

Abstract
Objectives: This study aimed to investigate molecular and cellular changes induced in human bone marrow mesenchymal stem cells (hMSCs) after treatment with microtubule‐interacting agents and to estimate damage to the bone marrow microenvironment caused by chemotherapy.
Materials and methods: Using an in vitro hMSC culture system and biochemical and morphological approaches, we studied the effect of nocodazole and taxol® on microtubule and nuclear envelope organization, tubulin and p53 synthesis, cell cycle progression and proliferation and death of hMSCs isolated from healthy donors.
Results and conclusions: Both nocodazole and taxol reduced hMSC proliferation and induced changes in the microtubular network and nuclear envelope morphology and organization. However, they exhibited only a moderate effect on cell death and partial arrest of hMSCs at G2 but not at M phase of the cell cycle. Both agents induced expression of p53, exclusively localized in abnormally shaped nuclei, while taxol, but not nocodazole, increased synthesis of β‐tubulin isoforms. Cell growth rates and microtubule and nuclear envelope organization gradually normalized after transfer, in drug‐free medium. Our data indicate that microtubule‐interacting drugs reversibly inhibit proliferation of hMSCs; additionally, their cytotoxic action and effect on microtubule and nuclear envelope organization are moderate and reversible. We conclude that alterations in human bone marrow cells of patients under taxol chemotherapy are transient and reversible.
Introduction
Taxol, a plant alkaloid that binds to β‐tubulin and acts by stabilizing microtubules rendering them rigid and less dynamic (1, 2), is one of the most effective chemotherapeutic drugs utilised to date, commonly used clinically in treatment of human carcinomas (3). Pharmacological effects of taxol vary, depending on the cell line, relative amounts of different β‐tubulin isotypes, dose and treatment scheme. At low (nanomolar) concentrations, taxol induces mitotic arrest (4, 5), inhibits protein prenylation (6), affects nuclear envelope organization and nucleo‐cytoplasmic transport through the nuclear pore complex (5) and leads, eventually, to apoptosis (4, 5, 6). At higher (micromolar) concentrations, in addition to mitotic arrest and nuclear envelope defects (7), taxol promotes microtubule polymerization and increases microtubule‐polymer mass (8). Moreover, it exerts other effects, which occur almost immediately after treatment – it promotes synthesis and release of cytokines (9, 10, 11) and induces ‘early response’ genes, including those that encode tumour suppressors (12). Although normal, non‐transformed cells have been reported to be more resistant to taxol activity than tumour cells (13, 14), the agent may exert its anti‐mitotic effect in renewing and dividing human cells, acting on their microtubule network. Nocodazole is another type of microtubule‐targeted agent that acts as a microtubule‐destabilizer. Although its action is opposite to that of taxol, it is also very effective in disturbing microtubule dynamics and therefore, arresting cell‐cycle progression at mitosis. Several drugs, including vincristine and colcemid, act in a similar way to nocodazole, interfering with microtubules and causing arrest in the G2/M phase of the cell cycle. Unlike nocodazole, however, the effects of these agents may not be completely or readily reversed.
Human mesenchymal stem cells (hMSC) are multipotent adult cells present in various tissues, including the bone marrow, that may differentiate towards a variety of cell lineages, including to haematopoiesis‐supporting stromal cells (15, 16, 17, 18, 19, 20). Although they represent only 0.01–0.001% of total bone marrow cells, they can be separated easily, as they adhere to plastic and are proliferate in culture (21). The effect of chemotherapy and/or radiotherapy on bone marrow hMSCs has not been extensively studied. There are studies suggesting quantitative and qualitative damage to bone marrow hMSCs after the conditioning regimen of bone marrow transplantation (15, 22, 23, 24), and data demonstrating irreversible damage in culture‐expanded hMSCs after taxol treatment (25). However, other studies have shown resistance of bone marrow hMSCs to chemotherapeutic agents (25, 26).
In the present report, we have investigated pharmacological alterations induced by nocodazole and various concentrations of taxol on bone marrow hMSCs isolated from healthy donors. Specifically, we have studied the effect of these drugs on cell cycle progression, proliferation and death, on microtubule and nuclear envelope organization, p53 synthesis and localization, and on β‐tubulin isotype synthesis. We show moderate and reversible effects of microtubule‐acting drugs on hMSCs, which are suggestive of relative safety for the bone marrow microenvironment of chemotherapeutically treated cancer patients.
Materials and methods
Isolation and culture of hMSCs
hMSCs were grown from posterior iliac crest aspirates taken from 10 healthy donors after informed consent, as previously described (27). In brief, bone marrow mononuclear cells isolated using density gradient (Histopaque‐1077; Sigma, St. Louis, MO, USA) were cultured in MSC medium containing Dulbecco's modified Eagle's medium–low glucose (DMEM‐LG; Gibco Invitrogen, Paisley, Scotland), 10% foetal calf serum (FCS; Hyclone, Logan, UT, USA), and 100 IU/ml penicillin‐streptomycin (Gibco) at a concentration of 2 × 105 cells/cm2 in 25‐cm2 culture flasks in a 37 °C/5% CO2 humidified atmosphere. One to three days after seeding, floating cells were removed and medium was replaced with fresh MSC medium. Thereafter, attached cells were fed with fresh medium every 3–4 days. Cells were passaged when 70–90% confluence was reached, using 0.25% trypsin–1 mm EDTA (Gibco). hMSCs were identified by their morphological and immunophenotypic characteristics and their potential to differentiate towards three different pathways, adipocytes, osteocytes and chondrocytes.
Immunophenotypic characteristics of hMSCs
Trypsinized MSCs from passage 2 (P2) were characterized by flow cytometry using anti‐human monoclonal antibodies against (anti‐) CD29 (4B4; Cyto‐Stat, Beckman‐Coulter, Miami, FL, USA), anti‐CD44 (J173; Immunotech/Coulter, Miami, FL, USA), anti‐CD73 (AD2; Becton Dickinson–Pharmingen, San Diego, CA, USA), anti‐CDw90 (F15.42; Immunotech/Coulter), anti‐CD105 (SN6; Caltag, Burlingame, CA, USA), anti‐CD146 (P1H12; Becton Dickinson–Pharmingen), anti‐CD45 (IMMU19.2; Immunotech/Coulter), anti‐CD14 (RMO52; Immunotech/Coulter) and anti‐CD34 (QBend10; Beckman‐Coulter). In brief, 0.5 × 106 cells were washed in phosphate‐buffered saline (PBS)/0.5% FCS, pre‐incubated with human γ‐globulins to minimize non‐specific binding, followed by specific monoclonal antibody incubation, according to the manufacturer's instructions. Cells were then fixed in 2% paraformaldehyde (Sigma). Data were processed using an Epics Elite flow cytometer (Coulter, Miami, FL, USA).
Differentiation potential of hMSCs
P2 hMSCs were induced to differentiate in vitro in specific media (given in details below) for 3 weeks. Specifically, adipogenic differentiation was induced using MSC medium supplemented with 10% FCS (20% final concentration), 0.5 mm 1‐methyl‐3‐butylisoxanthine, 1 µm dexamethasone, 0.2 µm indomethacin and 10 µg/ml insulin. Differentiation was assessed by Oil red O staining (Gronthos et al. 1998). Osteogenic differentiation was induced using MSC medium supplemented with 0.1 µm dexamethasone, 0.15 mm ascorbate‐2‐phosphate and 3 mm NaH2PO4. Differentiation was assessed by using alkaline phosphatase (ALP) and von Kossa stains (19). For chondrogenic induction, cells were pelleted in 15‐ml tubes and cultured in DMEM–High Glucose (Gibco), supplemented with 6.25 µg/ml insulin, 6.25 µg/ml transferrin, 1.33 µg/ml linoleic acid, 1.25 mg/ml bovine serum albumin, 1 mm sodium pyruvate, 0.17 mm ascorbate‐2‐phosphate, 0.1 µm dexamethasone, 0.35 mm l‐proline, 6.25 ng/ml selenous acid, and 0.01 µg/ml transforming growth factor‐β3 (R&D Systems, Minneapolis, MN, USA) (27). Differentiation was identified by using alcian blue and Masson's trichrome stains (19). All reagents were purchased from Sigma unless otherwise indicated.
For further identification of differentiation potential of hMSCs, total RNA was isolated from differentiated MSCs (RNeasy Mini kit; Qiagen GmbH, Hilden, Germany), reverse transcribed (SUPERSCRIPT II; Gibco) and amplified by polymerase chain reaction (PCR) for evaluation of specific, differentiation‐associated gene expression, using previously reported primer sequences (27). Adipose fatty acid‐binding protein (aP2) and peroxisome proliferator‐activated receptor‐gamma (PPARγ) for adipocytes, ALP and runt‐related transcription factor 2 (RUNX2) for osteocytes, and collagen type II (COL2A1) and aggrecan (AGC1) for chondrocytes. Specific PCR conditions were 94 °C for 30 s, melting temperature (Tm) for 30 s, and 72 °C for 40 s. Tm was 60 °C for aP2, PPARγ, ALP, RUNX2, and β2m, and 57 °C for COL2A1 and AGC1. All PCRs were stopped at 29 cycles, except β2m reactions, which were stopped at 26 cycles.
Cell treatment
Upon reaching 30–40% confluence, cells from P2 grown either on culture dishes or on coverslips were incubated with 325 nm nocodazole or paclitaxel (Taxol®, Bristol‐Myers Squibb, New York, NY, USA) 1 nm, 10 nm or 500 nm diluted in culture medium, at 37 °C, for 24, 48 or 72 h, washed with fresh medium and incubated in drug‐free medium at 37 °C for up to 6 days.
Cell proliferation assay
Cell cultures (2500 cells/well) were expanded for 3 days on 24‐well plates and after the various treatments cell proliferation was determined using the MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide; thiazolyl blue) reaction, as specified by the supplier (Sigma). Percentages of viable cells for each treatment were determined by measurement of MTT absorbance (630 nm), relative to the initial cell population before any treatment. All cell proliferation data provided are means of eight measurements from three independent experiments. For determination of IC50, cell proliferation was measured in untreated hMSCs and in cultures incubated with a variety of concentrations (1, 5, 10, 50, 100 and 500 nm) of taxol for 3 days, then data were analysed using Prism software of GraphPad Software Inc. (San Diego, CA, USA) by sigmoidal fitting.
Numbers of live and dead cells
Numbers of live and dead cells were determined by incubating a fraction of the total cells (adherent and detached) or only adherent cells, with trypan blue for 5 min, placing cell suspensions in a Neubauer haemocytometer and counting numbers of figures that either excluded (alive) or incorporated (dead), the dye.
Examination of nuclear morphology
Mitotic indices and morphology of interphase nuclei were assessed in cells growing on coverslips, after staining with DNA‐binding dye (DAPI). At least 500 nuclei per sample were scored as mitotic (cells in prophase, metaphase, anaphase or telophase), normally‐shaped, lobulate or multinucleate, using a conventional fluorescence microscope.
Indirect immunofluorescence microscopy
Indirect immunofluorescence was performed as previously described (5). Briefly, cells grown on coverslips were washed with PBS, fixed in 4% formaldehyde for 10 min at room temperature or with methanol for 5 min at – 20 °C and permeabilized with Triton X‐100. Fixed cells were incubated in blocking buffer (PBS, pH 7.4, 0.5% Triton X‐100 and 1% fish skin gelatin) and stained with primary and then with secondary antibodies diluted in blocking buffer for 45 min. Primary antibodies used included: anti‐α‐acetylated tubulin antibody and anti‐β‐, anti‐βI‐, anti‐βII‐, anti‐βIII‐ and anti‐βIV‐tubulin monoclonal antibodies, all obtained from Sigma; anti‐p53 monoclonal antibody purchased from Neomarkers (Fremont, CA, USA), anti‐lamin B polyclonal and anti‐emerin polyclonal antibodies ((5) and references therein); and anti‐HP1α polyclonal, anti‐HP1β and HP1γ rat monoclonal antibodies ((28) and references therein). Appropriate secondary antibodies were used, including goat anti‐rabbit, goat anti‐rat (Sigma) and donkey anti‐mouse (Calbiochem‐Novabiochem GmbH, San Diego, CA, USA) immunoglobulins conjugated to fluorescein isothiocyanate. Chromatin was decorated by the DNA‐binding dye DAPI, or by the DNA/RNA‐binding dye propidium iodide, following RNAse treatment. Specimens were visualized using a Leica SP confocal microscope (Leica Microsystems, Mannheim, Germany).
Cell‐cycle analysis
Cells were trypsinized, washed with PBS and treated in order to stain DNA with the DNA‐Prep Coulter Reagent Kit (Beckman Coulter). DNA flow cytometry was performed in triplicate using a FACScalibur flow cytometer (Beckman Coulter); for each sample, 20 000 events were stored in the list mode file. Cell‐cycle analysis was completed with WinMDI software (Windows Multiple Document Interface for Flow Cytometry). FS area vs. SS width gating was arranged to exclude doublets from the G2–M region. To avoid cell debris contamination due to necrotic cell death; cells were selected by side scatter (SSC) vs. DNA log signals (PMT4 log).
Immunoblotting
Cells resuspended in PBS were sonicated (3 times for 5 s) and their protein contents were determined (Bio‐Rad, Hercules, CA, USA). Equal amounts of protein were electrophoresed on sodium dodecyl sulphate–polyacrylamide gels and then transferred to nitrocellulose membranes (Protran, Schleicher and Schuell, Einbech, Germany). Membranes were incubated in blocking buffer (20 mm Tris‐HCl, pH 7.4, 155 mm NaCl, 0.1% Tween 20 and 1% fish skin gelatin) overnight, at 4 °C. Then, they were immunoblotted with the respective primary antibody diluted in blocking buffer, for 1 h at room temperature, washed three times with blocking buffer and incubated with horseradish peroxidase‐conjugated anti‐mouse immunoglobulin G secondary antibody (Amersham Biosciences, Piscataway, NJ, USA, NA931V, 1 : 10 000). Membranes were washed three times in blocking buffer and three times in washing buffer (20 mm Tris‐HCl, pH 7.4, 155 mm NaCl, 0.1% Tween 20) and signals were visualized by enhanced chemiluminescence (ECL kit, Amersham Pharmacia Biotech, Piscataway, NJ, USA). Antibodies used were anti‐p53 (1 : 200, Neomarkers), anti‐α‐acetylated tubulin (1 : 500), anti‐β‐ (1 : 1000), anti‐βI‐ (1 : 500), anti‐βII‐ (1 : 500), anti‐βIII‐ (1 : 500) and anti‐βIV‐ (1 : 500) tubulin monoclonal antibodies, all obtained from Sigma. For semiquantitative analysis of tubulin isotypes, band intensities were quantified by PC‐based Image Analysis (Image Analysis Inc., Columbia, KY, USA). For purposes of normalization, blots were also stained with monoclonal anti‐actin antibody (sc1616, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1 : 1000 dilution.
Results
Identification of hMSCs
MSCs at P2 were used in all experiments. Cells displayed characteristic spindle‐like morphology, while immunophenotypic analysis demonstrated them to be a homogenous population, positive for CD73, CDw90, CD146, CD105, CD29 and CD44, and negative for CD45, CD14 and CD34 surface antigens. Furthermore, these P2 hMSCs were able to differentiate towards adipogenic, osteogenic and chondrogenic lineages, as shown by their culture under specific conditions and respective cytochemical stains and differentiation‐specific gene expression (Fig. 1).
Figure 1.
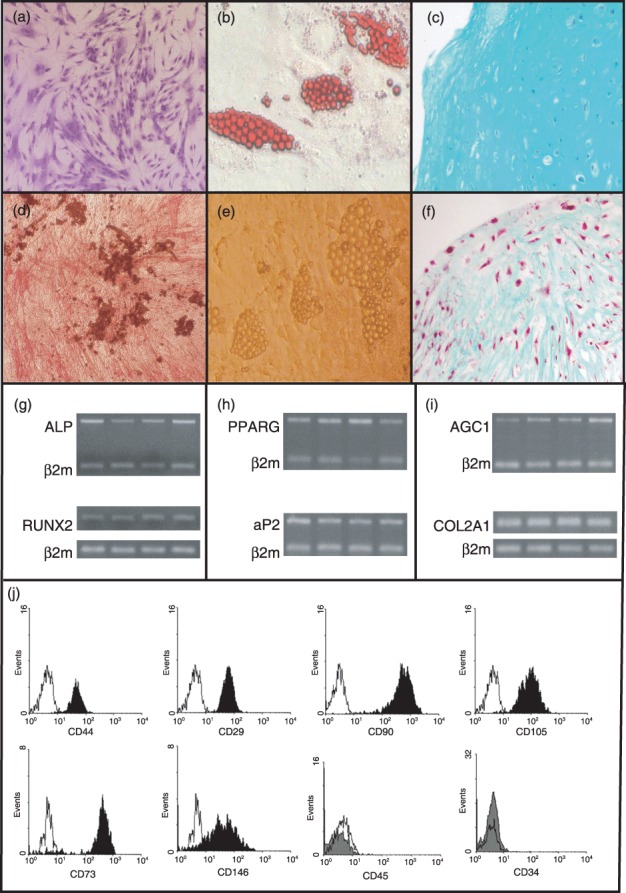
Differentiation potential and immunophenotypic characteristics of human mesenchymal stem cells (hMSCs). Culture expanded hMSCs from P2 stained with Giemsa, exhibiting characteristic spindle‐shaped morphology (a). Differentiated hMSCs from P2 towards osteogenic (d), adipogenic (b, e), and chondrogenic (c, f) lineages. Cell differentiation was identified with alkaline phosphatase (ALP)/Von Kossa (d), oil red O (b), Masson's (c) and alcian blue (f ) staining. Specific gene mRNA expression of P2 hMSCs upon differentiation towards the osteogenic (g), adipogenic (h), and chondrogenic (i) lineages. Lower panel ( j) shows representative plots from flow cytometric analysis of hMSCs at P2 stained with surface monoclonal antibodies. Black histogram bars indicate positive markers, and grey filled histogram bars depict the negative markers, in comparison to isotype‐matched controls (open histograms). Cells were also negative for CD14 (data not shown).
Since β‐tubulin and the nuclear envelope are main targets of taxol treatment, we examined by expression and localization of tubulin isotypes, lamin B, emerin and HP1 proteins in culture–expanded cells, by immunofluorescence. As shown in Fig. 2, β‐tubulin isotypes and acetylated α‐tubulin, as well as proteins of the nuclear lamina, inner nuclear membrane and heterochromatin, were expressed in a typical pattern for hMSCs. No differences were found in expression of these proteins from P2 up to P10 cells, when tested.
Figure 2.
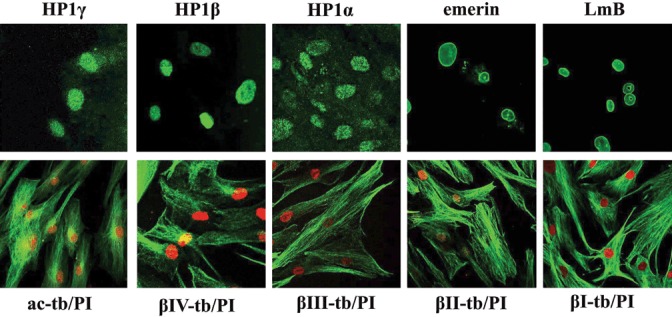
Expression and localization of tubulin isotypes, proteins of the nuclear envelope and heterochromatin. Immunofluorescence pattern of human mesenchymal stem cells (hMSC) stained with antibodies (green) for lamin B (LmB), emerin, heterochromatin 1 proteins (HP1α, HP1β, HP1γ), β‐tubulin isotypes (βI‐tb, βII‐tb, βIII‐tb, βIV‐tb) and acetylated α‐tubulin (ac‐tb). Nuclei (red) were stained with propidium iodide (PI).
Differential effects of taxol and nocodazole on cell proliferation and cell death
To evaluate cytotostatic and cytotoxic effects of taxol and nocodazole, we examined cell proliferation and cell death. Cell proliferation curves are shown in Fig. 3. The initial hMSC population, originated from cells cultured for 3 days, expanded with similar growth rates at 6 days and reached almost a plateau after 9 days in culture. Presence of taxol and nocodazole in media at concentrations 1–500 nm and 325 nm, respectively, inhibited cell proliferation; no significant increase in initial cell population was noted (Fig. 3, 3 days). IC50 calculated for taxol was 10 nm. When the cells were released from drug effects and cultured under normal conditions, cell proliferation was shown to depend on type of drug and concentration used. Cells that had been exposed to 10 nm or 500 nm taxol and thereafter cultured under normal conditions for 3 days were shown to be non‐proliferative (Fig. 3, 6 days), whereas cells that had been treated with 1 nm taxol recovered immediately and displayed growth rates similar to those of normal cultures (Fig. 3, 6 and 9 days). In cell cultures treated with nocodazole, viable cell populations remained unchanged for the first 3 days, then increased substantially during the next 3 days, indicating that cells had recovered from nocodazole treatment (Fig. 3, 6 and 9 days). To better characterize the effect of drugs on cell proliferation, we determined cell viability. Both adherent and floating cells were collected following treatment with the drugs for 3 days (3 days) and recovery from drugs for another 3 days (3 + 3 days), and stained with trypan blue to differentiate live from dead cells (Table 1). We observed a moderate increase in dead cells only in cultures treated with taxol at 10 or 500 nm for 3 days. Measurement of dead cells in the adherent cell population in cultures treated with taxol at 10 or 500 nm for 3 days showed that this cell population contained 47% and 65%, respectively, of total dead cells found both from adherent and floating cells. Therefore, dead cells in the adherent population may contribute, at least in part, to decrease in number of viable cells, shown 3 days after release of cells from taxol at 10 or 500 nm (Fig. 3, 6 days).
Figure 3.
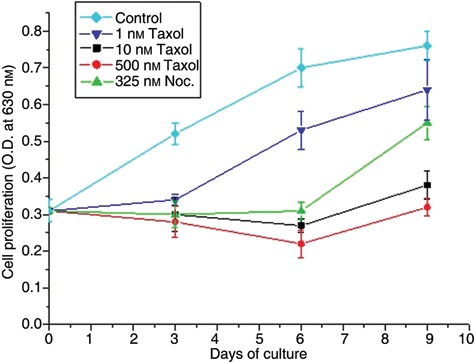
Effect of taxol and nocodazole on human mesenchymal stem cell (hMSC) proliferation. Proliferation curves of untreated (control) and hMSCs treated with 1, 10 and 500 nm taxol and 325 nm nocodazole (Noc.). At day 3, cells were transferred to drug‐free medium and cultured under normal conditions for 3 days (day 6) and 6 days (day 9). Cell proliferation was determined by measurement of MTT absorbance at 630 nm.
Table 1.
Cytotoxicity of taxol and nocodazole in human mesenchymal stem cells
| Drug | Concentration (nm) | Time (days) | Trypan blue‐positive cells (%) |
|---|---|---|---|
| No drug | 16.05 ± 2.19 | ||
| Taxol | 10 | 3 | 30.7 ± 1.31 |
| Taxol | 500 | 3 | 40.3 ± 1.84 |
| Nocodazole | 325 | 3 | 16.8 ± 1.41 |
| Taxol | 10 | 3 + 3 | 10.3 ± 0.23 |
| Taxol | 500 | 3 + 3 | 9.2 ± 0.85 |
| Nocodazole | 325 | 3 + 3 | 8.6 ± 1.20 |
Cells were cultured under normal conditions (No drug) or treated for 3 days with microtubule‐interacting drugs, then cultured without drugs for 3 days (3 + 3). Results are expressed as percentage of total cells. Values are means ± standard error of three measurements in three separate experiments.
Taxol and nocodazole alter the microtubule network, nuclear morphology and nuclear envelope organization
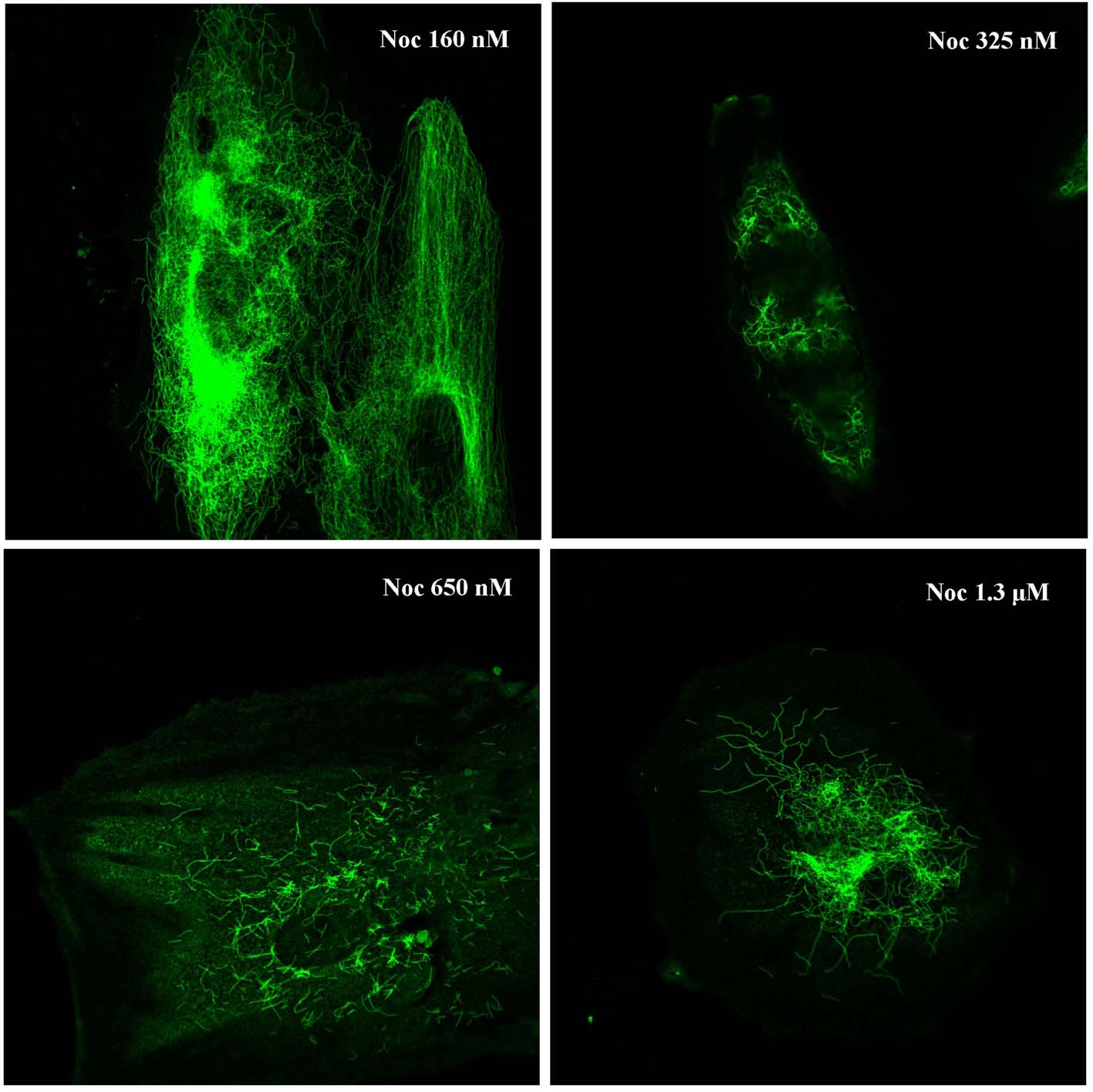
Cells treated with taxol or nocodazole usually have stabilized or depolymerized interphase microtubules, respectively. Typical images of the cells treated with either low (10 nm) or high (500 nm) taxol concentration or with nocodazole (325 nm) are presented in Fig. 4a (panel 3d). Both drugs, at all concentrations and time points tested, induced changes in organization of microtubules. After taxol treatment, we showed destabilization and aggregation of microtubules, which were no longer distributed uniformly in the cytoplasm but were concentrated at different sites, frequently at the cell periphery. This phenotype, although apparent at low concentrations (10 nm) of taxol traetment, became more pronounced when cells were treated with higher concentrations (500 nM; Fig. 4a, 3d–10 µm; not shown) of the drug. In cells treated for up to 3 days with 325 nm nocodazole, usual concentrations, microtubule depolymerization and cell‐cycle arrest at metaphase, disrupted microtubule networks were found. Specifically, microtubules were only partially depolymerized or fragmented and short components of polymer were found either dispersed or aggregated (Fig. 4a, 3d). A similar pattern was observed when different concentrations of nocodazole, ranged from 160 nm to 1.3 µm, were used (Fig. S1, Supporting Information).
Figure 4.
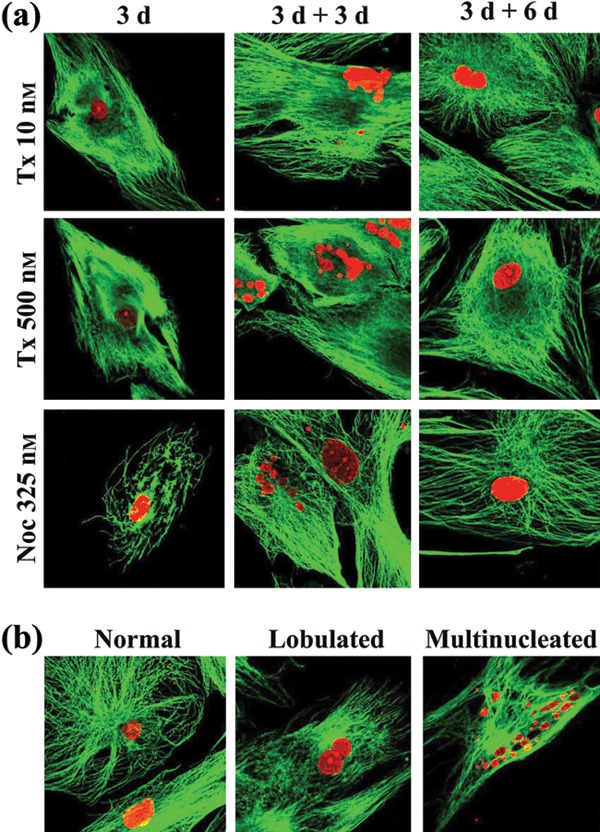
Effect of taxol and nocodazole on microtubule network and nuclear morphology. (a) Taxol‐ and nocodazole‐treated human mesenchymal stem cells (hMSC) for 3 days (3 d) then grown in drug‐free medium for 3 (3 d + 3 d) and 6 (3 d + 6 d) days, were stained with anti‐tubulin antibodies (microtubules, green) and propidium iodide (nuclei, red). (b) Representative images of multinucleate hMSCs and cells with normal‐looking and with lobulate nuclei (red).
Deformation and structural disorganization of nuclear envelopes are phenotypic alterations commonly observed in cancer cells treated with taxol. Thus, we examined nuclear morphology of our cells by immunofluorescence, after treatment with taxol or nocodazole. Many multinucleate cells and cells with lobulate nuclei were observed. Typical patterns of such phenotypes are presented in Fig. 4b. Although the microtubule network had been reorganized in cells with lobulate or micronucleate phenotype, it seems unlikely that this was the primary cause for altered nuclear shape, as intact or aggregated microtubules do not exert any tension at sites of nuclear deformation. We next determined the percentage of lobulate and multinucleate cells in cultures, in the presence or absence of taxol or nocodazole (Table 2, row 3d). Normally growing hMSC cultures had approximately 9% of cells with lobulate nuclei but no cells with the multinucleate phenotype. Percentages of cells with lobulate nuclei increased after treatment with either taxol or nocodazole. However the percentages of multinucleated cells that appeared as a result of taxol treatment, were significantly higher than those observed after nocodazole treatment.
Table 2.
Nuclear shape and nuclear envelope gaps in human mesenchymal stem cells growing under normal conditions (No drug) or treated for 3 days (3 d) with microtubule‐interacting drugs and then cultured without drugs for 3 (3 d + 3 d) and 6 (3 d + 6 d) days
| Treatment | Percentage of cells with nuclear shape | |||||
|---|---|---|---|---|---|---|
| Normal appearance | Lobulate | Multinucleate | ||||
| Total | Gaps | Total | Gaps | Total | Gaps | |
| No drug | 90.8 | 0.3 | 9.2 | 1.3 | 0.0 | |
| Taxol 10 nm | ||||||
| 3 d | 56.4 | 0.2 | 22.2 | 7.7 | 21.4 | 8.4 |
| 3 d + 3 d | 51 | 0.0 | 26.8 | 1.2 | 22.2 | 2.9 |
| 3 d + 6 d | 75 | 0.0 | 18.1 | 0.3 | 6.9 | 0.0 |
| Taxol 500 nm | ||||||
| 3 d | 68.7 | 0.0 | 16.6 | 2.1 | 14.7 | 6.9 |
| 3 d + 3 d | 57.8 | 0.0 | 21.6 | 0.3 | 20.6 | 5.9 |
| 3 d + 6 d | 75.1 | 0.0 | 13.4 | 0.3 | 11.5 | 3.3 |
| Nocodazole | ||||||
| 3 d | 73.5 | 0.3 | 21.2 | 1.5 | 5.3 | 1.3 |
| 3 d + 3 d | 81.3 | 0.0 | 18.0 | 0.7 | 0.7 | 0.0 |
| 3 d + 6 d | 86.0 | 0.0 | 13.2 | 0.3 | 0.8 | 0.3 |
Cells were immunostained for lamin B and nuclear envelope continuity was examined in more than 500 cells using a laser‐scanning confocal microscope. The number of cells with normal‐looking or abnormal‐looking (lobulate and multinucleate) nuclear phenotype (Total) with gaps (Gaps) are expressed as percentage of total interphase cells. Averages of two independent experiments are provided here, totalling at least 1000 cells for each time point.
Proceeding further, we determined the number of cells with normal‐ and abnormal appearing (lobulate and multinucleate) nuclei, that had defects in the nuclear lamina. As shown in Table 2, in cells whether treated with taxol or nocodazole or untreated cells with normal‐shaped nuclei, we found no cells nor counted minimal percentages of cells with nuclear lamina gaps. In contrast, cells with abnormally shaped nuclei presented gaps in the nuclear lamina. Percentages of cells with disrupted nuclear envelopes varied between treatments but did not exceed 31% of cells with abnormal nuclei. Taxol treatment was the most effective for production of lamina gaps, whereas percentages of abnormally‐shaped nuclei with gaps, during nocodazole treatment did not exceeded those found in normally grown cell cultures (Table 2).
Taxol‐ and nocodazole‐induced expression of p53 and partial cell arrest at G2 phase of the cell cycle
We examined expression and localization of p53 in cells after treatment with the drugs. Western blot experiments showed that expression of p53 was very low, almost undetectable, in untreated hMSCs (Fig. 5a). However, the p53 levels increased proportionally with time of cell exposure to taxol 10 nm or nocodazole, whereas in cells treated with taxol 500 nm, p53 levels increased and remained unchanged during drug exposure. We next examined localization of p53 by immunofluorescence. Figure 5b shows the pattern of lamin B, p53, and merging of the two in untreated cells, and in those treated for 72 h with nocodazole and taxol at 10 nm and 500 nm, respectively. Localization patterns shown in Fig. 5b are the same with those obtained when cells were incubated for 24 or 48 h and, therefore, are not shown here. p53 was not detected in untreated hMSCs, whereas in treated cells was it was consistently and exclusively localized in abnormally‐shaped nuclei, mainly in those of multinucleate cells (Fig. 5b).
Figure 5.
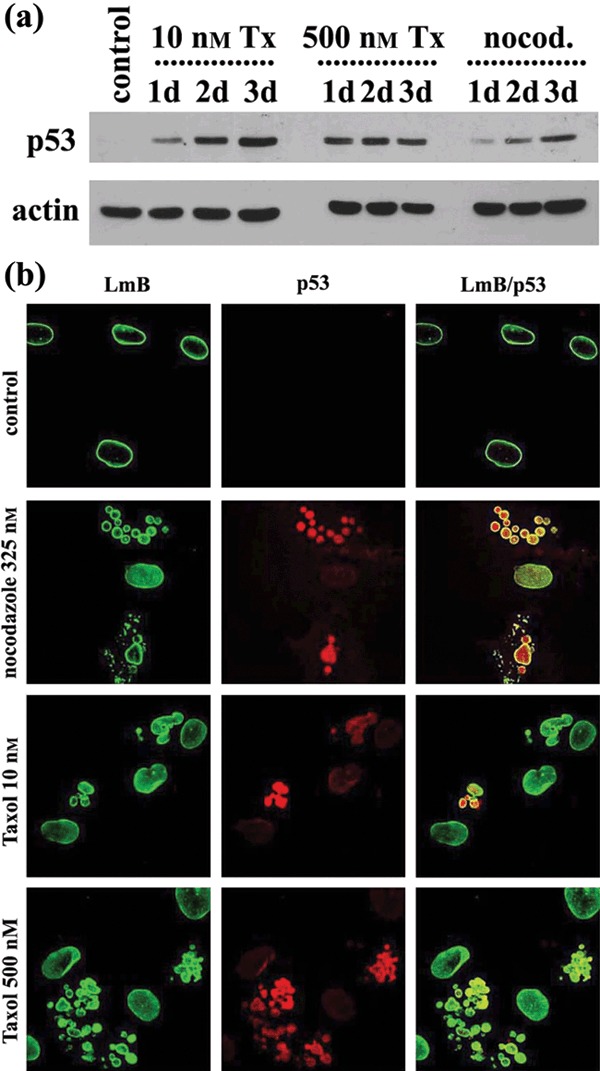
Expression and localization of p53. (a) Western blot analysis of whole lysates of untreated (control), treated with taxol (Tx) and nocodazole (nocod.) human mesenchymal stem cells (hMSC) for 1 day (1d), 2 days (2d) or 3 days (3d) with p53 and actin antibodies. (b) Typical images of lamin B (LmB) p53 and the merge of two (LmB/p53) in untreated (control) and treated with taxol and nocodazole hMSCs. Note the absence of p53 in untreated cells and its nuclear localization in multinucleate cells, following drug treatment.
Activation of p53 regulates genes involved in many cellular functions, the most important being cell‐cycle arrest and apoptosis. Therefore, we analysed proliferation cycles of cells treated with taxol and nocodazole for 3 days, by FACS analysis. We showed partial cell cycle arrest at G2 + M phase (4N content) when cells were treated with either taxol or nocodazole, and accumulation of cells in S phase, in cultures treated with 10 nm taxol (Fig. 6). To examine the ability of both drugs to induce mitotic arrest at the metaphase/anaphase transition of M phase, cells were labelled with DAPI and interphase and mitotic figures were counted using a fluorescence microscope. We showed that mitotic index after drug treatment for 3 days did not increase substantially (Fig. 6b). From the results we conclude that nocodazole and taxol are not able to induce significant mitotic arrest in hMSCs. This is not related to their cycle duration (doubling time) (Fig. 3; Bertani et al. (29)), but probably is associated with p53 induction and with structural or organizational features of the cells’ microtubular network. The latter is supported by our results presented in Fig. 4, which show that microtubules were partially sensitive to nocodazole treatment.
Figure 6.
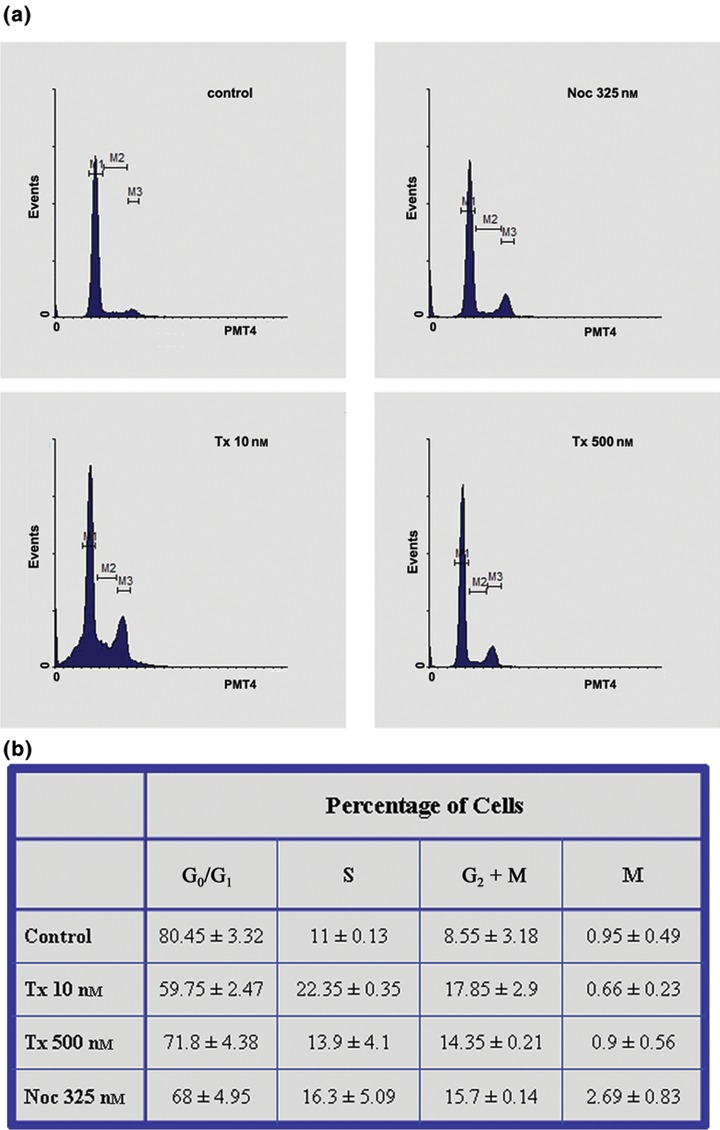
Effect of taxol and nocodazole on cell cycles. (a) Cell cycle analysis of untreated and treated hMSCs with taxol and nocodazole for 3 days. (b) Percentage of viable cells in the different phases of the cell cycle in untreated (control) and treated hMSCs with taxol and nocodazole for 3 days. Percentage of cells in M phase was determined by counting interphase and mitotic cells grown on coverslips and stained with DAPI. Values are means ± standard error of measurements in three separate experiments.
Taxol but not nocodazole modulates tubulin synthesis
We next examined, by immunoblotting, tubulin expression in the presence or absence of taxol or nocodazole (Fig. 7a). Following taxol treatment, with either 10 nm or 500 nm, we showed up to 9‐fold increase of total tubulin, which occurred more rapidly in cells treated with the higher taxol concentration. In contrast, no significant changes in tubulin levels were noted between non‐treated and nocodazole‐treated cells. These data and those presented in Fig. 4a (panel 3d) suggest that tubulin synthesis in cells treated with anti‐microtubule drugs is probably regulated by level of non‐polymerized tubulin. Most likely polymerization of microtubules and declining level of non‐polymerized tubulin would account for the increasing rate of tubulin synthesis in taxol‐treated cells. However, limited efficacy of nocodazole for microtubule depolymerization and therefore, mildly increasing levels of non‐polymerized tubulin could not result in a significant decrease of tubulin synthesis.
Figure 7.
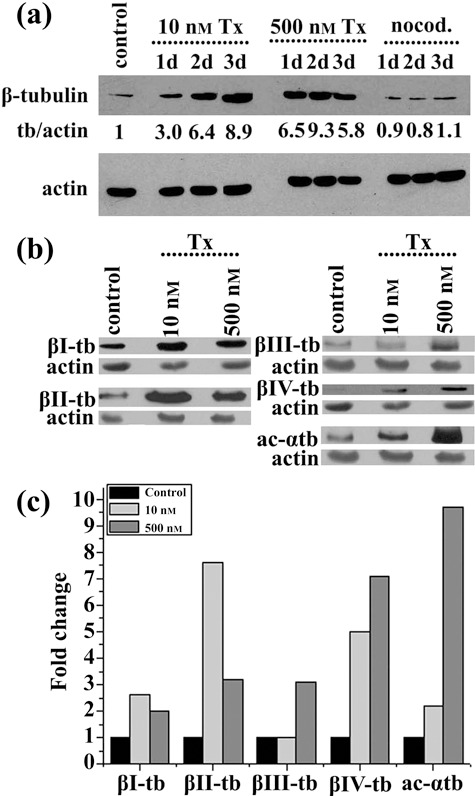
Effect of taxol and nocodazole on tubulin synthesis. (a) Western blot analysis of whole lysates of untreated (control), treated with taxol (Tx) and nocodazole (nocod.) human mesenchymal stem cells for 1 day (1d), 2 days (2d) or 3 days (3d) with β‐tubulin and actin antibodies. Tubulin/actin (tb/actin) ratios were calculated after determination of intensity of protein bands with image analysis. (b) Western blot of whole lysates of untreated (control) and treated hMSCs with taxol 10 nm or 500 nm, for 3 days with isotype‐specific β‐tubulin, acetylated α‐tubulin (ac‐αtb) and actin antibodies. (c) Histogram depicting changes in tubulin isotype/actin ratios in treated hMSCs with taxol 10 nm or 500 nm for 3 days. Ratios were calculated after determination of intensity of protein bands shown in panel (b) with image analysis.
Since all β‐tubulin isotypes are expressed in hMSCs (Fig. 2), we sought to examine the relative amounts of β‐tubulin isotypes under conditions of tubulin synthesis. Figure 7(b,c) shows acetylated α‐ and β‐tubulin isotype levels in cells treated with 10 nm or 500 nm taxol for 3 days. As expected, we found increased levels of acetylated tubulin, especially in cells treated with 500 nm taxol. Synthesis of β‐tubulin isotypes was differently increased, the higher ratio noted for classes β‐II and β‐IV, whereas the lowest was for β‐I tubulin. Since altered expression of β‐tubulin isotypes in cancer cell lines and tumours is related to taxol resistance (30), most likely differential β‐tubulin isotype synthesis following taxol treatment of the cells would account for reduced cell sensitivity to taxol.
Reorganization of microtubules and nuclear envelope in hMSCs released from drugs and cultured under normal conditions
We investigated whether the effect of the drugs on organization of microtubules and the nuclear envelope were reversible upon culture of our ells in drug‐free medium. For this, we examined the microtubule network, nuclear morphology and nuclear lamina structure of cells that had been cultured under normal conditions for up to 6 days after treatment with taxol, or for 3 days with nocodazole, by fluorescence microscopy. We showed that the effect of nocodazole on microtubules was fully reversed after 6 days culture in normal medium, whereas the microtubule network of cells that had been treated with taxol recovered at slower rates (Fig. 4a).
Determination of nuclear phenotype in nocodazole recovered cells showed that the number of cells with normal‐looking nuclei increased proportionally, while those with abnormal‐looking nuclei decreased in time of cell culture in normal conditions (Table 2). Moreover, cells with multinucleate phenotype represented less than 1% of total cells, and practically, there were no cells with nuclear lamina gaps. When cells were released from taxol treatment, we observed increase in numbers of cells with abnormally shaped nuclei after 3 days of cell culture (Table 2, 3d + 3d), whereas lower percentages of cells with such phenotype had been counted in cells cultured for 6 days (Table 2, 3d + 3d). Precisely, in cells recovered for 6 days from 10 nm taxol treatment, we noted an important decrease (by 67.6%) of multinucleate cells, whereas we found no gaps in the nuclear lamina. Recovery of cells treated with 500 nm taxol was slower as the percentage of cells with multinuclei was reduced by only 22.2%, although we noticed a significant drop (by 40%) in the number of multinucleate cells with nuclear envelope defects (Table 2). In conclusion, our results did not indicate major differences in rates of microtubule and nuclear envelope reorganization between hMSCs treated with low (10 nm) or high (500 nm) concentrations of taxol. Moreover, it is clearly shown that the cells sustained nocodazole treatment much better than taxol exposure, as 6 days after nocodazole treatment microtubules and nuclear envelope organization were similar to those of untreated cells (Fig. 4 and Table 2).
Discussion
In vitro observations have shown that hMSCs from chemotherapy‐exposed and unaffected bone marrow had similar phenotype, proliferation capacity and differentiation potential (26). Moreover, hMSCs from chemotherapy‐exposed bone marrow showed significant resistance to cisplatin, vincristine and etoposide compared to cancer cells, particularly at apoptosis‐inducing doses (26). hMSCs isolated from healthy donors were resistant to chemotherapeutic agents commonly used in bone marrow transplantation (busulphan, cyclophosphamide and methotrexate), but were sensitive to a panel of drugs commonly used for cancer treatment, such as paclitaxel, vincristine and etoposide. In addition, treated hMSCs were able to maintain their osteogenic and adipogenic differentiation potential, whereas vincristine‐ but not paclitaxel‐treated hMSCs significantly recovered from drug exposure (25).
In this study, we have found that nocodazole and taxol (paclitaxel) exerted cytostatic action on hMSCs isolated from healthy donors and cultured in vitro. Interestingly, low (10 nm) and high (500 nm) doses of taxol showed similar effects on cell proliferation, cell death and cell recovery. These concentrations are in the range of those measured in plasma of patients treated with paclitaxel (31, 32). After intavenous administration of 250 mg/m2 taxol to ovarian cancer patients for 24 h, plasma concentrations measured are 850 ± 210 nm (31), whereas plasma levels determined immediately after infusion of 60–90 mg/m2 taxol for 1.5 h in gastric cancer patients varied between 1.4 and 2.7 µm (32). Concentration of taxol in plasma decrease very rapidly in the hours following injection and, depending on initial dose, vary between 170–440 nm after 1.5 h, 75–170, 47–100 and 24–54 nm after 6, 12 and 24 h, respectively (32). IC50 value for taxol (10 nm) is similar (in the nanomolar range) to that generally reported for cancer cell lines treated with it for a cell‐cycle period (33, 34), and similar (4 nm) to that reported for normal human fibroblasts or hMSCs, respectively (34), but lower to that found (55 nm) for hMSCs in a conflicting study (25). hMSCs treated with very low concentration of taxol (1 nm) or nocodazole reverted to normal growth rates, immediately or after a lag period, respectively, when transferred to normal culture conditions. However, previous data indicating that hMSCs treated with taxol 1 µm are irreversibly damaged, do not recover and finally die (25), are not confirmed in this present study, showing that hMSCs treated with taxol 10 or 500 nm (Fig. 3) and 1 µm (Fig. S2, Supporting Information) were capable of recovery in drug‐free medium. This discrepancy might be attributed to physiology of hMSCs, which may depend on the donors and/or passage number of the hMSCs.
In this study, we present data suggesting that both taxol and nocodazole induce changes in microtubule and nuclear envelope organization and cause partial arrest of hMSCs in S and G2, but not in M phase of the cell cycle. Changes in nuclear shape (lobulate and polynucleate phenotype) and nuclear envelope organization (nuclear lamina ‘gaps’) in treated hMSCs are similar to those reported for cells of some cancers (5, 7, 35). However, prevalence of the abnormal nuclear phenotype was lower (26–43%) in the hMSCs in comparison to cancer cells treated with taxol (43–90%, depending on cell line). More importantly, prevalence of nuclear lamina defects in taxol treated hMSCs with abnormally shaped nuclei was much lower (30%) than those reported (94%) for cancer cells (5). Thus, our results show that hMSCs are less sensitive than cancer cells to taxol‐induced changes of nuclear envelope shape and organization.
Both low and standard concentrations of taxol can be equally cytostatic by inhibiting cell cycle progression of cancer cells regardless of the point of cell cycle arrest (36). It has been shown that MDM2 antagonists, which activate the p53 pathway, can protect (by arresting cells at the G1 and G2 phases) proliferating cancer and normal cells from mitotic block and apoptosis, caused by taxol (37). In our study, p53 was not detected in untreated hMSCs by immunofluorescence and Western blotting, whereas its synthesis was induced after cell treatment with either nocodazole or taxol. Absence or low expression of p53 has also been reported in undifferentiated low‐passage number hMSCs (38), in a human non‐transformed lymphoblastoid cell line (39), in primary human fibroblasts at early passage and in established, non‐transformed, Chinese hamster cell line at early passage (40). Increased p53 expression, independent of apoptosis induction, has been reported after in vitro exposure of hMSCs to cisplatin, vincristine and etoposide (26). Cellular localization of p53 depends on factors that influence its nuclear import and export and on its interaction with other proteins; this has been suggested to be an important parameter for p53 function in malignant cells (41). Specific accumulation of p53 in the nucleus of hMSCs with abnormal nuclear shape and disorganized nuclear envelope, after treatment with microtubule‐interacting drugs, could be a consequence of impaired nuclear transport and might be important for growth inhibition of these cells.
Colchicine and nocodazole, which both depolymerize microtubules in cultured fibroblasts lead to rapid inhibition of tubulin synthesis (42). Moreover, it has been reported that in many, but not all, cell types, tubulin synthesis decreases very rapidly in response to microtubule inhibitors, which increase the monomer pool, and that this decline in synthesis is associated with decline in amounts of both α‐ and β‐tubulin mRNAs (43). Taxol‐induced tubulin synthesis has been reported in the sea urchin (44) and in Tetrahymena (45), but not in mammalian cells. We found here that taxol induced differential synthesis of β‐tubulin isotypes in hMSCs. To the best of our knowledge there are no reports describing modulation in synthesis of tubulin isotypes after taxol treatment. Differential expression of β‐tubulin isotypes has been reported in taxol‐resistant cell lines and in human ovarian tumours, although it is not possible to make absolute judgement on the role that the β‐tubulin isotypes have in development of drug resistance ((46) and references therein).
In conclusion, our results have shown that hMSCs are moderately but reversibly affected by microtubule‐interacting drugs. Taxol, either at low or high concentration, affects cell‐cycle kinetics and organization of microtubules and nuclear envelopes, and induces p53 and differential β‐tubulin expression. Although most of these effects are very similar to those reported for taxol‐treated cancer cells, their prevalence in hMSCs is shown to be significantly lower. Thus, it seems that treatment of cancer patients with microtubule‐interacting drugs does not permanently damage hMSCs and may not irreversibly affect the bone marrow pool of hMSCs; this provides useful information for clinicians deliberating doses for cancer chemotherapy.
Supporting information
Figure S1. Effect of various concentrations of nocodazole on microtubule network. Representative images of human mesenchymal stem cells treated for 3 days with 160, 325, 650 and 1300 nm nocodazole.
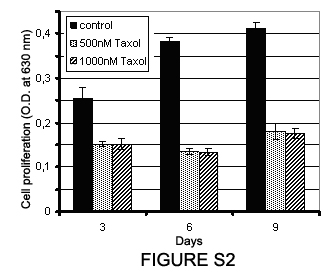
Figure S2. Effect of taxol on human mesenchymal stem cell (hMSC) proliferation. Histogram depicting proliferation of untreated (control) and hMSCs treated with 500 nM and 1000 nM taxol. At day 3, cells were transferred to drug‐free medium and cultured under normal culture conditions for 3 days (day 6) and 6 days (day 9). Cell proliferation was determined by measurement of MTT absorbance at 630 nm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgements
This work was supported by the Cretan Association for Biomedical Research and by an EPAN grant (EPAN ΣΠ‐ΥΒ 85) from the Greek Secretariat of Research and Technology. We thank Iordanis Pelagiadis for performing FACS analysis and Prof Elias Castanas for helpful comments on the manuscript.
References
- 1. Arnal I, Wade RH (1995) How does taxol stabilize microtubules? Curr. Biol. 5, 900–908. [DOI] [PubMed] [Google Scholar]
- 2. Mickey B, Howard J (1995) Rigidity of microtubules is increased by stabilizing agents. J. Cell Biol. 130, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rowinsky EK, Donehower RC (1995) Paclitaxel (taxol). N. Engl. J. Med. 332, 1004–1014. [DOI] [PubMed] [Google Scholar]
- 4. Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L (1996) Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 56, 816–825. [PubMed] [Google Scholar]
- 5. Theodoropoulos PA, Polioudaki H, Kostaki O, Derdas SP, Georgoulias V, Dargemont C et al (1999) Taxol affects nuclear lamina and pore complex organization and inhibits import of karyophilic proteins into the cell nucleus. Cancer Res. 59, 4625–4633. [PubMed] [Google Scholar]
- 6. Danesi R, Figg WD, Reed E, Myers CE (1995) Paclitaxel (taxol) inhibits protein isoprenylation and induces apoptosis in PC‐3 human prostate cancer cells. Mol. Pharmacol. 47, 1106–1111. [PubMed] [Google Scholar]
- 7. Michalakis J, Georgatos SD, Romanos J, Koutala H, Georgoulias V, Tsiftsis D et al (2005) Micromolar taxol, with or without hyperthermia, induces mitotic catastrophe and cell necrosis in HeLa cells. Cancer Chemother. Pharmacol. 56, 615–622. [DOI] [PubMed] [Google Scholar]
- 8. Schiff PB, Horwitz SB (1980) Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 77, 1561–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bogdan C, Ding A (1992) Taxol, a microtubule‐stabilizing antineoplastic agent, induces expression of tumor necrosis factor alpha and interleukin‐1 in macrophages. J. Leukoc Biol. 52, 119–121. [DOI] [PubMed] [Google Scholar]
- 10. Ding AH, Porteu F, Sanchez E, Nathan CF (1990) Shared actions of endotoxin and taxol on TNF receptors and TNF release. Science 248, 370–372. [DOI] [PubMed] [Google Scholar]
- 11. Lee LF, Schuerer‐Maly CC, Lofquist AK, Van Haaften‐Day C, Ting JP, White CM et al (1996) Taxol‐dependent transcriptional activation of IL‐8 expression in a subset of human ovarian cancer. Cancer Res. 56, 1303–1308. [PubMed] [Google Scholar]
- 12. Moos PJ, Fitzpatrick FA (1998) Taxane‐mediated gene induction is independent of microtubule stabilization: induction of transcription regulators and enzymes that modulate inflammation and apoptosis. Proc. Natl. Acad. Sci. USA 95, 3896–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abal M, Souto AA, Amat‐Guerri F, Acuna AU, Andreu JM, Barasoain I (2001) Centrosome and spindle pole microtubules are main targets of a fluorescent taxoid inducing cell death. Cell Motil. Cytoskeleton. 49, 1–15. [DOI] [PubMed] [Google Scholar]
- 14. Pushkarev VM, Starenki DV, Saenko VA, Namba H, Kurebayashi J, Tronko MD et al (2004) Molecular mechanisms of the effects of low concentrations of taxol in anaplastic thyroid cancer cells. Endocrinology 145, 3143–3152. [DOI] [PubMed] [Google Scholar]
- 15. Devine SM, Hoffman R (2000) Role of mesenchymal stem cells in hematopoietic stem cell transplantation. Curr. Opin. Hematol. 7, 358–363. [DOI] [PubMed] [Google Scholar]
- 16. Majumdar MK, Thiede MA, Haynesworth SE, Bruder SP, Gerson SL (2000) Human marrow‐derived mesenchymal stem cells (MSCs) express hematopoietic cytokines and support long‐term hematopoiesis when differentiated toward stromal and osteogenic lineages. J. Hematother. Stem Cell Res. 9, 841–848. [DOI] [PubMed] [Google Scholar]
- 17. Majumdar MK, Thiede MA, Mosca JD, Moorman M, Gerson SL (1998) Phenotypic and functional comparison of cultures of marrow‐derived mesenchymal stem cells (MSCs) and stromal cells. J. Cell. Physiol. 176, 57–66. [DOI] [PubMed] [Google Scholar]
- 18. Bianco P, Riminucci M, Gronthos S, Robey PG (2001) Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells 19, 180–192. [DOI] [PubMed] [Google Scholar]
- 19. Gronthos S, Graves SE, Simmons PJ (1998). Isolation, purification and in vitro manipulation of human bone marrow stromal precursor cells In: Beresford JN, Owen ME, eds. Marrow Stromal Cell Culture, pp. 26–42. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 20. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147. [DOI] [PubMed] [Google Scholar]
- 21. Colter DC, Class R, DiGirolamo CM, Prockop DJ (2000) Rapid expansion of recycling stem cells in cultures of plastic‐adherent cells from human bone marrow. Proc. Natl. Acad. Sci. USA 97, 3213–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Domenech J, Gihana E, Dayan A, Truglio D, Linassier C, Desbois I et al (1994) Haemopoiesis of transplanted patients with autologous marrows assessed by long‐term marrow culture. Br. J. Haematol. 88, 488–496. [DOI] [PubMed] [Google Scholar]
- 23. Domenech J, Roingeard F, Herault O, Truglio D, Desbois I, Colombat P et al (1998) Changes in the functional capacity of marrow stromal cells after autologous bone marrow transplantation. Leuk. Lymphoma. 29, 533–546. [DOI] [PubMed] [Google Scholar]
- 24. Galotto M, Berisso G, Delfino L, Podesta M, Ottaggio L, Dallorso S et al (1999) Stromal damage as consequence of high‐dose chemo/radiotherapy in bone marrow transplant recipients. Exp. Hematol. 27, 1460–1466. [DOI] [PubMed] [Google Scholar]
- 25. Li J, Law HK, Lau YL, Chan GC (2004) Differential damage and recovery of human mesenchymal stem cells after exposure to chemotherapeutic agents. Br. J. Haematol. 127, 326–334. [DOI] [PubMed] [Google Scholar]
- 26. Mueller LP, Luetzkendorf J, Mueller T, Reichelt K, Simon H, Schmoll HJ (2006) Presence of mesenchymal stem cells in human bone marrow after exposure to chemotherapy: evidence of resistance to apoptosis induction. Stem Cells 24, 2753–2765. [DOI] [PubMed] [Google Scholar]
- 27. Kastrinaki MC, Sidiropoulos P, Roche S, Ringe J, Lehmann S, Kritikos H et al (2008) Functional, molecular and proteomic characterisation of bone marrow mesenchymal stem cells in rheumatoid arthritis. Ann. Rheum. Dis. 67, 741–749. [DOI] [PubMed] [Google Scholar]
- 28. Kourmouli N, Theodoropoulos PA, Dialynas G, Bakou A, Politou AS, Cowell IG et al (2000) Dynamic associations of heterochromatin protein 1 with the nuclear envelope. EMBO J. 19, 6558–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bertani N, Malatesta P, Volpi G, Sonego P, Perris R (2005) Neurogenic potential of human mesenchymal stem cells revisited: analysis by immunostaining, time‐lapse video and microarray. J. Cell Sci. 118, 3925–3936. [DOI] [PubMed] [Google Scholar]
- 30. Kavallaris M, Kuo DY, Burkhart CA, Regl DL, Norris MD, Haber M et al (1997) Taxol‐resistant epithelial ovarian tumors are associated with altered expression of specific β‐tubulin isotypes. J. Clin. Invest. 100, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jamis‐Dow CA, Klecker RW, Sarosy G, Reed E, Collins JM (1993) Steady‐state plasma concentrations and effects of taxol for a 250 mg/m2 dose in combination with granulocyte‐colony stimulating factor in patients with ovarian cancer. Cancer Chemother. Pharmacol. 33, 48–52. [DOI] [PubMed] [Google Scholar]
- 32. Kobayashi M, Oba K, Sakamoto J, Kondo K, Nagata N, Okabayashi T et al (2007) Pharmacokinetic study of weekly administration dose of paclitaxel in patients with advanced or recurrent gastric cancer in Japan. Gastric Cancer 10, 52–57. [DOI] [PubMed] [Google Scholar]
- 33. Papas S, Akoumianaki T, Kalogiros C, Hadjiarapoglou L, Theodoropoulos PA, Tsikaris V (2007) Synthesis and antitumor activity of peptide‐paclitaxel conjugates. J. Pept. Sci. 13, 662–671. [DOI] [PubMed] [Google Scholar]
- 34. Teraishi F, Wu S, Sasaki J, Zhang L, Zhu HB, Davis JJ et al (2005) P‐glycoprotein‐independent apoptosis induction by a novel synthetic compound, MMPT [5‐[(4‐Methylphenyl) methylene]‐2‐(phenylamino)‐4 (5H)‐thiazolone]. J. Pharmacol. Exp. Ther. 314, 355–362. [DOI] [PubMed] [Google Scholar]
- 35. Michalakis J, Georgatos SD, De Bree E, Polioudaki H, Romanos J, Georgoulias V et al (2007) Short‐term exposure of cancer cells to micromolar doses of paclitaxel, with or without hyperthermia, induces long‐term inhibition of cell proliferation and cell death in vitro . Ann. Surg. Oncol. 14, 1220–1228. [DOI] [PubMed] [Google Scholar]
- 36. Giannakakou P, Robey R, Fojo T, Blagosklonny MV (2001) Low concentrations of paclitaxel induce cell type‐dependent p53, 21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel‐induced cytotoxicity. Oncogene 20, 3806–3813. [DOI] [PubMed] [Google Scholar]
- 37. Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT (2005) Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 65, 1918–1924. [DOI] [PubMed] [Google Scholar]
- 38. Park JS, Kim HY, Kim HW, Chae GN, Oh HT, Park JY et al (2005) Increased caveolin‐1, a cause for the declined adipogenic potential of senescent human mesenchymal stem cells. Mech. Ageing. Dev. 126, 551–559. [DOI] [PubMed] [Google Scholar]
- 39. Ciciarello M, Mangiacasale R, Casenghi M, Zaira Limongi M, D’Angelo M, Soddu S et al (2001) p53 displacement from centrosomes and p53‐mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 276, 19205–19213. [DOI] [PubMed] [Google Scholar]
- 40. Moro F, Ottaggio L, Bonatti S, Simili M, Miele M, Bozzo S et al (1995) p53 expression in normal versus transformed mammalian cells. Carcinogenesis 16, 2435–2440. [DOI] [PubMed] [Google Scholar]
- 41. O’Brate A, Giannakakou P (2003) The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist. Updat. 6, 313–322. [DOI] [PubMed] [Google Scholar]
- 42. Ben‐Ze’ev A, Farmer SR, Penman S (1979) Mechanisms of regulating tubulin synthesis in cultured mammalian cells. Cell 17, 319–325. [DOI] [PubMed] [Google Scholar]
- 43. Cleveland DW, Lopata MA, Sherline P, Kirschner MW (1981) Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell 25, 537–546. [DOI] [PubMed] [Google Scholar]
- 44. Gong ZY, Brandhorst B (1988) Autogenous regulation of tubulin synthesis via RNA stability during sea urchin embryogenesis. Development 102, 31–43. [DOI] [PubMed] [Google Scholar]
- 45. Stargell LA, Heruth DP, Gaertig J, Gorovsky MA (1992) Drugs affecting microtubule dynamics increase α‐tubulin mRNA accumulation via transcription in Tetrahymena thermophila . Mol. Cell. Biol. 12, 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burkhart CA, Kavallaris M, Band Horwitz S (2001) The role of β‐tubulin isotypes in resistance to antimitotic drugs. Biochim. Biophys. Acta 1471, O1–O9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of various concentrations of nocodazole on microtubule network. Representative images of human mesenchymal stem cells treated for 3 days with 160, 325, 650 and 1300 nm nocodazole.
Figure S2. Effect of taxol on human mesenchymal stem cell (hMSC) proliferation. Histogram depicting proliferation of untreated (control) and hMSCs treated with 500 nM and 1000 nM taxol. At day 3, cells were transferred to drug‐free medium and cultured under normal culture conditions for 3 days (day 6) and 6 days (day 9). Cell proliferation was determined by measurement of MTT absorbance at 630 nm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item