

Abstract
Abstract. It has been predicted that whole‐culture methods of synchronization cannot synchronize cells. We have tested whether thymidine block, one type of whole‐culture synchronization, can synchronize L1210 cells. We demonstrate experimentally that the thymidine block method cannot produce a synchronized culture. Although thymidine‐treated cells are arrested primarily with an S‐phase amount of DNA, there is no narrowing of the cell size distribution and there is no synchronized division pattern following release from the thymidine block. In contrast to a whole‐culture synchronization method, cells produced by a selective (i.e. non‐whole‐culture) method not only have a specific DNA content, but also have a narrow size distribution and divide synchronously. Generalizing the results to other cell lines, we suggest that these conclusions call into question experimental measurements of gene expression during the division cycle based on thymidine inhibition synchronization.
INTRODUCTION
In order to understand the passage of a cell through the cell cycle – from birth by division of a mother cell to the next division – it is important to know what events take place at different times during the cell cycle. Because the measurement of chemical events is difficult in single cells, much effort has been expended on methods to synchronize the population. A properly or well‐synchronized culture is one in which the cells move as a uniform cohort through the cell cycle. Such synchronized cells would and should accurately reflect the events occurring in growing unperturbed cells during normal passage through the phases. The sine qua non of synchronization is that the cells move uniformly through the cell cycle and divide synchronously over a relatively narrow span of time (Cooper & Shedden 2003; Cooper 2004a).
Synchronization methods can be divided into two classes: whole‐culture methods and selective methods (Cooper 1991). Whole‐culture methods are those in which treatment of all cells in the culture is proposed to lead to a group of cells that are arrested at a specific point in the cell cycle. Release of these growth‐arrested cells is presumed to produce a synchronized culture. Selective methods are those methods in which a subset of the original population is selected to produce a synchronized culture and the remaining cells are discarded.
It is important to distinguish between cells that have one property in common (e.g. all cells have a G1‐phase amount of DNA), and cells that are truly synchronized. Cells that are not synchronized but have one property in common should be referred to as ‘aligned’ for that property, but should not be referred to as ‘synchronized’. Others have observed (Gong et al. 1995) that cells that are ‘chemically’ treated exhibit growth imbalance and higher heterogeneity with respect to all parameters measured, including the expression of cyclins and the degree of refinoblastoma product phosphorylation.
What is lacking in proposed of synchronization methods is the generality of the rule that whole‐culture methods cannot synchronize cells. A whole‐culture method is a method whereby all of the cells in it are treated and no cells are discarded; presumably the entire population has been transformed into a synchronized cohort. To put the rule succinctly, whole‐culture methods cannot synchronize cells (Cooper 2003a). This rule does not imply that whole‐culture methods synchronize cells poorly, or weakly, or occasionally, but rather that such methods do not synchronize cells at all. These methods may align cells for some particular property (such as DNA content), but the cells do not reflect the totality of properties of cells at a particular cell age during the normal cell cycle. This is the case even though the cells may all have some particular property, such as DNA content, in common. The importance of this proposal, when put forward in a constructive way (i.e. thymidine block does not synchronize cells), is that methods that are widely used to study the cell cycle do not yield information regarding the normal cell cycle. Observations of cyclical patterns and other changes following whole‐culture synchronization must perforce be results of, or artifacts of, the treatment and not related to the normal, unperturbed cell cycle.
A study of lovastatin, a compound that was originally proposed to be a general whole‐culture synchronizing agent (Keyomarsi et al. 1991), demonstrated that lovastatin does not synchronize cells. Time‐lapse analysis of cell division following lovastatin arrest has shown that the cells do not divide synchronously (Cooper 2002). Furthermore, reconsideration of the published data showing synchronized S‐phases and initial arrest with a G1‐phase amount of DNA demonstrates that the S‐phases are not synchronized, and that the cells are not even arrested with a G1‐phase amount of DNA (Cooper 2002).
A further experimental example of problems with whole‐culture synchronization is the use of nocodazole. Nocodazole is widely used as a synchronizing agent (see, for example, Jansen‐Durr et al. 1993; Ludlow et al. 1993; Ouyang et al. 1998; Harper 2005), but cells treated with it are arrested prior to mitosis. Support for this proposal is the widely reported observation that nocodazole treatment produces cells with a G2‐phase amount of DNA (Kung et al. 1990). Experiments have clearly demonstrated that even when a culture inhibited with nocodazole has almost all cells with a G2‐phase amount of DNA, these cells are not synchronized (Cooper et al. 2006). The two criteria on which this conclusion was based are the observations that there was no narrowing of the cell‐size distribution after inhibition with nocodazole, and that there is no synchronized cell division following release from nocodazole inhibition (Cooper et al. 2006).
Experiments have also been presented showing that serum starvation, a widely used method to presumably synchronize cells, does not work (Liliensiek et al. 2006).
We now describe experiments on the use of thymidine block to produce ‘synchronized’ cells. We have studied the effect of thymidine block on L1210 mouse cells and have measured the size distribution of cell populations treated with inhibitory concentrations of thymidine and the pattern of growth following release from thymidine arrest. Comparison of DNA and size measurements of thymidine‐arrested cells with cells produced by a selective method (membrane elution) support the proposal that thymidine‐arrested cells are not synchronized.
We argue that the results presented here are not restricted to the cells and conditions used in this study. Rather, these results apply broadly and generally to mammalian cells and other growth systems. It might be suggested that our results here are applicable only to the particular cell line studied, and cannot be extended to other cell lines. This viewpoint suggests that in order to prove that a thymidine block cannot synchronize cells one must look at essentially every (or perhaps the majority) of cell lines and show that a thymidine block does not synchronize them. However, these experiments are strongly buttressed by theoretical considerations rigorously supporting the proposal that whole‐culture methods cannot synchronize cells (Cooper 1998, 2003a, 2003b, 2004a, 2004b, 2006; Cooper & Shedden 2003). The prohibition against the experimental demonstration of a universal negative, in other words, that whole‐culture methods cannot synchronize cells, does not apply in this case.
We propose that the fit of theory and experiment allows the extension of these results to other cell lines. To the theoretical concept that these methods ‘cannot synchronize cells’, we present experimental results supporting the proposal that such methods ‘do not synchronize cells’.
MATERIALS AND METHODS
Chemicals
Thymidine was prepared in a stock solution of 0.4 m and was used at a 1/100 dilution to produce a 4 mm thymidine concentration.
Cells
L1210 cells, a mouse laeukemic line (ATCC designation CCL219) were used for all experiments. These cells are non‐adherent and grow with a doubling time of approximately 9–10 h.
Growth media
Liebovitz's L‐15 medium (Cellgro by Mediatech, Herndon, VA 20171) was supplemented with 2 mg/mL glucose, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% cosmic calf serum (CCS). CCS prepared by Hyclone Inc. (Logan, UT, USA), is a modified calf serum that substitutes for foetal bovine serum. The cost of CCS is between one‐fifth and one‐tenth that of foetal bovine serum. Buffering in L‐15 medium allows cell growth and pH maintenance without a CO2 atmosphere. Cells were grown at 37 °C in sealed flasks prior to a membrane‐elution experiment. They were kept below 200 000 cells/mL during exponential growth. Under these growth conditions, they were in steady‐state growth and did not approach overgrowth conditions.
DNA analysis
Cells were collected by centrifugation, washed once in phosphate‐buffered saline (PBS, pH 7.4), and suspended in 70% ethanol. After ethanol treatment in the cold for at least 20 min, they were collected by centrifugation, ethanol was aspirated off, and cells were suspended in PBS containing propidium iodide (50 µg/mL) and RNase A (100 µg/mL). After at least 20 min incubation on ice, cells were analysed in a Becton‐Dickinson FACScan analyser using CellQuest software (Becton Dickinson, San Jose, CA, USA). Further analysis of the data was carried out with WINMdi software (J. Trotter, Scripps Institute, La Jolla, CA, USA). Data were transferred to an Excel spreadsheet program for plotting. Data were smoothed using a running average over three Excel points, these smoothed the data but did not alter the basic result.
Cell counting and cell sizing
Cells were counted and sized using a Coulter Z2 electronic cell counter with a 70‐micron orifice (Coulter Electronics Inc., Miami, FL, USA). Data were collected and analysed with the Z2 AccuComp program from Beckman Coulter version 3.01 (Fullerton, CA, USA). Cells were counted directly in L‐15 medium. Analyses of size distributions were performed using an Excel spreadsheet program. A three‐box running average was used to smooth the data from the Coulter Counter.
Membrane elution
The membrane‐elution apparatus and method have been colloquially referred to as the ‘baby machine’, as it continuously produces newborn cells. This apparatus has been described in detail previously (Thornton et al. 2002; Helmstetter et al. 2003; Eward et al. 2004), but some details will be presented here. A support screen (Millipore, catalogue number YY3014234; Bedford, MA, USA) was secured in a holder with rubber gaskets so that a membrane (Millipore, catalogue number GSWP14250; 142 mm nitrocellulose membrane, 0.22 micron pores) lay directly on the support screen. A Lucite ring confined the liquid to the top of the membrane. Rubber gaskets between the membrane and Lucite ring prevented leakage. The support screen was atop a funnel that could be inserted into a side‐arm flask to allow suction to pull medium through the membrane.
Cells were grown to a concentration of less than 200 000 cells per mL to obtain approximately 60–70 million cells. For example, 600 mL of cells at 100 000 cells per mL gave 60 million cells. All experiments were carried out in a warm room. The membrane holder was in a full‐view incubator, within the warm room, to ensure constant temperature. To start the production of newborn cells, 50 mL of warm (37 °C) PBS with 10 µg/mL concanavalin A was filtered through the membrane. Upon completion of filtration, no residual liquid remained. Warm PBS (100 mL) was filtered through the membrane to remove unbound concanavalin A; again no residual liquid remained. Cells in 300–600 mL of medium were then passed slowly on the membrane with gentle suction over 3–5 min. When 20–30 mL of liquid remained above the membrane, the liquid was poured off so that the cells never dried nor were exposed to air. The membrane apparatus was inverted and filled with fresh medium. Medium from a 4‐L reservoir was pumped through the membrane. After 5–10 min unbound and weakly bound cells had been removed. Cells obtained from this initial flow of medium through the membrane (along with cells in the residual medium) were collectively referred to as the ‘wash‐off’ that was in the order of 10–20% of input cells. This means that over 80% of the initial cells were bound to the membrane. After wash‐off was collected, the membrane was placed over a large funnel, connected by tubing to a peristaltic pump. Fresh medium was pumped into the membrane holder at a rate of 2.5–3.0 mL/min. The pump connected to the bottom of the funnel pumped liquid at 4.0–10.0 mL/min; this prevented pooling of cells in the funnel. In a standard experiment, 84 sterile glass vials (40 mL capacity) in a Pharmacia fraction collector received eluate from the membrane‐elution apparatus. Although the entire apparatus (medium reservoir, pumps, membrane holder and fraction collector) were in a warm room, an incubator box built around the fraction collector maintained a constant temperature. The incubator box contained a thermocouple‐controlled heater with a fan. Thus, even when the warm room door was occasionally opened, there was no change in the temperature of the collected cells.
Cell fractions were collected for 15 min, 35–40 mL per vial. At the end of a collection period (12–18 h), vials were removed to an ice bath for subsequent analysis of cell size and DNA content. Because the last vial contained newborn cells and the previous vials were incubated for different lengths of time until collection to ice, vials contained cells of different ages. After overnight collection, the last vial contained newborn cells, the vial previous to that had cells that had grown for 15 min and cells before that had grown for 30 min, and so on.
RESULTS
DNA distribution following inhibition by thymidine
Thymidine was added to exponentially growing L1210 cells at 4 mm, and cells were incubated for various periods of time. A typical result of thymidine treatment is shown in Fig. 1. Cells were arrested when an S‐phase level of DNA was achieved. By mixing exponentially growing cells with inhibited cells, it was possible to delineate inhibited cells as having more DNA than G1‐phase amounts. The general observation was that on thymidine treatment, cells were arrested with the DNA content consistent for cells early in the S phase of the normal cell cycle.
Figure 1.
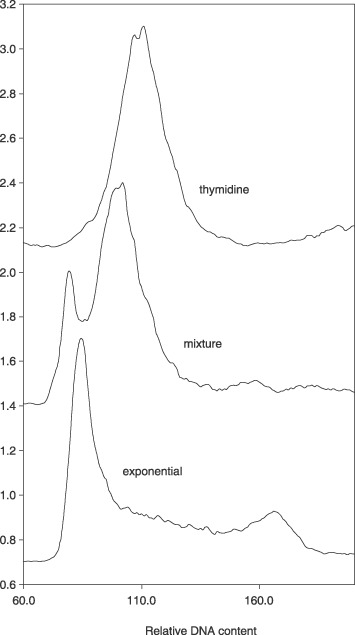
DNA content of thymidine‐inhibited cells compared to DNA distribution of exponential cells. Top line, DNA distribution of thymidine‐inhibited cells (19 h). There is a single peak that can be shown to not be equivalent to G1‐phase DNA content when a mixture of exponential DNA (bottom line) and thymidine‐inhibited cells are analysed (middle line). The thymidine peak is clearly different from the G1‐phase amount of DNA.
Size distribution of thymidine‐treated cells
The size distributions of thymidine‐treated cells and exponentially growing cells are compared in Fig. 2. The former were larger and had a broad cell size distribution, cell size distribution being inconsistent with sizes during the normal S phase of the cell cycle. For comparison, cells produced by membrane elution are compared to exponential cells in Fig. 3. Newborn cells had a narrow size distribution compared to the exponential culture. In addition, it is shown in Fig. 5 that cells produced by elution, without inhibition from the membrane‐elution apparatus, showed that size distribution of newborn cells was consistent with distribution expected from a truly synchronized culture, and that they grew synchronously. As will be discussed in detail below, the criterion of requiring a narrow size distribution for true synchronization indicates that thymidine‐inhibited cells are perforce not synchronized and do not represent cells of a particular age during the normal cell cycle.
Figure 2.
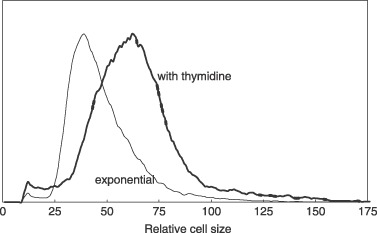
Size distribution of thymidine‐inhibited cells compared to exponential control cells. L1210 cells were treated with thymidine for 19 h. The cell size distribution of inhibited cells was compared to untreated control cells. Inhibited cells had a size distribution clearly larger than the exponential cells, and size distribution was as wide as that of exponential cells. There is no narrowing of size distribution as would be expected for truly synchronized cells.
Figure 3.
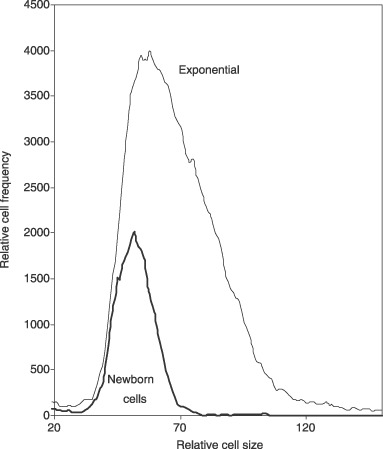
Size distribution of newborn membrane‐eluted cells compared to exponential control cells. Newborn cells were obtained by membrane elution. Cell size distribution of inhibited cells was compared to that of untreated control cells. Newborn cells have a size distribution that is narrow and consistent with the size distribution of the smallest cells in the exponential size distribution.
Figure 5.
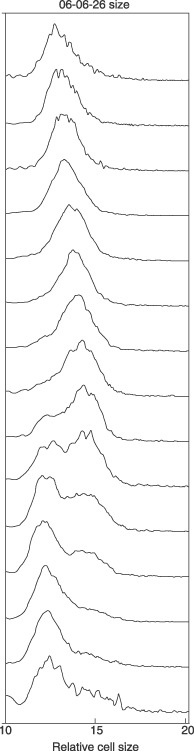
Size distribution of cells at different cell ages using membrane‐elution. Newborn cells were grown for various time periods and sizes were analysed using a Coulter Counter Z2. Notice that cells are born small, grow larger, and then give a bimodal peak at the time of division.
Cell division following release from thymidine inhibition
On release from thymidine block, cells did not exhibit synchronized division but rather exhibited a slow return to smaller cell size distribution of the exponential culture (Fig. 4), and cell size distribution is given at each hour after release from inhibition. As time progressed (from bottom up), there was a slow decrease in cell size. No bimodal distribution was observed indicating synchronized division. This sequence of size change is explained by having the largest cells in the culture divide first. Then, there was continuous removal of larger cells to produce smaller cells providing continuous reduction in cell size. In contrast to growth of thymidine‐inhibited cells, the size change of cells from a valid synchronization method such as membrane‐elution (a selective method) are shown in Fig. 5.
Figure 4.
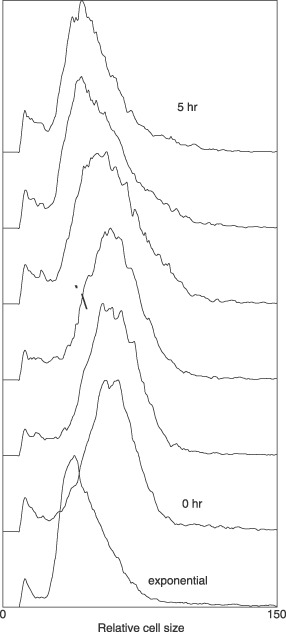
Size distributions of cells on release from thymidine inhibition. Bottom line, size distribution of exponential cells and line immediately above, thymidine‐inhibited cells. Note that as in Fig. 2 cell sizes of inhibited cells are larger and not narrowed as expected. When the thymidine‐inhibited cells were now allowed to grow without thymidine the pattern did not indicate synchronized divisions.
Numerous experiments with thymidine inhibition have not shown any appearance of synchronized division. In addition, use of successive thymidine blocks, in order to have a ‘double‐thymidine block method’, has uniformly provided results similar to that with a single thymidine block. This suggests that even the double‐thymidine method does not synchronize cells.
DISCUSSION
Synchronization with thymidine inhibition
The results reported here indicate that while thymidine inhibits cells, to produce an L1210 cell population with a primarily S‐phase amount of DNA, these cells are not synchronized. Not only is the cell‐size distribution of inhibited cells not narrowed, but also on release from thymidine block the cells do not progress normally through the cell cycle. Most importantly, these thymidine‐inhibited experiments support theoretical predictions that whole‐culture methods cannot synchronize cells (Cooper 1998; Cooper & Shedden 2003; Cooper 2003a, 2004a, 2004c).
There have been debates regarding the relationship of cell size and cell passage through the cell cycle (Cooper 2004a,b; Spellman & Sherlock 2004a,b), but the point made here is irrelevant to precise controls regulating passage through the cell cycle. Irrespective of how cells cycle, it is argued here that there is a criterion that must be satisfied for cells to be called ‘synchronized’, and that is that the starting cells reflect the cell size of cells at a particular cell age. In order for this to occur, cell size distribution must be narrower than size distribution of the starting cells.
Purpose of synchronization
The purpose of synchronization is to produce cells that can be used to measure and comprehend events occurring during the normal, unperturbed cell cycle. A truly synchronized culture is one that mimics the passage of cells through this normal cell cycle. A newborn cell has a number of different properties (e.g. DNA content, cell size, protein composition, internal cellular architecture). As a cell passes through the cycle, each of these must necessarily change in order to produce a dividing cell at the end of the cycle. At a minimum, each component of the cell must double so that the pre‐division cell has twice as much of everything as the newborn cell. At each time point during the cycle, a cell has a particular constellation of properties that are on the trajectory from newborn to dividing cell. A synchronized culture should, at each point during passage through the cycle, have cells with the specific properties associated with each cell cycle age during the growth of an unperturbed cell.
If we consider two properties of a growing cell, for example cell size and DNA content, a newborn cell of age 0.0 has a size of 1.0 and a DNA content of the G1‐phase. Size increases continuously during the cell cycle so that at division (age 1.0), the parent cell size is 2.0. DNA also increases during the cell cycle so that dividing cells have DNA content that reflects the G2‐phase amount of DNA. Just prior to division, for example at age 0.8, a cell may have a G2‐phase amount of DNA, and the cell size could be approximately size 1.8. If a synchronized culture truly reflected the normal cell cycle, one would expect to find that cells arrested with an S‐phase amount of DNA would have a relatively narrow cell size distribution of about size 1.5. With biological and statistical variation considered, the size distribution could vary from 1.4 to 1.6, or even be as wide as 1.3–1.7.
To summarize this analysis, size distribution of cells at any time in a truly synchronized culture should be narrower than the size distribution of the original culture. If size distribution is as wide or wider than the exponential cell size distribution, such a cell size distribution would indicate that the cells were not synchronized and did not reflect the normal size at any point during the division cycle. Such unsynchronized cells are presumably arrested with only one particular property common to all cells. In the case of thymidine arrest, this property is an S‐phase amount of DNA. Other properties would be reflective of cells of all ages, as the size distribution in this case would include cells of all different cell cycle sizes.
Criteria for synchronization
Several criteria have been proposed for recognizing a truly synchronized culture (Cooper & Shedden 2003; Cooper 2004a). From the list of 11 proposed criteria, those that are relevant to experimental analysis presented here are:
-
1
If newborn cells are produced by the synchronization method, there should be a minimal increase in cell number for a period of time covering a significant fraction of the interdivision time.
-
2
DNA distribution of cells should be narrow in synchronized cells and these distributions should then reflect the movement of cells through the division cycle. Thus, newborn cells should all have a G1‐phase amount of DNA. The DNA content should then move through the S‐phase content to a period of time when cells have only G2‐phase DNA content, followed by a return to an essentially pure G1‐phase DNA content.
-
3
The size distribution of newly synchronized cells should be narrower than the size distribution of the original population and cell size should increase as the cells move through the cell cycle. During the period of cell division, there should be a bimodal distribution of cell sizes.
A more general consideration of the problem of thymidine or whole‐culture synchronization is that alignment of cells so that all cells have a particular property in common (e.g. all cells having an S‐phase DNA content or a G2‐phase DNA content) does not mean that the cells are synchronized. Synchronized divisions are the sine qua non of synchrony.
Application of these criteria to thymidine treatment experiments described here indicates that thymidine‐inhibited cells are not synchronized. The cell size distribution is not narrow, the cells do not move through the division cycle with a DNA content pattern indicative of normal cell cycle passage, and there is no indication that cells divide synchronously.
Published work on synchronization following thymidine treatment
Literature on the use of high concentrations of thymidine to induce synchronization is enormous; even the subset dealing only with the cell cycle is extremely large. We cannot re‐analyse each and every paper that states the use of thymidine to synchronize cells, but a general conclusion can be derived from those instances in which cells have been analysed after release from thymidine block. The overwhelming result is that, after thymidine is removed, cells do not fit the criteria of a synchronized culture. For example, Whitfield et al. (2002) produced cells that, following release from thymidine treatment, did not have a pattern of DNA that indicated that cells were synchronized. It is true that the cells produced by Whitfield et al. were called synchronized, but a look at the data supporting this claim indicates that the cells are not synchronized. Rather, they start out with a DNA content that is not repeated in any subsequent cycles, they did not display the correct patterns of synchrony expected for DNA content, and no indication of cell size is presented. Analysis of micro‐array data on cells treated with a double‐thymidine block have also indicated that this method does not synchronize cells (Shedden & Cooper 2002).
Generalization of results to other cell lines and growth conditions
One critique of extension of the results presented here to other cells and growth conditions might be that our results are merely related to particular cells and growth conditions that we have used, in other words, L1210 cells grown in L‐15 medium with cosmic calf serum and glucose (cells and conditions used here). However, this argument ignores the theoretical generalization that has led to experiments presented here. Theory predicts the results obtained here – that thymidine block does not synchronize cells – and the theory is independent of cell type or cell line (Cooper 1998, 2003a, 2004a,c; Cooper & Shedden 2003).
Rather than placing the burden of proof on the proposal that all cell lines cannot be synchronized by whole‐culture methods, we now shift the burden of proof to those who propose using such whole‐culture methods, including thymidine block inhibition, as synchronization methods. We reject the argument proposing that perhaps ‘just around the corner’ there exists a cell line or a cell situation that can be synchronized by such whole‐culture methods, or as in the study here, by thymidine block. Theory predicts (Cooper 2003a) that whole‐culture synchronization cannot work and the experiments presented here support this theory.
The simple message regarding thymidine block methodology
The massage of this paper is that thymidine block does not synchronize cells, our results according with theoretical considerations. It is important not to confuse this conclusion with discussions concerning the way the cell passes through the cell cycle. The single question answered here is whether thymidine block works.
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation (grant MCB–0323346) and (in part) by the National Institutes of Health (University of Michigan's Cancer Center, support grant 5 P30 CA46592). K.C. and S.R. are associated with the Undergraduate Research Opportunity Program of the University of Michigan, which also supported this research. M. Gonzalez‐Hernandez was very helpful with some of the experiments. Alexandra Cooper was invaluable as an editor of this paper.
REFERENCES
- Cooper S (1991) Bacterial Growth and Division. San Diego, CA: Academic Press. [Google Scholar]
- Cooper S (1998) Mammalian cells are not synchronized in G1‐phase by starvation or inhibition: considerations of the fundamental concept of G1‐phase synchronization. Cell Prolif. 31, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S (2002) Reappraisal of G1‐phase arrest and synchronization by lovastatin. Cell Biol. Int. 26, 715–727. [DOI] [PubMed] [Google Scholar]
- Cooper S (2003a) Rethinking synchronization of mammalian cells for cell‐cycle analysis. Cell. Mol. Life Sci. 6, 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S (2003b) Reappraisal of serum starvation, the restriction point, G0, and G1‐phase arrest points. FASEB J. 17, 333–340. [DOI] [PubMed] [Google Scholar]
- Cooper S (2004a) Is Whole‐culture synchronization biology's ‘perpetual motion machine’? Trends Biotechnol. 26, 266–269. [DOI] [PubMed] [Google Scholar]
- Cooper S (2004b) Whole‐culture synchronization can not, and does not, synchronize cells. Trends Biotechnol. 22, 274–276. [DOI] [PubMed] [Google Scholar]
- Cooper S (2004c) Rejoinder: whole‐culture synchronization cannot, and does not, synchronize cells. Trends Biotechnol. 22, 274–276. [DOI] [PubMed] [Google Scholar]
- Cooper S (2006) Bacterial and eukaryotic checkpoints and restriction points. Bioessays 28, 1035–1039. [DOI] [PubMed] [Google Scholar]
- Cooper S, Shedden K (2003) Microarray analysis of gene expression during the cell cycle. Cell Chromosome 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Iyer G, Tarquini M, Bissett P (2006) Nocodazole does not synchronize cells: implications for cell‐cycle control and whole‐culture synchronization. Cell Tissue Res. 324, 237–242. [DOI] [PubMed] [Google Scholar]
- Eward KL, Van Ert MN, Thornton M, Helmstetter CE (2004) Cyclin mRNA stability does not vary during the cell cycle. Cell Cycle 3, 1057–1061. [PubMed] [Google Scholar]
- Gong J, Traganos F, Darzynkiewicz Z. (1995) Growth imbalance and altered expression of cyclins B1, A, E, and D3 in MOLT‐4 cells synchronized in the cell cycle by inhibitors of DNA replication. Cell Growth Differ. 6, 1485–1493. [PubMed] [Google Scholar]
- Harper JV (2005) Synchronization of cell populations in G1/S and G2/M phases of the cell cycle. Methods Mol. Biol. 296, 157–166. [DOI] [PubMed] [Google Scholar]
- Helmstetter CE, Thornton M, Romero A, Eward KL (2003) Synchrony in human, mouse and bacterial cell cultures – a comparison. Cell Cycle 2, 42–45. [DOI] [PubMed] [Google Scholar]
- Jansen‐Durr P, Meichle A, Steiner P, Pagano M, Finke K, Botz J, Wessbecher J, Draetta G, Eilers M (1993) Differential modulation of cyclin gene expression by MYC. Proc. Natl. Acad. Sci. USA 90, 3685–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyomarsi K, Sandoval L, Band V, Pardee AB (1991) Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 51, 3602–3609. [PubMed] [Google Scholar]
- Kung AL, Sherwood SW, Schimke RT (1990) Cell line‐specific differences in the control of cell cycle progression in the absence of mitosis. Proc. Natl. Acad. Sci. USA 87, 9553–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliensiek SJ, Schell K, Howard E, Nealey P, Murphy CJ (2006) Cell sorting but not serum starvation is effective for SV40 human corneal epithelial cell cycle synchronization. Exp. Eye Res. 83, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow JW, Glendening CL, Livingston DM, Decaprio JA (1993) Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol. Cell. Biol. 13, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang B, Lan Z, Meadows J, Pan H, Fukasawa K, Li W, Dai W (1998) Human Bub1: a putative spindle checkpoint kinase closely linked to cell proliferation. Cell Growth Differ. 9, 877–885. [PubMed] [Google Scholar]
- Shedden K, Cooper S (2002) Analysis of cell‐cycle‐specific gene expression in human cells as determined by microarrays and double‐thymidine block synchronization. Proc. Natl. Acad. Sci. USA 99, 4379–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G (2004a) Final words: cell age and cell cycle are unlinked. Trends Biotechnol. 22, 277–278. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G (2004b) Reply: whole‐culture synchronization – effective tools for cell cycle studies. Trends Biotechnol. 22, 270–273. [DOI] [PubMed] [Google Scholar]
- Thornton M, Eward KL, Helmstetter CE (2002) Production of minimally disturbed synchronous cultures of hematopoietic cells. Biotechniques 32, 1098–1105. [DOI] [PubMed] [Google Scholar]
- Whitfield M, Sherlock G, Saldanha A, Murray JI, Ball CA, Alexnder KE, Matese JC, Perou CM, Hurt MM, Brown PO, Botstein D (2002) Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]