

Abstract
Objectives: The aim of this study was to investigate mechanisms involved in the growth inhibitory effect of silymarin, in humanhepatocellular carcinoma.
Materials and Methods: The human hepatocellular carcinoma cell line HepG2 was utilized and the MTT assay was performed to study the antiproliferative effect of silymarin. Dual staining was undertaken for ethidium bromide/acridine orange, propidium iodide staining and DNA fragmentation studies were executed to confirm the presence of apoptosis. Cell‐cycle analysis was revealed by flow cytometry and mitochondrial transmembrane potential was measured by uptake of the mitochondrial‐specific lipophilic cationic dye rhodamine 123. Western blotting analysis for cytochrome c, p53, Bax, Bcl‐2, APAF‐1, caspase‐3, survivin, β‐catenin, cyclin D1, c‐Myc and PCNA was carried out.
Results: Silymarin inhibited population growth of the hepatocellular carcinoma cells in a dose‐dependent manner, and the percentage of apoptotic cells was increased after treatment with 50 and 75 µg/ml silymarin for 24 h. Silymarin treatment increased the proportion of cells with reduced DNA content (sub‐G0/G1 or A0 peak), indicative of apoptosis with loss of cells in the G1 phase. Silymarin also decreased mitochondrial transmembrane potential of the cells, thereby increasing levels of cytosolic cytochrome c while up‐regulating expression of pro‐apoptotic proteins (such as p53, Bax, APAF‐1 and caspase‐3) with concomitant decrease in anti‐apoptotic proteins (Bcl‐2 and survivin) and proliferation‐associated proteins (β‐catenin, cyclin D1, c‐Myc and PCNA).
Conclusions: Our results demonstrate that silymarin treatment inhibited proliferation and induced apoptosis in the human hepatocellular carcinoma cell line HepG2.
Introduction
Hepatocellular carcinoma is the fifth most common cancer worldwide and the third most common cause of cancer mortality (1). Although hepatocellular carcinoma is significantly more prevalent in Asia and Africa, rising incidence has been reported in Western countries (2). Most patients with hepatocellular carcinoma present at an advanced stage when successful surgical treatment is no longer feasible, and current therapeutic options achieve clinical responses in only a small percentage of people. As a consequence, effective approaches for prevention and treatment of hepatocellular carcinoma need to be established. Extract from the seeds of milk thistle (Silybum marianum) is a widely used traditional herbal/dietary supplement around the world (3). Chemically, the active constituent of this extract is a flavolignan, silymarin, which in itself represents the mixture of four isomeric flavonoids: silibinin, isosilibinin, silydianin and silychristin. Recently, using a combination of liquid chromatography and electrospray ionization mass spectrometry, six main active constituents of silymarin have been separated, which include silydianin, silychristin, silibinin A, silibinin B, isosilibinin A and isosilibinin B (4). Silibinin is the major component (70–80%) found in silymarin and is thought to be the most biologically active. Experimental evidence from several in vitro and in vivo cancer models suggests that there is no significant difference between silymarin and silibinin in terms of chemopreventive or biological activity (5, 6). Pharmacological studies have revealed that silymarin is nontoxic even at relatively high physiological doses, which suggests that it is safe to use for treatment of various diseases (7). Some studies have shown that silymarin is a strong antioxidant and hypolipidaemic agent with a potent anticarcinogenic effect (8, 9), and also have the ability to inhibit malondialdehyde–DNA adduct formation (10).
Although silymarin has been used for centuries and its anticancer effects on various malignancies has been studied, the mechanism by which it exerts its activity in hepatocellular carcinoma is not known. This notion of ours is in line with a recent review by Kaur & Agarwal (11), which says that there is a dearth of evidence concerning silymarin against liver cancer and more efforts are needed in this direction in the near future. Earlier studies of silibinin on hepatocellular carcinoma have shown that it exerts strong anticancer activity (12, 13); these studies concentrated mainly on cell‐cycle control‐associated proteins and on the phosphatase and tensin homologue deleted on chromosome 10 (PTEN) pathway. In the present study, we have evaluated the mechanism of action by which silymarin inhibiting proliferation and induces apoptosis in the human hepatocellular carcinoma cell line HepG2.
Materials and methods
Chemicals
Dulbecco's modified Eagle's medium (DMEM), 0.25% trypsin‐EDTA solution, sodium bicarbonate solution, bovine serum albumin (BSA), 3‐[4,5‐dimethythiazol‐2‐yl]2,5‐diphenyl tetrazolium bromide (MTT), propidium iodide, ethidium bromide, acridine orange, rhodamine 123, agarose, β‐actin antibody and silymarin were purchased from Sigma Chemicals Co. (St. Louis, MO, USA). Fetal bovine serum (FBS) and antibiotic/antimycotic solution were from Gibco (Gibco, USA). Sodium phosphate (monobasic and dibasic), sodium chloride, sodium hydroxide, sodium carbonate, hydrochloric acid and methanol were purchased from Sisco Research Laboratories (Mumbai, India). Primary antibodies against apoptotic protease‐activating factor 1 (APAF‐1), cyclin D1 and c‐Myc were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Bcl‐2 and Bax (Santa Cruz Biotechnology) were kind gift from Prof. C. M. Elinos‐Baez, Universidad Nacional Autónoma de México, Mexico; p53 was a kind gift from Dr C. Lazzari, Regina Elena Cancer Institute, Italy; survivin and β‐catenin were kind gifts from Prof. L. L. Muzio, University of Foggia, Italy; cytochrome c was a kind gift from Prof. R. Jemmerson, University of Minnesota, USA; caspase‐3 was a kind gift from Prof. Hyder Raza, United Arab Emirates University, UAE; and proliferative cell nuclear antigen (PCNA) was a kind gift from Prof. Micheal Aschner, Vanderbilt University, USA.
Cell culture
The HepG2 cell line was procured from the National Centre for Cell Science (Pune, India). Cells were grown in T75 culture flasks containing DMEM supplemented with 10% FBS. Upon reaching confluence, cells were detached using Trypsin‐EDTA solution.
Cell proliferation assay
Proliferation of HepG2 cells was assessed by MTT assay (14). Cells were plated in 24‐well plates at a concentration of 5 × 104 cells/well 24 h after plating; they were washed twice with 500 µl of serum‐free medium and were starved by incubation in serum‐free medium for an hour at 37 °C. After starvation, cells were treated with silymarin of different concentrations for 24 h. At the end of treatment, media from control and silymarin‐treated cells were discarded and 500 µl of MTT containing DMEM (0.5 mg/ml) was added to each well. Cells were then incubated for 4 h at 37 °C in a CO2 incubator. MTT‐containing medium was then discarded and the cells were washed with 1× phosphate‐buffered saline (PBS; 1 ml). Crystals were then dissolved by adding 500 µl of solubilization solution and this was mixed effectively by pipetting up and down. Spectrophotometrical absorbance of the purple blue formazan dye was measured using a microplate reader at 620 nm. Optical density of each sample was then compared with control optical density and graphs were plotted. Based on MTT assay, we selected doses 50 and 75 µg/ml silymarin treatment for 24 h in further studies because these are doses below the IC50 (50% inhibitory concentration) value of silymarin in HepG2.
Ethidium bromide/acridine orange (dual staining)
Ethidium bromide/acridine orange staining was carried out by the method of Gohel et al. (15). HepG2 cells were plated at a density of 5 × 104 in 6‐well plates containing sterile coverslips. They were allowed to grow at 37 °C in a humidified CO2 incubator until they were 70–80% confluent. Then cells were treated with silymarin (50 and 75 µg/ml) for 24 h. The culture medium was aspirated from each well and cells were gently rinsed twice with PBS at room temperature. Then the cover slips were taken and put on glass slides to be stained with 100 µl of dye mixture (1 : 1 of ethidium bromide and acridine orange) and were viewed immediately by fluorescence microscopy. Viable cells had green fluorescent nuclei with organized structure, early apoptotic cells had yellow chromatin in nuclei that were highly condensed or fragmented; apoptotic cells also exhibited membrane blebbing. Late apoptotic cells had orange chromatin with nuclei that were highly condensed and fragmented; necrotic cells had bright orange chromatin in round nuclei. Only cells with yellow, condensed, or fragmented nuclei were counted as apoptotic cells in a blinded, nonbiased manner. For each sample, at least 500 cells/well and 4 wells/condition were counted, and the percentage of apoptotic cells was determined [% of apoptotic cells = (total number of apoptotic cells/total number of cells counted) × 100].
Assessment of nuclear morphology after propidium iodide staining
Propidium iodide staining was carried out by the method of Chandramohan et al. (16). HepG2 cells were plated at a density of 5 × 104 in 6‐well plates containing sterile coverslips. They were allowed to grow at 37 °C in a humidified CO2 incubator until they were 70–80% confluent. Then cells were treated with silymarin (50 and 75 µg/ml) for 24 h. Culture medium was aspirated from each well and cells were gently rinsed twice with PBS at room temperature, before fixing in methanol:acetic acid (3 : 1 v/v) for 10 min, and stained with 50 µg/ml propidium iodide for 20 min. Nuclear morphology of apoptotic cells with condensed/fragmented nuclei was examined by fluorescence microscopy and at least 1 × 103 cells were counted for assessing apoptotic cell death.
Flow cytometric analysis of the cell cycle
Flow cytometric analysis was carried out as described by Rasola & Geuna (17). Briefly, 1 × 106 cells were plated in 100‐mm Petri dishes with DMEM containing 10% FBS. Cells were incubated for 24 h in 5% CO2 and 95% air at 37 °C. Control cells received 0.1% dimethyl sulphoxide (DMSO) containing DMEM, and silymarin‐treated cells received 50 and 75 µg/ml of silymarin containing DMEM. After 24 h, the cells were trypsinized and combined with floating cells in the medium they were used for flow cytometry assay. The treatment protocol is as follows: 1 × 106 cells were taken from control and from silymarin‐treated plates and were centrifuged at 1000 g for 5 min. Supernatant was removed and cells were washed twice with PBS. The pellet was resuspended in approximately 500 µl of ice‐cold PBS and cells were mixed by aspiration 20 times using a pipette. Cells were fixed by adding 5 ml of cold ethanol drop by drop and were kept at –20 °C overnight. After overnight fixation, ethanol was removed by centrifuging at 1000 g for 10 min. The pellet was washed twice with PBS + 1% BSA (ethanol‐fixed cells were difficult to pellet; adding BSA or serum to the wash medium overcame this). The pellet was resuspended in 800 µl of PBS containing 1% BSA. One hundred microlitre of 10× propidium iodide solution was added (500 µg/ml propidium iodide in PBS, pH 7.4) and one hundred microlitre of RNase A was added (10 mg/ml prepared in 10 mm Tris‐Cl, pH 7.5) and incubated at 37 °C for 30 min. Cell‐cycle analysis was performed using a Beckman vantage flow cytometer and quantification of cell cycle distribution was performed using MultiCycle software (Phoenix Flow System, San Diego, CA, USA). Percentage of cells in the different cell‐cycle phases was assessed.
Determination of mitochondrial membrane potential
Changes in mitochondrial transmembrane potential were measured by uptake of the mitochondrial‐specific lipophilic cation dye rhodamine 123, by the method of Chandramohan et al. (16). Approximately 1 × 106 cells were plated in 100‐mm Petri dishes with DMEM containing 10% FBS, then they were incubated for 24 h in 5% CO2 at 37 °C. Control cells received 0.1% DMSO containing DMEM and silymarin‐treated cells received 50 and 75 µg/ml of silymarin‐containing DMEM. After 24 h, cells were trypsinized and used for mitochondrial membrane potential analysis using flow cytometry. Briefly, 1 × 106 cells were taken from control and silymarin‐treated plates, and were centrifuged at 1000 g for 5 min. Supernatant was removed, and cells were washed twice with PBS. The pellet was resuspended in approximately 900 µl of ice‐cold PBS and cells were mixed by aspiration 20 times using a pipette. To this 100 µl of rhodamine 123 (100 µg/ml) was added and all were incubated at room temperature for 30 min in the dark. Cells were pelleted again and the supernatant was discarded (to remove excess rhodamine 123) and cells were resuspended in PBS. The samples 104 events were then immediately subjected to flow cytometric analysis at an excitation wavelength of 488 nm and emission wavelength of 545 nm. Mean fluorescence intensities were recorded and compared.
DNA agarose gel electrophoresis
DNA extraction and agarose gel electrophoresis were performed using the following method. Briefly, 1 × 106 cells were plated in 100‐mm Petri dishes with DMEM containing 10% FBS. Cells were incubated for 24 h in 5% CO2 and 95% air at 37 °C. Control cells received 0.1% DMSO containing DMEM, and silymarin‐treated cells received 50 and 75 µg/ml of silymarin‐containing DMEM. After 24 h, the cells were trypsinized and combined with the cells in the medium by centrifugation at 1500 r.p.m. for 5 min, then they were washed twice with PBS. The resulting pellet was resuspended in 0.25 ml of lysis buffer, transferred to a microfuge tube, and incubated for 1 h at 37 °C. To this 4 µl of proteinase K was added and tubes were then incubated at 50 °C for 3 h. To each tube, 0.5 ml of phenol:chloroform:isoamyl alcohol (25 : 24 : 1) was added, mixed and centrifuged at 13 000 r.p.m. for 30 min at 4 °C to separate the DNA containing upper aqueous phase. To the resultant aqueous phase, two volumes of ice‐cold absolute ethanol and 1/10 the volume of 3 m sodium acetate were added and kept at –20 °C overnight to precipitate DNA. The DNA was pelleted by centrifuging at 13 000 r.p.m. for 10 min at 4 °C and the supernatant was aspirated and the pellet washed in 1 ml of 70% ethanol. After repeating the above centrifugation step and removing last traces of the supernatant fraction, the pellet was allowed to dry at room temperature for approximately 30 min before being resuspended in 50 µl of Tris‐EDTA buffer. DNA was quantified by ultraviolet‐visible spectroscopy and 10 µg of DNA was electrophoresed in 1.5% agarose gel containing ethidium bromide in a mini gel tank containing Tris‐borate‐EDTA buffer for 2 h at 90 V. The gel was then examined under ultraviolet light and photographed.
Western blot analysis
Western blot analysis for protein expression in our HepG2 cells was assessed using the following method. Approximately 50 µg of protein was mixed with an equal volume of 2× sample buffer, boiled for 5 min at 95 °C, cooled, loaded on each lane of 8–15% polyacrylamide gel, and separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) at room temperature. Resolved proteins were electrophoretically transferred to nitrocellulose membranes which were then blocked in 5% nonfat milk in Tris‐buffered saline with 0.1% Tween 20 for 1 h at room temperature, and then probed with the following primary antibodies: APAF‐1 (dilution 1 : 500), Bcl‐2 (rabbit polyclonal antibody at a dilution of 1 : 500), Bax (rabbit polyclonal antibody at a dilution of 1 : 500), p53 (rabbit polyclonal antibody at a dilution of 1 : 1000), cytochrome c (mouse monoclonal antibody at a dilution of 1 : 500) survivin (mouse monoclonal antibody at a dilution of 1 : 500), active caspase‐3 (goat polyclonal antibody at a dilution of 1 : 250), β‐catenin (mouse monoclonal antibody at a dilution of 1 : 1000), cyclin D1 (mouse monoclonal antibody at a dilution of 1 : 500), PCNA (mouse monoclonal antibody at a dilution of 1 : 1000), β‐actin (mouse monoclonal antibody at a dilution of 1 : 2000); this was performed overnight at 4 °C. Blots were then extensively washed with Tris‐buffered saline with 0.1% Tween 20 (TBS‐T) and were incubated with respective (anti‐goat, anti‐rabbit and anti‐mouse) horseradish peroxidase‐labelled secondary antibodies (Genei, Bangalore, India) at dilution of 1 : 2000 for 1 h at room temperature. After thorough washes in TBS‐T, bands were visualized by treating the membranes with 3,3′‐diaminobenzidine tetrahydrochloride (Sisco Research Laboratories). Membranes were photographed and quantified with ImageJ image analysis software (National Institutes of Health, Bethesda, MD, USA). Densitometry data presented in bar graphs are ‘fold change’ as compared with control in each case.
Statistical analysis
All grouped data were significantly evaluated using SPSS/10 software. Hypothesis‐testing methods included one‐way analysis of variance (anova) followed by least significant difference test. P‐values of less than 0.05 were considered statistically significant. All these results were expressed as mean ± standard deviation (n = 3).
Results
Silymarin treatment for 24 h dose‐dependently inhibited population growth of HepG2 cells
Figure 1(a) shows the effect of silymarin at various doses (50, 75, 100 and 200 µg/ml) on HepG2 cells for 24 h by MTT assay. All doses of silymarin inhibited cell population growth and inhibition was directly proportional to dose [that is, increase in dose resulted in increased inhibition (P < 0.05) of proliferation]. Based on this study, we selected 50 and 75 µg/ml silymarin for further procedures. Figure 1(b) shows the morphological changes of control and silymarin‐treated HepG2 cells as seen by light microscopy after treating them with 50 and 75 µg/ml of silymarin. Silymarin treatment resulted in decreased number of attached cells and increased number of floating cells.
Figure l.
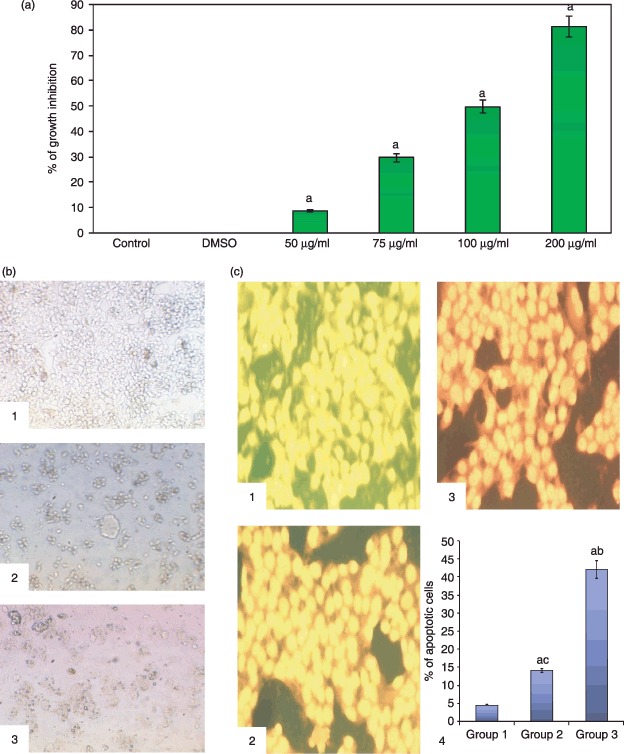
(a) MTT assay showing silymarin treatment for 24 h dose‐dependently inhibited population growth of HepG2 cells. (b) Morphological changes of HepG2 by light microscopy (×20): (1) HepG2 controls, (2) treatment with 50 mg/ml of silymarin and (3) treatment with 75 µg/ml of silymarin. (c) Silymarin increased percentage of apoptosis of HepG2 cells as viewed by fluorescence microscopy (ethidium bromide/acridine orange staining, ×25): (1) HepG2 control (showing viable green fluorescent nuclei), (2) treatment with 50 µg/ml of silymarin (early apoptotic cells are yellow fluorescent nuclei), (3) treatment with 75 µg/ml of silymarin (late apoptotic cells with orange fluorescent nuclei), and (4) representative bar chart showing percentage of apoptotic cells. Results are expressed as mean ± standard deviation (n= 3). P < 0.05 compared with aHepG2 control, b50 µg/ml of silymarin and c75 µg/ml of silymarin.
Silymarin‐induced apoptosis in HepG2 cells
Figure 1(c) shows by fluorescence microscopy morphological changes in control and silymarin‐treated HepG2 cells after staining with ethidium bromide/acridine orange. The percentage of apoptotic cells after treatment with 50 and 70 µg/ml of silymarin increased (P < 0.05) drastically to 14% and 42%, respectively. Figure 2(a) shows morphological changes of control and silymarin‐treated cells by fluorescence microscopy after staining with propidium iodide. The percentage of apoptotic nuclei after treatment with 50 and 75 µg/ml of silymarin increased enormously (P < 0.05) to 22% and 59%, respectively, as revealed by nuclear condensation and fragmentation. Figure 2(b) reveal agarose gel electrophoretic pattern of nuclear DNA from control and silymarin‐treated HepG2 cells. Silymarin treatment resulted in apoptosis, as evidenced by specific shearing of DNA, which is considered to be the hallmark of apoptosis.
Figure 2.
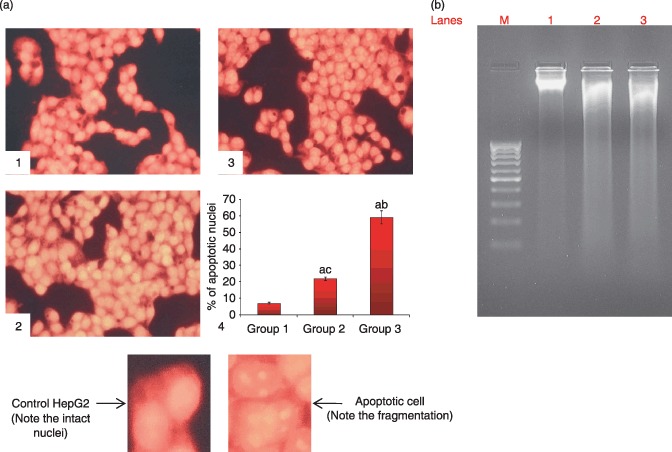
(a) Silymarin increased percentage of apoptosis in HepG2 cells as viewed by fluorescence microscopy (propidium iodide staining, ×25). (1) HepG2 control (normal nuclear pattern), (2) treatment with 50 µg/ml silymarin (nuclear condensation and fragmentation), (3) treatment with 75 µg/ml silymarin (nuclear condensation and fragmentation) and (4) representative bar chart showing percentage of apoptotic nuclei. Results are expressed as mean ± standard deviation (n= 3). P < 0.05 compared with aHepG2 control, b50 µg/ml of silymarin and c75 µg/ml of silymarin. (b) Silymarin‐induced apoptosis in HepG2 cells as revealed by agarose gel electrophoresis pattern of nuclear DNA. Lanes 1, 2, 3 and M representing HepG2 control, 50 µg/ml silymarin, 75 µg/ml silymarin and marker, respectively.
Silymarin inhibited cell‐cycle progression and down‐regulated expression of proliferation‐associated proteins β‐catenin, cyclin D1, c‐Myc and PCNA in HepG2 cells
Figure 3(a) shows the effect of silymarin on cell‐cycle regulation in HepG2 cells. Treatment with 50 and 75 µg/ml of silymarin for 24 h significantly increased the proportion of cells with a reduced DNA content (sub‐G0/G1 or A0 peak), indicative of apoptosis with loss of cells in the G1 phase. Loss of DNA occurs as a result of diffusion of degraded DNA from the cells after endonuclease cleavage. After staining with propidium iodide, these cells would have taken up less stain and appear in sub‐G0/G1 or A0 peak (that is to the left of the G0/G1 peak). Incubation of the cells with 50 and 75 µg/ml of silymarin for 24 h significantly increased the proportion of cells with a reduced DNA content from 6.08% (control) to 46.86% (50 µg/ml silymarin) and 67.14% (75 µg/ml of silymarin). Figure 3(b) illustrates the protein expression analysis of β‐catenin, cyclin D1, cMyc and PCNA in control and silymarin‐treated cells. Silymarin treatment significantly decreased (P < 0.05) the expression of β‐catenin, cyclin D1, cMyc and PCNA dose dependently as is evident from immunoblotting and corresponding densitometry data.
Figure 3.

(a) Silymarin inhibited cell‐cycle progression in HepG2 cells: (a), (b) and (c) showing representative histograms of HepG2 control, 50 µg/ml of silymarin, and 75 µg/ml of silymarin, respectively. (d) Quantitative analysis of apoptotic cell population. P < 0.05 compared with aHepG2 control, b50 µg/ml silymarin and c75 µg/ml silymarin. (b) Silymarin down‐regulated expression of proliferation‐associated proteins β‐catenin, cyclin D1, cMyc and PCNA in HepG2 cells. Lanes l, 2 and 3 correspond to lysates of HepG2 control, 50 µg/ml of silymarin, and 75 µg/ml of silymarin, respectively.
Silymarin depolarized mitochondrial membrane potential, thereby aiding release of cytochrome c from mitochondria into the cytosol
Figure 4(a) shows the effect of silymarin on mitochondrial membrane potential of control and silymarin‐treated cells as assessed by flow cytometry. Silymarin treatment depolarized mitochondrial membranes as is evident from the significant decrease (P < 0.05) in mean fluorescence of rhodamine 123 (shifting the peak towards the left side). Figure 4(b) is of levels of mitochondrial and cytosolic cytochrome c of control and silymarin‐treated HepG2 cells as assessed by immunoblotting and by their respective densitometry data. Silymarin treatment resulted in release of cytochrome c from mitochondria to cytosol.
Figure 4.
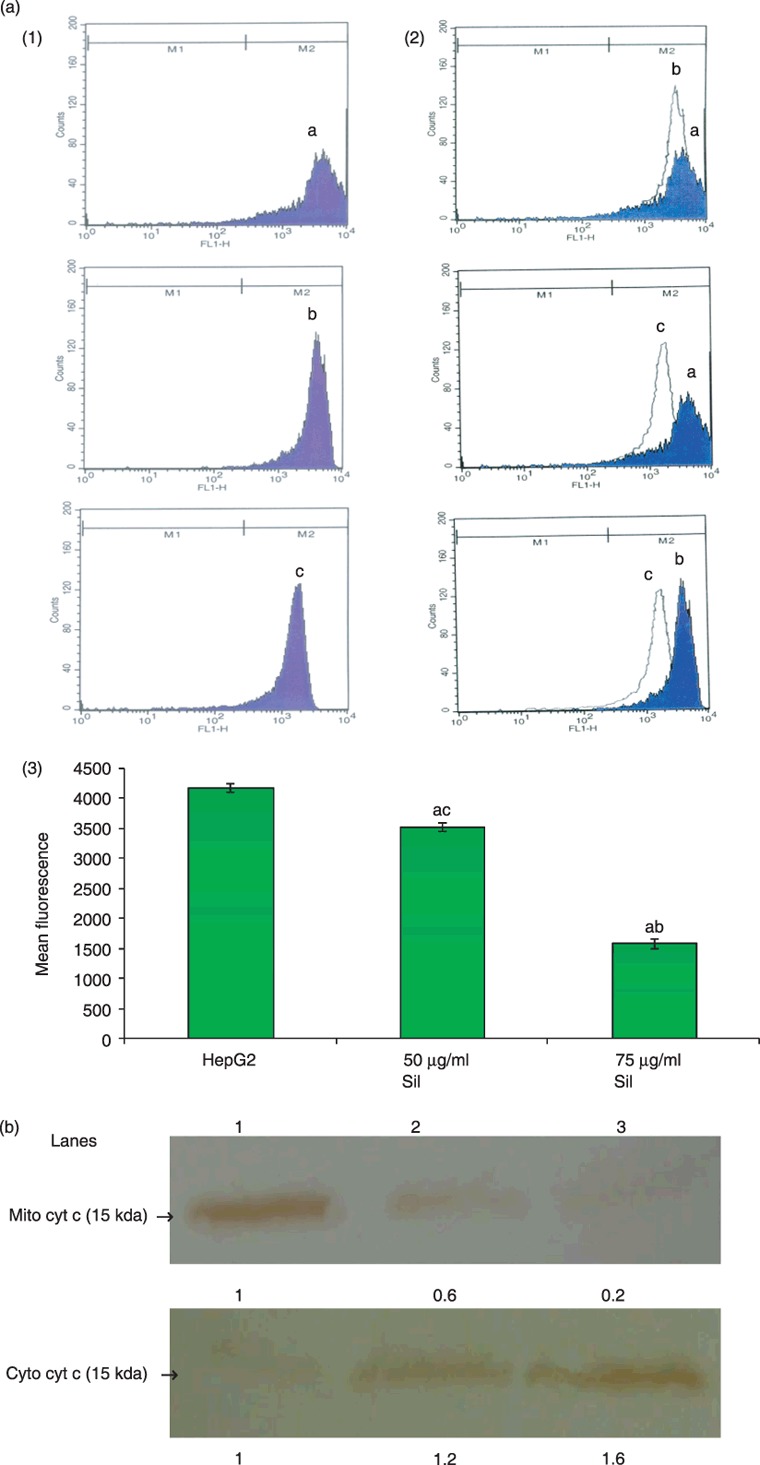
(a) Silymarin treatment decreased mitochondrial transmembrane potential of HepG2 cells. (1) Mitochondrial membrane potential as assessed by staining with rhodamine 123. (a), (b) and (c) representing control HepG2 cells, treatment with 50 µg/ml of silymarin, and treatment with 75 µg/ml silymarin, respectively. (2) Overlay. (3) Bar chart representing mean fluorescence of rhodamine 123 in HepG2 control, treatment with 50 µg/ml silymarin, and treatment with 75 µg/ml silymarin. (b) Silymarin treatment released cytochrome c from mitochondria into the cytosol in HepG2 cells. Lanes l, 2 and 3 correspond to the lysates of HepG2 control, 50 µg/ml silymarin and 75 µg/ml silymarin, respectively. P < 0.05 compared with aHepG2 control, b50 µg/ml of silymarin and c75 µg/ml of silymarin.
Silymarin treatment up‐regulated expression of pro‐apoptotic proteins p53, Bax, APAF‐1 and caspase‐3 with concomitant decrease in levels of anti‐apoptotic proteins Bcl‐2 and survivin
Figure 5 demonstrates protein expression analysis of APAF‐1, Bcl‐2, Bax, p53, survivin and caspase‐3 in control and silymarin‐treated HepG2 cells, as assessed by immunoblotting and by their respective densitometry data. Silymarin treatment significantly increased (P < 0.05) expression of pro‐apoptotic proteins APAF‐1, Bax, p53 and caspase‐3 and subsequently decreased (P < 0.05) expression of anti‐apoptotic proteins Bcl‐2 and survivin dose dependently as is evident from immunoblotting and corresponding densitometry data.
Figure 5.

Immunoblotting analysis of APAF‐1, Bcl‐2, Bax, p53, survivin and caspase‐3 in control and silymarin‐treated HepG2 cells. Lanes l, 2 and 3 correspond to the lysates of HepG2 control, 50 µg/ml silymarin and 75 µg/ml silymarin, respectively.
Discussion
In the present study, induction of apoptosis by silymarin in HepG2 cells was assessed by procedures, such as ethidium bromide/acridine orange staining, propidium iodide staining and DNA fragmentation. All the above clearly indicated that silymarin treatment induced apoptosis. Substantial evidence implicates mitochondria in apoptotic cell death and there is a direct relationship between mitochondrial depolarization and cytochrome c release. Cytochrome c is undoubtedly one of the prominent actors in the apoptotic scene. It can escape from a cell's power plant, the mitochondrion, when it receives instruction to break down its outer membrane. Once unleashed from its usual context, cytochrome c can be recruited into the death squad and contribute to apoptotic dismantling of the cell (18). Here, silymarin treatment significantly decreased mitochondrial membrane potential of HepG2 cells, thus, leading to opening of mitochondrial permeability transition pores and consequently, release of cytochrome c from the intermembrane space into the cytosol. This initiates the apoptotic cascade, which is well correlated with earlier studies of silymarin on other types of cancer cell (19, 20). APAF‐1 is a 130‐kD protein that plays a central role in mitochondrial control of apoptosis and APAF‐1 also appears to be an essential component of p53‐mediated apoptosis (21). In response to apoptotic stimuli, APAF‐1 binds to cytochrome c and procaspase‐9 in the presence of adenosine triphosphate to form a multiprotein complex called the apoptosome. This results in activation of procaspase‐9 by autocatalytic cleavage, initiating a cascade of downstream effector caspases, especially caspase‐3, which cleave several cell proteins, ultimately leading to apoptosis (22). APAF‐1 deficiency has been shown in cancer cells, which ends in diminished caspase activation (23). Here, silymarin treatment resulted in increased APAF‐1 and activated caspase‐3 in the hepatocellular carcinoma cells. Treatment with silymarin inducing expression of APAF‐1, might have interacted with released cytochrome c‐forming apoptosomes and thereby might have activated the caspase‐3.
p53 protein inhibits growth of tumours by arresting cell proliferation and inducing apoptosis. It induces apoptosis by activating many pro‐apoptotic genes, which ultimately activate APAF‐1 and caspase‐9 through several pathways (24). It has been estimated that in excess of 100 genes are regulated by p53, many of which can promote population growth arrest or apoptosis. Alteration in p53 gene is the most frequently identified mutation in human cancers. Loss of p53 function allows cells with damaged DNA to continue to proliferate and, therefore, it is associated with tumour progression (25). Silymarin treatment dose dependently increased the level of p53 in our experiment and it has been found that p53 might directly facilitate cytochrome c release (26), thereby increasing expression of p53 after silymarin treatment might directly facilitate release of cytochrome c from mitochondria and initiate apoptosis. Bcl‐2 and Bax protein are members of a large family of proteins called ‘the Bcl‐2 family’ and both are important regulators of apoptosis. Bcl‐2 can prolong cell survival by suppressing apoptosis, and Bax can enhance apoptosis. Bcl‐2 can block mitochondrial permeability transition pores opening, thereby preventing release of caspase activators from mitochondria (27). Overexpression of Bax has been shown to accelerate apoptosis, whereas Bcl‐2 represses the death function of Bax (28). Thus, ratio between Bcl‐2/Bax might be one of the critical factors of a cell's threshold for undergoing apoptosis (29). In the present study, there was increased expression of Bcl‐2 and a subsequent decreased in expression of Bax was seen in HepG2 cells; hence, an increase in the Bcl‐2/Bax ratio, which is an indication of diminished apoptosis. Silymarin treatment decreased the Bcl‐2/Bax ratio which might be due to the ability of silymarin to induce p53 expression because p53 is a positive transcriptional activator for Bax and a negative transcriptional activator for Bcl‐2 (30), thus the activation of the p53 pathway by silymarin might lead to the down‐regulation of Bcl‐2 and up‐regulation of Bax. Survivin protein is a recently described member of the inhibitor of apoptosis family of anti‐apoptotic proteins, which may act simultaneously with Bcl‐2 family proteins to inhibit apoptosis (31). It is the smallest member of the inhibitor of apoptosis family and expression of survivin is closely related to cell proliferation. Survivin inhibits apoptosis mainly through targeting terminal effector caspase‐3 activity in the apoptotic protease cascade (32). Cancer‐specific expression of survivin, coupled with its importance in inhibiting cell death and in regulating cell division, makes it a useful diagnostic marker of cancer and a potential target for cancer treatment (33). Recently, studies have set out to evaluate the possibility of targeting survivin function in vivo as an anticancer strategy in which it has been shown that inhibition of survivin could effectively inhibit de novo tumour formation and progression (33, 34). Here, silymarin treatment significantly decreased the level of this protein, which might be due to the ability of silymarin to up‐regulate the level of p53 as survivin is negatively regulated by p53 (35).
Uncontrolled tumour cell proliferation plays the most crucial role in hepatocellular carcinoma growth. PCNA is a useful marker of proliferative activity, it functions as a cofactor of DNA polymerase and an important marker for evaluating the proliferation of several cancers, including hepatocellular carcinoma (36). Silymarin treatment significantly decreased the proliferative index as revealed by down‐regulated expression of PCNA. β‐catenin plays a critical role as a component of the cell adhesion complex and as a co‐activator of the T‐cell transcription factor/lymphoid enhancer binding factor (TCF/LEF) family of an transcription factors. As a regulator of an intracellular signalling network involved in oncogenic process, activated β‐catenin signalling favours cell proliferation as well as exerts anti‐apoptotic effects in various cancers (37). Several studies have documented positive correlation between β‐catenin accumulation and cancer cell proliferation including for hepatocellular carcinoma (38, 39, 40). It has been found that protein products of the target genes of β‐catenin‐TCF pathway were growth‐promoting factors such as cyclin D1 and c‐Myc (41, 42). Cyclin D1 is a major regulator of progression of cells through the cell cycle (43); it is rate limiting for the G1/ S cell‐cycle checkpoint. Recently, it has been shown that chronic cyclin D1 overexpression in transgenic mice is associated with rapidly progressing development of hepatocellular adenomas and carcinomas (44). In normal cells, c‐Myc is induced upon growth factor stimulation, whereas it is constitutively high in transformed cells. Some degree of c‐Myc overexpression is estimated to occur in 70% of human tumours (45). Constitutive expression of the proto‐oncogene c‐Myc results in oncogenic activation and contributes to progression of a wide range of human and animal tumours. c‐Myc activation promotes cell proliferation by up‐regulating one of its target genes, which codes for ornithine decarboxylase. This is a rate‐limiting enzyme of polyamine biosynthesis and has been shown to be required for entry into and progression through the cell cycle (46). Down‐regulation of c‐Myc by antioxidants has been proposed as a promising pharmacological treatment for cancer (47). Silymarin is a potent antioxidant and has been shown to have an inhibitory effect on ornithine decarboxylase activity (48, 49). In our work, silymarin administration significantly decreased levels of b‐catenin and protein products of its target genes cyclin D1 and c‐Myc. This ability of silymarin to suppress levels of β‐catenin and its target genes/proteins might be attributed to its antioxidant property (8) as antioxidants have been shown to suppress β‐catenin mutation in hepatocellular carcinomas (50). Thus, it can be concluded that silymarin suppresses proliferation of hepatocellular carcinoma HepG2 cells by inhibiting β‐catenin accumulation and, hence, its target genes for cyclin D1 and c‐Myc and this may be the underlying mechanism of its antiproliferative effect. In conclusion, it has been found that the mechanism by which silymarin exhibits its growth inhibitory effect on human hepatocellular carcinoma cells in vitro are by inhibiting cell proliferation and by inducing apoptosis.
Acknowledgements
The authors wish to thank Prof. Lorenzo Lo Muzio, University of Foggia, Italy, for his kind gift of antibodies againist β‐catenin and survivin; Prof. Carmen Martha Elinos‐Baez, Universidad Nacional Autónoma de México, Mexico for her kind gift of antibodies against Bcl‐2 and Bax; Prof. Michael Aschner, Vanderbilt University, USA, for his kind gift of anti‐PCNA; Prof. Ron Jemmerson, University of Minnesota, USA, for his kind gift of the antibody against cytochrome c; Prof. Hyder Raza, UAEU, UAE, for his kind gift of the antibody against caspase‐3; and Dr C. Lazzari, Regina Elena Cancer Institute, Italy, for his kind gift of anti‐p53. The authors wish to thank Prof. M. Michael Aruldhas and faculty members of the Department of Endocrinology, University of Madras, Taramani Campus, Chennai, for allowing us to utilize their laboratory facilities. The authors wish to thank Dr A. Raja, Department of Biotechnology, TANUVAS (Tamilnadu University of Veterinary and Animal Sciences), Chennai, for his help during flow cytometric analysis. The authors wish to thank Mr Albert (Loyola College, Chennai), Mr M. Sridhar, Mr Senthil, Mr R. C. Vignesh and Mr G. Krishnamurthy for their help during the study.
References
- 1. El‐Serag HB, Rudolph KL (2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gasteroenterology 132, 2557–2576. [DOI] [PubMed] [Google Scholar]
- 2. El‐Serag HB, Mason AC (1999) Rising incidence of hepatocellular carcinoma in the United States. N. Engl. J. Med. 340, 745–750. [DOI] [PubMed] [Google Scholar]
- 3. Wellington K, Jarvis B (2001) Silymarin: a review of its clinical properties in the management of hepatic disorders. BioDrugs 15, 465–489. [DOI] [PubMed] [Google Scholar]
- 4. Lee JI, Narayan M, Barrett JS (2007) Analysis and comparison of active constituents in commercial standardized silymarin extracts by liquid chromatography‐electrospray ionization mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 845, 95–103. [DOI] [PubMed] [Google Scholar]
- 5. Katiyar SK (2005) Silymarin and skin cancer prevention: anti‐inflammatory, antioxidant and immunomodulatory effects (review). Int. J. Oncol. 26, 169–176. [PubMed] [Google Scholar]
- 6. Bhatia N, Zhao J, Wolf DM, Agarwal R (1999) Inhibition of human carcinoma cell growth and DNA synthesis by silibinin, an active constituent of milk thistle: comparison with silymarin. Cancer Lett. 147, 77–84. [DOI] [PubMed] [Google Scholar]
- 7. Ferenci P, Dragosics B, Dittrich H, Frank H, Benda L, Lochs H, Meryn S, Base W, Schneider B (1989) Randomized controlled trial of silymarin treatment in patients with cirrhosis of the liver. J. Hepatol. 9, 105–113. [DOI] [PubMed] [Google Scholar]
- 8. Ramakrishnan G, Raghavendran HRB, Vinodhkumar R, Devaki T (2006) Suppression of N‐nitrosodiethylamine induced hepatocarcinogenesis by silymarin in rats. Chem. Biol. Interact. 161, 104–114. [DOI] [PubMed] [Google Scholar]
- 9. Ramakrishnan G, Elinos‐Baez CM, Jagan S, Augustine TA, Kamaraj S, Anandakumar P, Devaki T (2008) Silymarin downregulates COX‐2 expression and attenuates hyperlipidemia during NDEA‐induced rat hepatocellular carcinoma. Mol. Cell. Biochem. 313, 53–61. [DOI] [PubMed] [Google Scholar]
- 10. Ramakrishnan G, Augustine TA, Jagan S, Vinodhkumar R, Devaki T (2007) Effect of silymarin on N‐nitrosodiethylamine induced induced hepatocarcinogenesis in rats. Exp. Oncol. 29, 39–44. [PubMed] [Google Scholar]
- 11. Kaur M, Agarwal R (2007) Silymarin and epithelial cancer chemoprevention: how close we are to bedside? Toxicol. Appl. Pharmacol. 224, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Varghese L, Agarwal C, Tyagi A, Singh RP, Agarwal R (2005) Silibinin efficacy against human hepatocellular carcinoma. Clin. Cancer Res. 11, 8441–8448. [DOI] [PubMed] [Google Scholar]
- 13. Lah JL, Cui W, Hu KQ (2007) Effects and mechanisms of silibinin on human hepatoma cell lines. World J. Gastroenterol. 13, 5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Safadi FF, Xu J, Smock SL, Kanaan RA, Selim AH, Odgren PR, Marks SC Jr, Owen TA, Popoff SN (2003) Expression of connective tissue growth factor in bone: its role in osteoblast proliferation and differentiation in vitro and bone formation in vivo . J. Cell. Physiol. 196, 51–62. [DOI] [PubMed] [Google Scholar]
- 15. Gohel A, McCarthy MB, Gronowicz G (1999) Estrogen prevents glucocorticoid‐induced apoptosis in osteoblasts in vivo and in vitro . Endocrinology 140, 5339–5347. [DOI] [PubMed] [Google Scholar]
- 16. Chandramohan KVP, Gunasekaran P, Varalakshmi E, Hara Y, Nagini S (2007) In vitro evaluation of the anticancer effect of lactoferrin and tea polyphenol combination on oral carcinoma cells. Cell Biol. Int. 31, 599–608. [DOI] [PubMed] [Google Scholar]
- 17. Rasola A, Geuna M (2001) A flow cytometry assay simultaneously detects independent apoptotic parameters. Cytometry 45, 151–157. [DOI] [PubMed] [Google Scholar]
- 18. Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G (2006) Mechanisms of cytochrome c release from mitochondria. Cell Death Diff. 13, 1423–1433. [DOI] [PubMed] [Google Scholar]
- 19. Tyagi A, Singh RP, Agarwal C, Agarwal R (2006) Silibinin activates p53‐caspase 2 pathway and causes caspase‐mediated cleavage of Cip1/p21 in apoptosis induction in bladder transitional‐cell papilloma RT4 cells: evidence for a regulatory loop between p53 and caspase 2. Carcinogenesis 27, 2269–2280. [DOI] [PubMed] [Google Scholar]
- 20. Katiyar SK, Roy AM, Baliga MS (2005) Silymarin induces apoptosis primarily through a p53‐dependent pathway involving Bcl‐2/Bax, cytochrome c release, and caspase activation. Mol. Cancer Ther. 4, 207–216. [PubMed] [Google Scholar]
- 21. Zlobec I, Vuong T, Compton CC (2006) The predictive value of apoptosis protease‐activating factor 1 in rectal tumors treated with preoperative high dose rate brachytherapy. Cancer 106, 284–286. [DOI] [PubMed] [Google Scholar]
- 22. Hickman ES, Helin K (2002) The regulation of APAF1 expressionduring development and tumorigenesis. Apoptosis 7, 161–171. [DOI] [PubMed] [Google Scholar]
- 23. Wolf BB, Schuler M, Li W, Eggers‐Sedlet B, Lee W, Tailor P, Fitzgerald P, Mills GB, Green DR (2001) Defective cytochrome c‐dependent caspase activation in ovarian cancer cell lines due to diminished or absent apoptotic protease activating factor‐1 activity. J. Biol. Chem. 276, 34244–34251. [DOI] [PubMed] [Google Scholar]
- 24. Jin S, Levine AJ (2001) The p53 functional circuit. J. Cell Sci. 114, 4139–4140. [DOI] [PubMed] [Google Scholar]
- 25. Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutation in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54, 4855–4878. [PubMed] [Google Scholar]
- 26. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11, 577–590. [DOI] [PubMed] [Google Scholar]
- 27. Kluck RM, Bossy‐Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl‐2 regulation of apoptosis. Science 275, 1132–1136. [DOI] [PubMed] [Google Scholar]
- 28. Zhao L, Guo QL, You QD, Wu ZQ, Gu HY (2004) Gambogic acid induces apoptosis and regulates expressions of Bax and Bcl‐2 protein in human gastric carcinoma MGC‐803 cells. Biol. Pharm. Bull. 27, 998–1003. [DOI] [PubMed] [Google Scholar]
- 29. Cory S, Adams JM (2005) Killing cancer cells by flipping the Bcl‐2/Bax switch. Cancer Cell 8, 5–6. [DOI] [PubMed] [Google Scholar]
- 30. Xiao EH, Li JQ, Huang JF (2004) Effects of p53 on apoptosis and proliferation of hepatocellular carcinoma cells treated with transcatheter arterial chemoembolization. World. J. Gasteroenterol. 10, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu H, Chen XP, Zhang WG, Luo SF, Zhang BX (2005) Expression and significance of new inhibitor of apoptosis protein surviving in hepatocellular carcinoma. World. J. Gasteroenterol. 11, 3855–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki A, Ito T, Kawano H, Hayashida M, Hayasaki Y, Tsutomi Y, Akahane K, Nakano T, Miura M, Shiraki K (2000) Survivin initiates procaspase 3/p21 complex formation as a result of interaction with cdk4 to resist fas mediated cell death. Oncogene 19, 1346–1353. [DOI] [PubMed] [Google Scholar]
- 33. Kanwar JR, Shen WP, Kanwar RK, Berg RW, Krissansen CW (2001) Effects of survivin antagonists on growth of established tumors and B7‐1 immunogene therapy. J. Natl. Cancer Inst. 93, 1541–1552. [DOI] [PubMed] [Google Scholar]
- 34. Mesri M, Wall NR, Li J, Kim RW, Altieri DC (2001) Cancer gene therapy using a survivin mutant adenovirus. J. Clin. Invest. 108, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M (2002) Transcriptional repression of the anti‐apoptotic surviving gene by wild type p53. J. Biol. Chem. 277, 3247–3257. [DOI] [PubMed] [Google Scholar]
- 36. Wu WY, Xu Q, Shi LC, Zhang WB (2000) Inhibitory effects of Curcuma aromatica oil on proliferation of hepatoma in mice. World J. Gastroenterol. 6, 216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Porter AC, Vaillancourt RR (1998) Tyrosine kinase receptor‐activated signal transduction pathways which lead to oncogenesis. Oncogene 17, 1343–1352. [DOI] [PubMed] [Google Scholar]
- 38. Derksen PW, Tjin E, Meijer HP, Klok MD, Gillavry HDM, Van Oers MHJ, Lokhorst HM, Bloem AC, Clevers H, Nusse R, Neut RV, Spaargaren M, Pals ST (2004) Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci. USA 101, 6122–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saldanha G, Ghura V, Potter L, Fletcher A (2004) Nuclear β‐catenin in basal cell carcinoma correlates with increased proliferation. Br. J. Dermatol. 151, 157–164. [DOI] [PubMed] [Google Scholar]
- 40. Calvisi DF, Ladu S, Factor VM, Thorgeirsson SS (2004) Activation of β‐catenin provides proliferative and invasive advantages in c‐myc/TGF‐α hepatocarcinogenesis promoted by phenobarbital. Carcinogenesis 25, 901–908. [DOI] [PubMed] [Google Scholar]
- 41. Tetsu O, McCormick F (1999) β‐Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 298, 422–426. [DOI] [PubMed] [Google Scholar]
- 42. He T, Sparks A, Rago C, Hermeking H, Zawel L, Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c‐MYC as a target of the APC pathway. Science 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- 43. Sherr CJ (1996) Cancer cell cycles. Science 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- 44. Deane N, Parker M, Aramandla R, Diehl L, Lee WJ, Washington MK, Nanney LB, Shyr Y, Beauchamp RD (2001) Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 61, 5389–5395. [PubMed] [Google Scholar]
- 45. Gordan JD, Thompson CB, Simon MS (2007) HIF and c‐Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peckham G, Cleveland JL (1995) c‐Myc and apoptosis. Biochim. Biophys. Acta 1242, 11–28. [DOI] [PubMed] [Google Scholar]
- 47. Nigris FD, Sica V, Herrmann J, Condorelli G, Chade AR, Tajana G, Lerman A, Lerman LO, Napoli C (2003) c‐Myc oncoprotein: cell cycle‐related events and new therapeutic challenges in cancer and cardiovascular diseases (Review). Cell Cycle 2, 325–328. [PubMed] [Google Scholar]
- 48. Agarwal R, Katiyar SK, Lundgren DW, Mukhtar H (1994) Inhibitory effect of silymarin, an anti‐hepatotoxic flavonoid, on 12‐O‐tetradecanoylphorbol‐13‐acetate‐induced epidermal ornithine decarboxylase activity and mRNA in SENCAR mice. Carcinogenesis 15, 1099–1103. [DOI] [PubMed] [Google Scholar]
- 49. Katiyar SK, Korman NJ, Mukhtar H, Agarwal R (1997) Protective effects of silymarin against photocarcinogenesis in a mouse skin model. J. Natl. Cancer Inst. 89, 556–565. [DOI] [PubMed] [Google Scholar]
- 50. Hara A, Sakata K, Yamada Y, Kuno T, Kitaori N, Oyama T, Hirose Y, Murakami A, Tanaka T, Mori H (2005) Suppression of β‐catenin mutation by dietary exposure of auraptene, a citrus antioxidant, N,N‐diethylnitrosamine – induced hepatocellular carcinoma in ras. Oncol. Report 14, 345–351. [PubMed] [Google Scholar]