When confluence reached more than 90%, IOSE‐C21 cells showed contact inhibition and an increase in p15INK4B expression. When serum was withdrawn from the culture medium, IOSE‐C21 cells underwent population growth arrest accompanied by a dramatic increase of p15INK4B protein expression. However, p16INK4A protein levels remained unchanged with a very weak signal (Fig. 4d).
Figure 4.
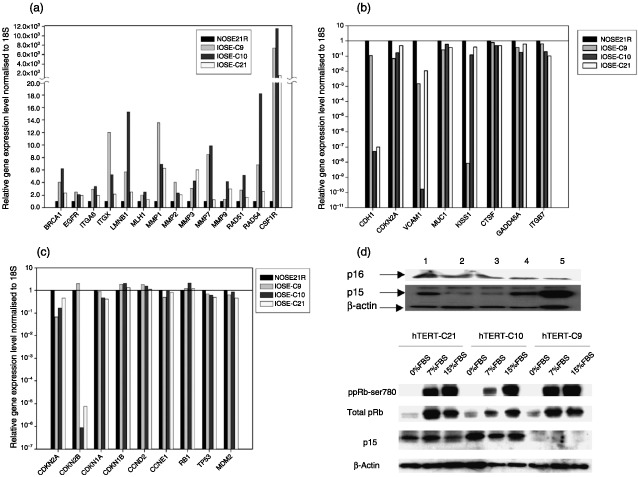
Semiquantitative real‐time RT‐PCR analysis of 94 cancer related genes. (a) Genes with relative expression levels increased in all three IOSE lines compared to parental NOSE21R cells. (b) Genes with relative expression levels decreased in all three IOSE lines compared to parental NOSE21R cells. The expression levels of genes in parental NOSE21R cells were calibrated to 1 (1.00E+00). (c) Relative expression levels of Rb, TP53 and cell cycle control pathway‐associated genes. Expression levels of genes in parental NOSE21R cells were calibrated to 1 (1.00E+00). (d) Western blot analysis confirmed the low protein expression levels of p15 in IOSE‐C21 line when cultured at low density but significant increases in expression in confluent culture; however, p16 protein levels remained undetectable. 1: NOSE21R parental cells; 2: IOSE‐C21 passage 9 at 70% confluence; 3: IOSE‐C21 passage 14 at 80% confluence; 4: IOSE‐C21 passage 18 at 95% confluence; 5: IOSE‐C21 cells in 5% serum medium. (e) When serum was deprived, all three IOSE lines showed a decreased ratio of phospho‐Rb to total Rb protein and two lines (IOSE‐C21, IOSE‐C10) showed increased p15 expression.
pRb G1/S cell cycle checkpoint remains intact in hTERT immortal lines
We analysed mRNA expression levels of other genes involved in the pRb G1/S cell cycle checkpoint pathway, in addition to p16INK4A and p15INK4B (Fig. 4c). We found no significant changes in mRNA expressions for CDKN1A (p21Cip1), CDKN1B (p27Kip1), Cyclin D2, Cyclin E and Rb1. In order to confirm that the function of pRb was intact, we decreased the serum levels in culture medium from 15% to zero. As a consequence, all three immortal lines showed a decrease in the ratio of phosphorylated to total pRb protein (Fig. 4e), leading to G1/S cell cycle arrest that was confirmed by 5’‐bromo‐2’‐deoxyuridine fluorescence‐activated cell sorter analysis (data not shown). IOSE‐C21 and IOSE‐C10 lines showed increasing p15INK4B protein levels during serum deprivation (Fig. 4e), but p16INK4A protein was almost undetectable. IOSE‐C9 cells had undetectable p16INK4A and p15INK4B protein expressions despite relatively abundant mRNA expression levels (described above). It is possible that there is an interruption in protein translation or decreased protein stability for these molecules in IOSE‐C9 cells.
p53 function is maintained in immortalized cells
The mRNA expression levels for TP53 and MDM2 are similar between all IOSE cell lines and the parental cells (Fig. 4c). We further examined p53 activity in immortalized cells following exposure to UV radiation. In‐cell Western assays showed a significant increase in p53 protein levels 8 h after exposure (P < 0.01) (Fig. 5a). A time course analysis at 4, 8, 24 and 36 h after exposure showed the same p53 up‐regulation after 8 h with levels remaining high up to the 36 h time point (data not shown).
Figure 5.
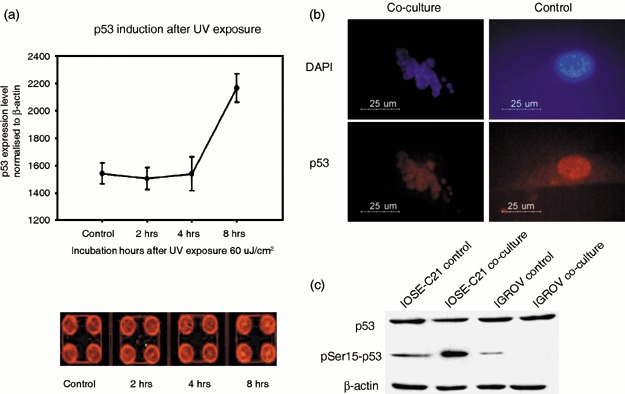
hTERT immortal lines maintain intact p53 function. (a) In‐cell Western assay indicates that after exposure of IOSE‐C21 (passage 18) cells to ultraviolet (UV) damage, p53 protein levels increased significantly 8 h after UV exposure (P < 0.01) as indicated by the mean intensity of p53 signal normalized to β‐actin signal (p53: red fluorescence staining; actin: green fluorescent staining) in four replicates. (b) IOSE‐C21 cells underwent apoptosis with highly expressed p53 in the nucleus when co‐cultured with human macrophages. (c) Phospho‐p53 is up‐regulated in IOSE‐C21 cells but not in the cancer line IGROV‐3 cells when co‐cultured with human macrophages.
Nitric oxide released by macrophages can activate non‐mutated p53 by increasing its serine15 phosphorylation level and engaging the G2/M checkpoint (Hofseth et al. 2003). Thus, we co‐cultured IOSE‐C21 cells with macrophages as previously described (Hagemann et al. 2005). After 48 h of co‐culture, IOSE‐C21 cells showed positive p53 nuclear staining and up‐regulation of serine15 phosphorylated p53 by Western blotting. In contrast, the cancer line IGROV‐3 (which expresses mutated p53) showed depletion of serine15 phosphorylated p53 (Fig. 5b,c). These results were reproducible in the other two immortalized lines. We were unable to perform the same experiments on primary normal OSE cells due to the limited cell numbers available from a primary cell culture.
DISCUSSION
Having immortalized normal human somatic cells greatly facilitates studies of carcinogenesis in vitro. It has been proposed that telomerase activation is the only step required for immortalization of somatic cells (Jiang et al. 1999; Morales et al. 1999) and that hTERT is sufficient for immortalization when culture conditions are optimal (Herber et al. 2002). Here, in this study, we successfully established hTERT‐immortalized human normal ovarian surface epithelial cell lines with intact pRb and p53 functions under optimum culture conditions. This has confirmed that fully requisite culture conditions are an essential criterion for hTERT immortalization, and that intact pRb and p53 functions indicate that although immortalized, these cell lines maintain relatively normal characteristics during long‐term culture. Thus, compared to previously established immortal cell lines in which pRb and p53 functions have been disrupted by SV40 or E6/E7, the hTERT‐immortalized cells we generated are likely to be a more representative model of ‘normal’ ovarian surface epithelial cells.
Previous studies have also suggested that immortalization of human epithelial cells is highly dependent on both Rb/p16INK4A inactivation and telomerase activity (Kiyono et al. 1998). In this study, we could not detect altered expression of p16INK4A at mRNA or protein levels, while we observed differential expression of p15INK4B during G1/M cell cycle arrest in two of the IOSE lines. Similar to p16INK4A, p15INK4B is one of the INK4A gene products, and it prevents CDK4/6 forming a complex with CyclinD to abrogate cyclinD‐induced pRb phosphorylation and engage G1/M cell cycle arrest. Our data suggest that p15INK4B may play a role similar to that of p16INK4A in the absence of p16INK4A function in IOSE‐C10 and C21 cell lines. Studies on malignant cell lines have shown that p15INK4B and p16INK4A have a similar ability to inhibit cell proliferation, trigger senescence and inhibit telomerase activity, but only when functional pRb protein is present (Fuxe et al. 2000; Latres et al. 2000). We observed the efficient switch of phosphorylation status of pRB in response to serum starvation, which confirmed the intact function of this essential cell cycle control molecule in all three immortal lines. IOSE‐C9 line also showed pRb hypo‐phosphorylation under serum starvation conditions despite the absence of both p15INK4B and p16INK4A proteins, which may suggest an involvement of further molecules in the signal transduction of pRb to facilitate the cell cycle control response.
Since the discovery of hTERT by Meyerson (Meyerson et al. 1997) and Nakamura (Nakamura et al. 1997), studies have revealed that several proto‐oncogenes and tumour suppressor genes, such as c‐myc, Bcl‐2, p21WAF1, Rb, TP53, PKC, AKT/PKB are involved in hTERT regulation (Liu 1999; Garcia‐Cao et al. 2002); the SIP1/TGF‐β pathway and Menin protein (potentially through the JunD or nuclear factor kappa B pathway) repress hTERT (Lin & Elledge 2003); c‐myc directly binds to the hTERT promoter to activate its transcription, while its counterpart, Mad1, represses hTERT by repressing the transcription activity of c‐myc (Wang et al. 1998; Xu et al. 2001). In our study, we found that hTERT immortalization of OSE cells did not significantly change mRNA transcription of c‐myc, p21WAF1, Rb and TP53. However, we did see low‐level increases in mRNA transcription of BRCA1, MLH1, RAD51, RAD54. This could be due to the direct association of hTERT with these molecules that has been described before (Li et al. 2002; Wei et al. 2002). We observed a significant overexpression of CSF1R in immortal OSE cells, which may suggest a potential association between CSF1R and hTERT immortalization. Binding of the CSF1 ligand to CSF1R triggers multiple intracellular pathways that are associated with cell survival, proliferation and differentiation (Sherr et al. 1988). Constitutive activation of CSF1R leads to the loss of cell membrane E‐cadherin, disruption of epithelial cell–cell adhesion and an increase of cell motility (Wrobel et al. 2004). The inverse expression pattern of CSF1R and CDH1 that we observed in the immortal cells not only mirrors this finding, but may also imply an alteration of cell–cell adhesion and mobility in the immortal cells. Meanwhile, increased transcription of a series of genes that are associated with extracelluar matrix modification and cell mobility, such as MMPs, integrins and Lamin B, may suggest an increased potential for migration and invasion of the immortal cells, which is consistent with the role of immortalization as a prerequisite for carcinogenesis. However, these findings will need further and more detailed investigation to confirm them.
In conclusion, we have successfully immortalized human normal OSE cells by causing retroviral overexpression of the hTERT gene, alone. All three immortal lines derived retain normal epithelial cell characteristics and intact functions of pRb and p53. These hTERT‐immortalized human OSE cell lines will provide a useful resource, not only for mechanistic studies of hTERT immortalization, but also for studies of the cell biology of human OSE.
Supporting information
Table S1. Gene list of TaqMan real‐time RT‐PCR low‐density array
Supporting info item
ACKNOWLEDGEMENTS
This project was funded by Cancer Research UK and a project grant from the Eve Appeal. We would like to thank Jon Strefford for his help with M‐FISH analysis, and the staff of the Cancer Research UK FACS Laboratory. Dr. Simon Gayther is a Higher Education Funding Council for England funded Senior Lecturer.
The first two authors (N. F. Li, S. Broad) contributed equally to this paper.
REFERENCES
- Alvero AB, Fishman DA, Qumsiyeh MB, Garg M (2004) Telomerase prolongs the lifespan of normal human ovarian surface epithelial cells without inducing neoplastic phenotype. J. Soc. Gynecol. Investig. 11, 553–561. [DOI] [PubMed] [Google Scholar]
- Auersperg N, Maines‐Bandiera SL, Dyck HG, Kruk PA (1994) Characterization of cultured human ovarian surface epithelial cells: phenotypic plasticity and premalignant changes. Lab. Invest. 71, 510–518. [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB (1998) Extension of life‐span by introduction of telomerase into normal human cells. Science 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Bond JA, Haughton MF, Rowson JM, Smith PJ, Gire V, Wynford‐Thomas D (1999) Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol. Cell. Biol. 19, 3103–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BR, Steele IA, Edmondson RJ, Zwolinski SA, Saretzki G, Von Zglinicki T (2003) Immortalisation of human ovarian surface epithelium with telomerase and temperature‐sensitive SV40 large T antigen. Exp. Cell Res. 288, 390–402. [DOI] [PubMed] [Google Scholar]
- Drayton S, Peters G (2002) Immortalisation and transformation revisited. Curr. Opin. Genet. Dev. 12, 98–104. [DOI] [PubMed] [Google Scholar]
- Duncan EL, Reddel RR (1997) Genetic changes associated with immortalisation. A review. Biochemistry 62, 1263–1274. [PubMed] [Google Scholar]
- Fuxe J, Akusjarvi G, Goike HM, Roos G, Collins VP, Pettersson RF (2000) Adenovirus‐mediated overexpression of p15INK4B inhibits human glioma cell growth, induces replicative senescence, and inhibits telomerase activity similarly to p16INK4A. Cell. Growth. Differ. 11, 373–384. [PubMed] [Google Scholar]
- Garcia‐Cao M, Gonzalo S, Dean D, Blasco MA (2002) A role for the R6 family of proteins in controlling telomere length. Nat. Genet. 32, 415–419. [DOI] [PubMed] [Google Scholar]
- Gregorie L, Rabah R, Schmelz EM, Munkarah A, Roberts PC, Lancaster W (2001) Spontaneous malignant transformation of human ovarian surface epithelial cells in vitro . Clin. Cancer Res. 7, 4280–4287. [PubMed] [Google Scholar]
- Hagemann T, Wilson J, Kulbe H, Charles K, Li NF, Klemm F, Trümper L, Binder C, Balkwill FR (2005) Co‐culture of Macrophages with tumour cells cause TNF‐α dependent activation of c‐Jun, NF‐kappaB in the malignant cells. J. Immunol. 175, 1197–1205. [DOI] [PubMed] [Google Scholar]
- Hahn WC (2002) Immortalisation and transformation of human cells. Mol. Cells 13, 351–361. [PubMed] [Google Scholar]
- Harley CB (1991) Telomere loss: mitotic clock or genetic time bomb? Mutat. Res. 256, 271–282. [DOI] [PubMed] [Google Scholar]
- Hendrix ND, Wu R, Kuick R, Schwartz DR, Fearon ER, Cho KR (2006) Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer Res. 66, 1354–1362. [DOI] [PubMed] [Google Scholar]
- Herbert BS, Wright WE, Shay JW (2002) p16INK4A inactivation is not required to immortalise human mammary epithelial cells. Oncogene 21, 7897–7900. [DOI] [PubMed] [Google Scholar]
- Hofseth LJ, Saito SS, Hussain SP, Espey MG, Miranda KM, Araki Y, Jhappan C, Higashimoto Y, He P, Linke SP, Quezado MM, Zurer I, Harris CC (2003) Nitric oxide‐induced cellular stress and p53 activation in chronic inflammation. Proc. Natl Acad. Sci. USA 100, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, Kaur P, Smith PP, Perez‐Reyes N, Blanton RA, McDougall JK (1991) Progression of human papillomavirus type 18‐immortalized human keratinocytes to a malignant phenotype. Proc. Natl Acad. Sci. USA 88, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huschtscha LI, Reddel RR (1999) p16INK4A and the control of cellular proliferative life span. Carcinogenesis 20, 921–926. [DOI] [PubMed] [Google Scholar]
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tisty TD, Chiu CP (1999) Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet. 21, 111–114. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ (1998) Both Rb/p16INK4A inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84–88. [DOI] [PubMed] [Google Scholar]
- Latres E, Mulumbres M, Sotillo R, Martin J, Ortega S, Martin‐Caballero J, Flores JM, Cordon‐Cardo C, Barbacid M (2000) Adenovirus‐mediated overexpression of p15INK4A. EMBO. J. 19, 3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lee TH, Averaham H (2002) A novel tricomplex of BRCA1, Nmi, and c‐Myc inhibits c‐Mye‐induced human telomerase reverse transcriptase gene (hTERT) promote activity in breast cancer. J. Biol. Chem. 277, 20965–20973. [DOI] [PubMed] [Google Scholar]
- Li NF, Willbank G, Balkwill F, Jacobs IJ, Dafou D, Gayther SA (2004) A modified medium that significantly improves the growth of human normal ovarian surface epithelial (OSE) cells in vitro . Lab. Invest. 84, 923–931. [DOI] [PubMed] [Google Scholar]
- Lin SY, Elledge SJ (2003) Multiple tumor suppressor pathways negatively regulate telomerase. Cell 113, 881–889. [DOI] [PubMed] [Google Scholar]
- Liu JP (1999) Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J. 13, 2091–2104. [DOI] [PubMed] [Google Scholar]
- Lu Y‐J, Birdsall S, Osin P, Gusterson B, Shipley J (1997) Phyllodes tumors of the breast analyzed by comparative genomic hybridization and association of increased 1q copy number with stromal overgrowth and recurrence. Genes Chromosomes Cancer 20, 275–281. [PubMed] [Google Scholar]
- Maeda T, Tashiro H, Katabuchi H, Begum M, Ohtake H, Kiyono T, Okamura H (2005) Establishment of an immortalised human ovarian surface epithelial cell lines without chromosome instability. Br. J. Cancer 93, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AP, Mes‐Masson AM, Seymour RJ, Tetrault M, Provencher DM, Tonin PN (1999) Expression of FHIT in primary cultures of human epithelial ovarian tumors and malignant ovarian ascites. Mol. Carcinog. 24, 218–225. [DOI] [PubMed] [Google Scholar]
- Markowitz D, Goff S, Bank A (1988) Construction of a safe and efficient line. Virology 167, 400–406. [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA (1997) hEST2, the putative human telomerase catalytic subunit gene, is up‐regulated in tumor cells and during immortalization. Cell 90, 785–795. [DOI] [PubMed] [Google Scholar]
- Morales CP, Holt SE, Quellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW (1999) Absence of cancer‐associate changes in human fibroblasts immortalised with telomerase. Nat. Genet. 21, 115–118. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper‐free packaging cell line. Nucleic. Acids Res. 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin GB (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521–529. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Depinho RA (2000) Cellular senescence: mitotic clock or culture shock? Cell 102, 407–410. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roussel MF, Rettenmier CW (1988) Colony‐stimulating factor‐1 receptor (c‐fms). J. Cell. Biochem. 38, 179–187. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Weinberg RA (2002) Senescence: does it all happen at the ends? Oncogene 21, 627–630. [DOI] [PubMed] [Google Scholar]
- Swift SE, Lorens JB, Achacoso P, Nolan GP (1999) Rapid production of retroviruses for efficient gene delivery to mammalian cells In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, eds. Current Protocols in Immunology, Vol. 10.17C, p. 1–17. New York: Wiley. [DOI] [PubMed] [Google Scholar]
- Tsao SW, Mok SC, Fey E, Fletcher JA, Wan TK, Chew EC (1995) Characterization of human ovarian surface epithelial cells immortalized by human papilloma virall oncogenes (HPV‐E6E7 ORFs). Exp. Cell Res. 218, 499–507. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Benchimol S (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life‐span. Curr. Biol. 8, 279–282. [DOI] [PubMed] [Google Scholar]
- Wang AS, Auersperg N (2002) Normal ovarian surface epithelium. Cancer Treat. Res. 107, 161–183. [DOI] [PubMed] [Google Scholar]
- Wang J, Xie LY, Allan S, Beach D, Hannon GJ (1998) Myc activates telomerase. Genes Dev. 12, 1769–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Skopp R, Takata S, Price CM (2002) Effects of double‐strand break repair proteins on vertebrate telomere structure. Nucleic. Acids Res. 30, 2862–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright W, Shay JW (2000) Telomere dynamics in in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat. Med. 6, 849–851. [DOI] [PubMed] [Google Scholar]
- Wrobel CN, Debnath J, Lin E, Beausoleil S, Roussel MF, Brugge JS (2004) Autocrine CSF‐1R activation promotes Src‐dependent disruption of mammary epithelial architecture. J. Cell Biol. 165, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Popov N, Hou M, Wang Q, Bjorkholm M, Gruber A, Menkel AR, Henriksson M (2001) Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentation of HL60 cells. Proc. Natl. Acad. Sci. USA 98, 3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalvide J, Stubdal H, Decaprio JA (1998) The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol. Cell. Biol. 18, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene list of TaqMan real‐time RT‐PCR low‐density array
Supporting info item