

Abstract
Objectives
To investigate the function and regulatory mechanism of Krüppel‐like factor 3 (KLF3) in lung cancer.
Materials and Methods
KLF3 expression was analysed by qRT‐PCR and Western blot assays. The proliferation, migration, invasion, cycle and apoptosis were measured by CCK‐8 and EdU, wound‐healing and Transwell, and flow cytometry assays. The tumour growth was detected by nude mouse tumorigenesis assay. In addition, the interaction between KLF3 and Sp1 was accessed by luciferase reporter, EMSA and ChIP assay. JAK2, STAT3, PI3K and p‐AKT levels were evaluated by Western blot and IHC assays.
Results
The results indicated that KLF3 expression was elevated in lung cancer tissues. Knockdown of KLF3 inhibited lung cancer cell proliferation, migration and invasion, and induced cell cycle arrest and apoptosis. In addition, the downregulation of KLF3 suppressed tumour growth in vivo. KLF3 was transcriptionally activated by Sp1. miR‐326 could bind to 3′UTR of Sp1 but not KLF3 and decreased the accumulation of Sp1, which further indirectly reduced KLF3 expression and inactivated JAK2/STAT3 and PI3K/AKT signaling pathways in vitro and in vivo.
Conclusions
Our data demonstrate that miR‐326/Sp1/KLF3 regulatory axis is involved in the development of lung cancer, which hints the potential target for the further therapeutic strategy against lung cancer.
Keywords: KLF3, lung cancer, microRNA‐326, SP1
1. INTRODUCTION
Lung cancer represents the major cause of cancer‐related deaths among men and ranks as the second leading cause of cancer among women worldwide.1 Based on histology, lung cancer consists of non–small‐cell lung cancer (NSCLC) and small‐cell lung cancer (SCLC).2 NSCLC accounts for 83% of total lung cancer and can be further divided into squamous carcinoma, adenocarcinoma and large cell carcinoma.3, 4 The main risk factors for NSCLC are cigarette smoke and air pollution.5, 6 Surgical resection is an effective approach in maintaining long‐term survival and thus widely used in lung cancer. In spite of the significant improvement in surgery and chemotherapy, the prognosis of metastatic lung cancer is still unsatisfactory, and the 5‐year survival rate for patients with NSCLC is lower than 21%.7 Therefore, favourable biomarkers are of great importance in the prediction of early metastasis of patients with lung cancer.
MicroRNAs (miRNAs), a group of small non‐coding RNAs, contribute to regulate gene expression at the posttranscriptional level through perfect or partial base‐pairing, mainly at the 3′UTR of the target messenger RNA. MiR‐326, for instance, was first identified among a set of miRNAs with remarkable high expression in neurons.8 It has been implicated that abnormal miR‐326 expression participates in a variety of pathological process, including multiple sclerosis, endometrial cancer, gastric cancer, lung cancer, osteosarcoma, pulmonary fibrosis and breast tumours.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 Of note, recent evidence revealed that miR‐326 regulated HbF synthesis by targeting erythroid Krüppel‐like factor (KLF), indicating the targeting effect of miR‐326 on KLF.9 Alternatively, miR‐326 reverses chemoresistance in human lung adenocarcinoma cells by targeting specificity protein 1 (Sp1).12
Krüppel‐like factor 3 (KLF3/BKLF), a member of the KLF family, was initially cloned from erythroid cells in a screen for factors with homology to the DNA‐binding domain of EKLF.22, 23 KLF3 encodes a sequence‐specific transcriptional repressor with three highly conserved C‐terminal C2H2 zinc finger motifs that bind CACCC boxes and other GC‐rich elements in the promoter and enhancer of target genes.23, 24 The N‐terminal domains are less conserved, and individual KLFs integrate different regulators to function as transcriptional activators or repressors.25 For example, recruitment of the acetyltransferases p300/CBP by KLF1 potentiates its activation of the β‐globin gene.26 However, KLF3 functions as a transcriptional repressor via recruitment of C‐terminal binding proteins 1 and 2 (CtBP1/2),27, 28 which in turn facilitates assembly of a potent silencing complex that drives epigenetic modification of gene regulatory regions.25, 27, 28, 29 Moreover, KLF3 is widely expressed and involved in multiple processes, such as erythropoiesis, metabolism and B lymphopoiesis.25, 30, 31, 32, 33, 34, 35 Although the roles of KLF3 have been broadly investigated in developmental processes, little is known about the function of KLF3 in lung cancer progression.
Specificity protein 1 (Sp1) is a transcription factor that is ubiquitously expressed in many mammalian cell types.36 Clinical evidences showed that Sp1 was overexpressed in a number of human cancers, such as gastric cancer,37 pancreatic adenocarcinoma38 and hepatocellular carcinoma.39 Emerging evidences showed that Sp1 accelerates tumour growth and metastasis through regulating gene transcription.40, 41 For example, overexpression of Sp1 increased the invasiveness of glioma cells via upregulating the expression and activity of MMP‐2.42 Choi et al also reported that Sp1 could bind to the promoter region of Slug and repressed EMT process.43 However, given many potential physiologic roles of Sp1, the target genes of Sp1 remain to be clearly characterized. Whether Sp1 could affect the expression of KLF3 in lung cancer has never been reported. Our preview studies have revealed that miR‐326 overexpression inhibited cell proliferation and migration in lung cancer.44 The potential targets, KLF and Sp1, were previously reported involved in the growth of lung cancer cells. 45, 46, 47 In this study, we aim to explore the possibility as well as related mechanisms of miR‐326‐Sp1‐KLF3 axis in the regulation of lung cancer development.
2. MATERIALS AND METHODS
For detailed Materials and methods, please see Supporting Information. For related sequences in the experiment please see Tables 1, 2, 3.
Table 1.
Sequences of relative primers
| ID | Forward sequence (5′‐3′) | Reverse sequence (5′‐3′) |
|---|---|---|
| U6 | CTCGCTTCGGCAGCACA | AACGCTTCACGAATTTGCGT |
| GAPDH | CCTCGTCTCATAGACAAGATGGT | GGGTAGAGTCATACTGGAACATG |
| miR‐326 | ACACTCCAGCTGGGCCTCTGGGCCCTTCCTCC | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCTGGAGG |
| KLF3 | GCCTCTCATGGTCTCCTTATC | TAATCTGTCCTCTGTGGTTCG |
| SP1 | TGGCAGCAGTACCAATGGC | CCAGGTAGTCCTGTCAGAACTT |
Table 2.
siRNA sequences
| Name | Sense (5′‐3′) | Antisense (3′‐5′) |
|---|---|---|
| Negative control | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT |
| siKLF3‐534 | GCAUGCAAGUACCUGUAAUUU | UUCGUACGUUCAUGGACAUUA |
| siKLF3‐772 | GAUACACAGAUGUGAUUAUUU | UUCUAUGUGUCUACACUAAUA |
| siKLF3‐902 | GUCUGAUGAACUAACAAGAUU | UUCAGACUACUUGAUUGUUCU |
| siSp1‐491 | CCUGGAGUGAUGCCUAAUAUU | UUGGACCUCACUACGGAUUAU |
| siSp1‐996 | CCAGCAACAUGGGAAUUAUUU | UUGGUCGUUGUACCCUUAAUA |
| siSp1‐1648 | GCCGUUGGCUAUAGCAAAUUU | UUCGGCAACCGAUAUCGUUUA |
Table 3.
Sequences of relative primers for recombinant vector construction
| ID | Sequence (5′‐3′) |
|---|---|
| SP1‐XhoI F | CCGCTCGAGGATCAGGCACCCGGGG |
| SP1‐NotI R | ATTTGCGGCCGCCAGCCAACCCCTGTGAATG |
| SP1 MUT‐F | GATCAGGCACCCGGGGTTCACATCATATGGGCCATACC |
| SP1 MUT‐R | GGTATGGCCCATATGATGTGAACCCCGGGTGCCTGATC |
| KLF3‐KpnI ‐F | GGGGTACCTTCCCGCTGGAGCCG |
| KLF3‐XhoI R | CCGCTCGAGTGCCCGCTCCGGG |
| KLF3 MUT‐F | CCTGGCAGCCCGCGCTATAACGGCTTGCTCGCCCGCGAGCC |
| KLF3 MUT‐R | GGCTCGCGGGCGAGCAAGCCGTTATAGCGCGGGCTGCCAGG |
3. RESULTS
3.1. KLF3 is overexpressed in human lung cancer tissues
To explore the potential role of KLF3 in lung cancer progression, we firstly assessed the expression level of KLF3 in lung cancer tissues. Forty lung cancer tissues and paired tumour‐adjacent normal tissues were collected from Jiangsu Province Hospital, and the mRNA level of KLF3 was examined by real‐time PCR. The results showed that KLF3 was overexpressed in lung cancer tissues compared with that in adjacent normal tissues (Figure 1A, P < 0.001). Furthermore, we performed Western blot to evaluate the protein level of KLF3 in lung cancer tissues. Similarly, the KLF3 expression was highly upregulated in lung cancer tissues compared to that in the adjacent normal control (Figure 1B). The result of IHC indicated that the amount of KLF3 protein in lung cancer tissues was remarkably increased compared to that in normal lung tissues (Figure 1E). Then, we examined the expression of KLF3 in different lung cancer cell lines by real‐time PCR and Western blot. As shown in Figure 1C and D, the mRNA and protein levels of KLF3 were also significantly higher in lung cancer cell lines than that in control HBECs, especially in A549 and 95D cells (P < 0.01). Together, these results suggested that the highly expressed KLF3 may be associated with lung cancer progression.
Figure 1.
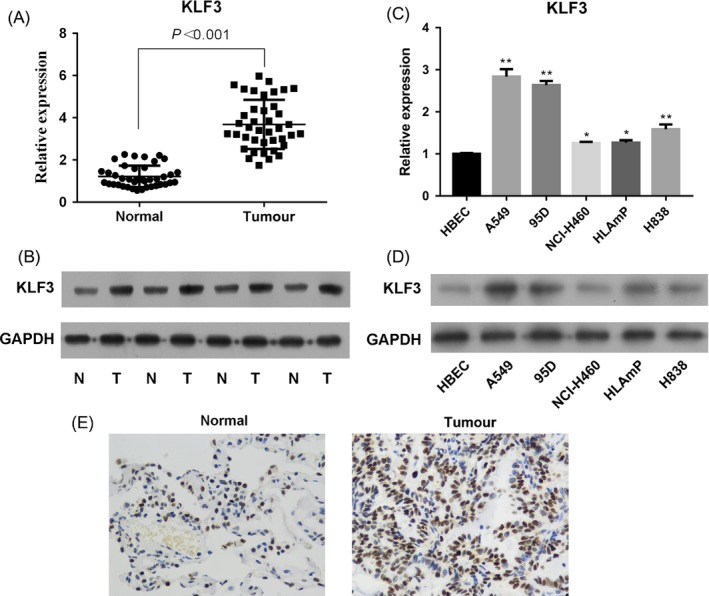
KLF3 is overexpressed in lung cancer tissues and cell lines. A, The mRNA level of KLF3 was measured by real‐time PCR. The results showed that KLF3 was upregulated in lung cancer tissues compared with paired adjacent normal tissues (N = 40), P < 0.001. B, Western blot analysis revealed that KLF3 was upregulated in lung cancer tissues. C and D, The mRNA and protein level of KLF3 was measured by real‐time PCR and Western blot in the human bronchial epithelial cell line HBEC and various lung cancer cell lines (A549, 95D, NCI‐H460, HLAmp and H838). E, Immunohistochemical analysis indicated that KLF3 was highly expressed in lung cancer tissues compared to that in normal tissues (×400). *indicates that P < 0.05 vs HBECs. **indicates that P < 0.001 vs HBECs
3.2. Effects of suppression of KLF3 on lung cancer cells
To better understand the functional significance of KLF3 in regulating the biological processes of lung cancer cells, we performed loss‐of‐function experiments of KLF3 by transfection with specific siRNA targeting KLF3. The transfection efficiency was confirmed by Western blot in Figure S2, and the results showed that siRNA targeting KLF3 successfully downregulated the expression of KLF3 in both A549 and 95D cells (Figure 2A). Next, the proliferations of A549 and 95D cells were assessed by CCK‐8 assay. As shown in Figure 2B, downregulation of KLF3 significantly inhibited cell proliferation of A549 cells compared with the negative control (NC) cells (P < 0.001). Similar results were also obtained in 95D cells (Figure 2C, P < 0.01). Furthermore, we performed 5‐ethynyl‐2′‐deoxyuridine (EDU) staining to assess cell proliferation ability at 48 hours after transfection of KLF3 siRNA. Figure 2D and E also validated that knockdown of KLF3 inhibited cell proliferation in A549 and 95D cells (P < 0.05), consistent with the results obtained from CCK‐8 assay.
Figure 2.
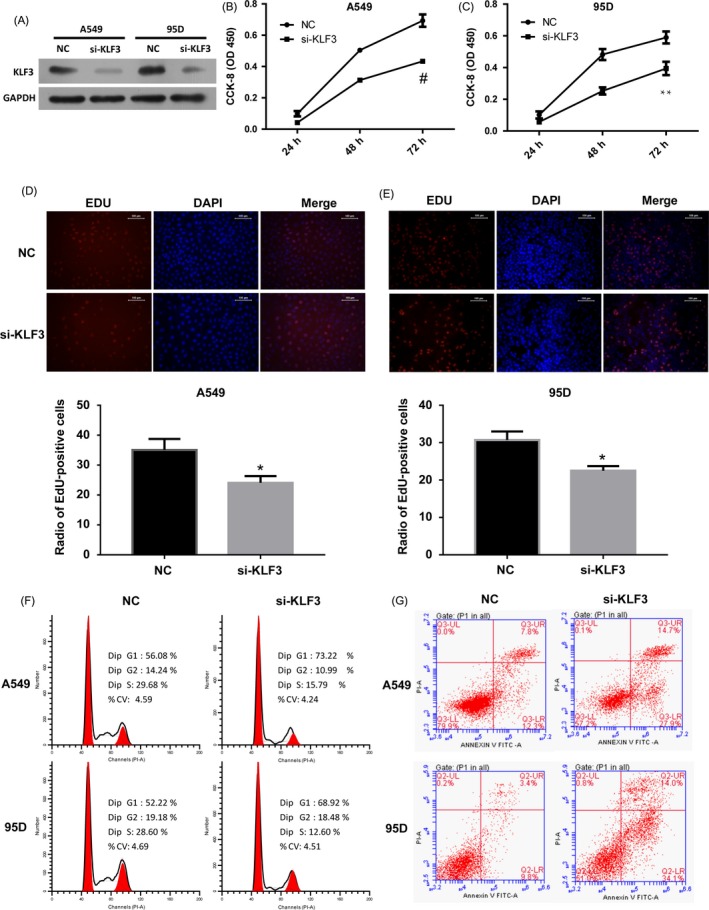
Effects of KLF3 downregulation on lung cancer cells. (A) The knockdown efficiency of KLF3 in lung cancer cells was confirmed by Western blot. (B and C) Cell proliferation was evaluated by CCK‐8 assay. The results showed that downregulate the expression of KLF3 inhibited cell proliferation in A549 and 95D cells. Experiments were performed three times; Mean ±SD. # indicates that P < 0.001; **indicates that P < 0.01. (D and E) EDU staining also revealed that silencing the expression of KLF3 inhibited cell proliferation in A549 (D) and 95D cells (E) (×200). Quantitative analysis of EDU‐positive A549 and 95D cells were shown as histograms. *indicates that P < 0.05. (F) Cell cycle analysis was performed by flow cytometry. Knockdown the expression of KLF3 resulted in cell cycle arrest at G1 phase. (G) Cell apoptosis was stained with PI and Annexin V double staining and analysed by flow cytometry. Statistical analysis revealed that siRNA‐mediated silencing of KLF3 induced cell apoptosis in A549 and 95D cells
In order to investigate the mechanisms underlying the effect of KLF3 on cell proliferation of lung cancer cells, we examined cell cycle and apoptosis by flow cytometry. We observed that KLF3 knockdown increased the proportion of cells in the G1 phase and decreased the cells in S phase compared with the control (Figure 2F), suggesting that KLF3 could accelerate the cell cycle during lung cancer progression. Cell apoptosis was determined by using Annexin V‐fluorescein isothiocyanate (FITC) and propidium iodide (PI) double staining. The silencing of KLF3 induced cell apoptosis of A549 and 95D cells (Figure 2G), based on the observations of the percentage of the Annexin V‐positive cells (increased from 20.1% to 42.6% in A549 cells and 13.2% to 48.1% in 95D cells). Together, these results suggest that KLF3 facilitates the proliferation of lung cancer cells and may function as an oncogene during lung cancer progression.
3.3. Downregulation of KLF3 inhibited cell migration and invasion
A critical feature of lung cancer is aggressive behaviour in distant metastasis sites including lymph node. Cell migration and invasion are important steps in the process of metastasis. To determine whether KLF3 affects inhuman lung cancer cell migration and invasion, wound healing and cell invasion assays were performed. The results showed that in untreated A549 cells, wound closure occurred gradually at 48 hours after scratch, whereas this effect was significantly reduced by the treatment of si‐KLF3 (Figure 3A). Similar results were obtained in 95D cells (Figure 3B). In line with this finding, suppression of KLF3 significantly repressed the invasive ability of A549 and 95D cells (P < 0.01) (Figure 3C and D). In summary, these results indicated that KLF3 participated in the regulation of lung cancer cell proliferation, migration and invasion.
Figure 3.
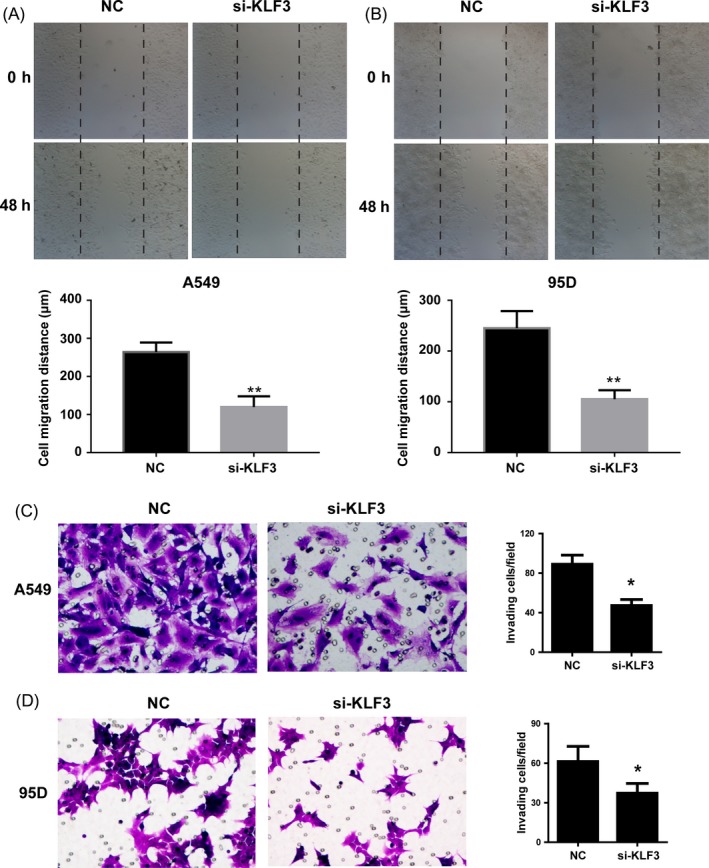
The decrease in KLF3 expression reduces cell migration and invasion. (A and B) Wound‐healing assay was used to detect cell migration ability in A549 and 95D cells. Compared with NC, si‐KLF3 cells showed weaker migration ability. Quantitative analysis of cell migration distance was shown as histograms. **indicates that P < 0.01. (C and D) Transwell assays showed that knockdown the expression of KLF3 significantly inhibited cell invasion in A549 (C) and 95D cells (D) (×200). Experiments were performed 3 times. Representative images of invaded cells were shown. Statistical analysis showed the effect of KLF3 on cell invasion. Mean ± SD. *indicates that P < 0.05
3.4. The block of KLF3 suppressed the activation of JAK2/STAT3 and PI3K/AKT signalling pathways in vitro
Considering that the promoting effect of KLF3 on cell growth and migration, we performed Western blot assay to measure which signalling pathways were involved by KLF3. As revealed in Figure 4, the phosphorylation levels of JAK2, STAT3, PI3K and AKT were downregulated in the absence of KLF3 in both A549 and 95D cells in vitro, suggesting that JAK2/STAT3 and PI3K/AKT signalling pathways may be involved in lung cancer progression regulated by KLF3.
Figure 4.
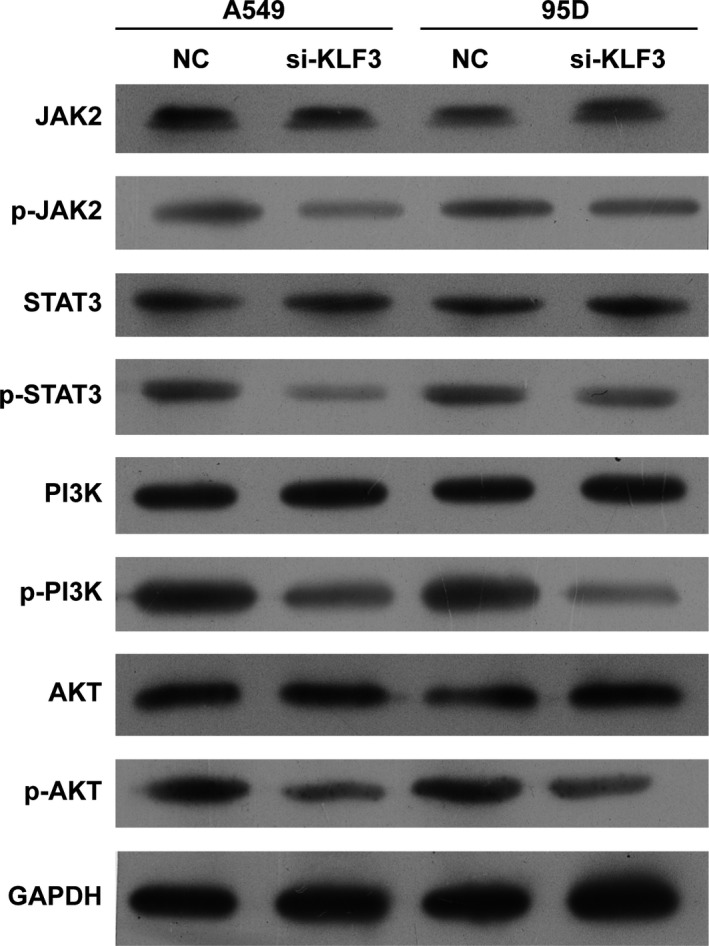
Knockdown of KLF3 suppressed the activation of JAK2/STAT3 and PI3K/AKT signalling pathways in vitro. Western blot analysis demonstrated that knockdown the expression of KLF3 significantly decreased the expression of JAK2, STAT3, PI3K, and AKT phosphorylation levels in A549 and 95D cells in vitro
3.5. Sp1 is positively correlated with the expression of KLF3
Based on the above findings, the function of KLF3 in promoting cell proliferation, migration and invasion have been well demonstrated. However, little is known about the regulatory mechanism of KLF3 in lung cancer cells. Through bioinformatics analysis of the proximal promoter of KLF3, we found that there are some putative binding sites of Sp1 (Figure S1). Therefore, we performed experiments to investigate the possible relation between KLF3 and Sp1. Firstly, we detected the expression of Sp1 in lung cancer tissues through real‐time PCR and Western blot. The results showed that Sp1 was upregulated in lung cancer tissues compared with that in the adjacent normal tissues (Figure 5A and B, P < 0.001), which correlated with the expression of KLF3 in lung cancer tissues (Figure 1A and B). The IHC data also indicated that the level of Sp1 in lung cancer tissues was evidently elevated compared to that in normal lung tissues (Figure 5C). As expected, the mRNA and protein levels of Sp1 were also elevated in different lung cancer cell lines compared with that in the HBECs (Figure 5D and E). Next, we examined the expression of KLF3 after reducing of Sp1 level in A549 and 95D cells. Figure 5F showed that the mRNA level of KLF3 was downregulated after downregulation of Sp1 by siRNA in A549 cells compared with that in the NC group (P < 0.01). Similarly, the mRNA expression of KLF3 was also decreased in 95D cells (Figure 5G, P < 0.01). Moreover, Western blot analysis revealed that the protein level of KLF3 was obviously decreased after expression of Sp1 declined in A549 and 95D cells (Figure 5H). These data indicated Sp1 positively correlated with the expression of KLF3 in lung cancer cells.
Figure 5.
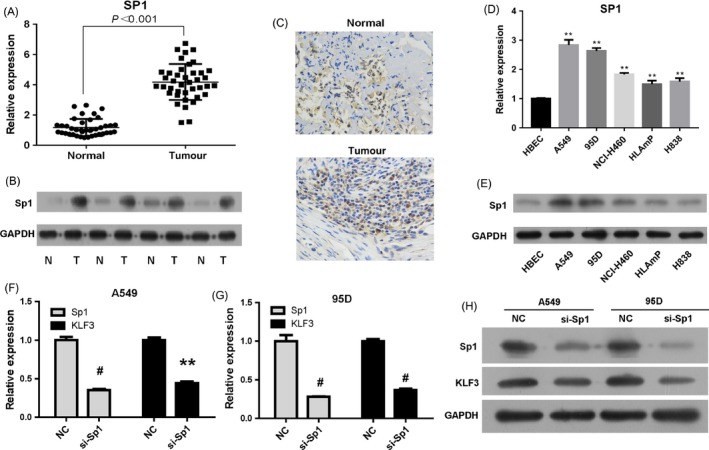
KLF3 expression is positively regulated by Sp1. A, The mRNA level of Sp1 was detected by real‐time PCR. Compared with the normal tissues, Sp1 was upregulated in lung cancer tissues, P < 0.001. B, Western blot analysis revealed that Sp1 was upregulated in lung cancer tissues. C, Immunohistochemical analysis indicated that the level of Sp1 was increased in lung cancer tissues compared to that in normal tissues (×400). D and E, The mRNA and protein levels of Sp1 were examined by real‐time PCR and Western blot. The results showed that Sp1 was overexpressed in lung cancer cell lines. ** indicates that P < 0.001 vs HBECs. F and G, Real‐time PCR results showed that KLF3 expression was downregulated after silencing the expression of Sp1 in A549 and 95D cells. Mean ± SD. **indicates that P < 0.01; # indicates that P < 0.001. H, Western blot analysis confirmed that knockdown the expression of Sp1 significantly reduced KLF3 expression
3.6. KLF3 is a direct target of Sp1
To further explore the molecular basis for this regulation, we amplified the promoter region of KLF3 and performed dual‐luciferase reporter assay. The wild‐type (WT) and mutant promoters were transfected alone or with the Sp1 expression vector into HEK293T cells. The results showed that overexpression of Sp1 markedly increased the relative luciferase activity of KLF3‐WT (Figure 6A, lanes 1 and 2), but it did not activate the relative promoter activity of the mutant KLF3 (Figure 6A, lanes 3 and 4), suggesting the interaction between promoter region of KLF3 and Sp1 putative binding sites. To further determine whether KLF3 is a direct target gene of Sp1, we performed electrophoretic mobility shift assays (EMSAs) in vitro using the nuclear extracts from A549 cells and the oligonucleotide probes containing the predicted Sp1 binding sites. The results demonstrated that shift bands were observed when probes were incubated with nuclear extracts (Figure 6B). Moreover, as expected, super‐shift bands were observed when Sp1 antibody was added to the binding system (Figure 6B). In addition, chromatin immunoprecipitation (ChIP) assay was initially employed to further determine whether Sp1 could bind to the promoter of KLF3. The pull‐down DNA was identified by RT‐PCR and the results showed that KLF3 was markedly amplified from the Sp1‐immunoprecipitated A549 chromatins, but absent from chromatins immunoprecipitated by IgG (Figure 6C). In summary, these results demonstrate that KLF3 is a direct target gene of Sp1.
Figure 6.
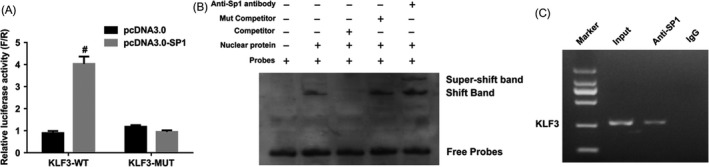
KLF3 is a direct target gene of Sp1. A, The luciferase activity of KLF3 was detected by Dual‐luciferase report assay after transfection of the WT and Mutant constructs alone or together with the Sp1 expression vector. The results showed that KLF3 promoter activity was significantly enhanced by Sp1. #indicates that P < 0.001. B, EMSAs were used to investigate the binding of Sp1 and KLF3 promoter in vitro. Nuclear protein was extracted from A549 cells. When DNA probes were incubated with nuclear extracts, a specific DNA‐protein complex was observed and was supershifted by the anti‐Sp1 antibody. C, ChIP analysis of Sp1 at the KLF3 promoter region in A549 cells. The results suggested that Sp1 can directly bind to the promoter region of KLF3
3.7. Sp1 is a downstream target of miR‐326
In our preview studies, we have found that miR‐326 inhibited lung cancer cell proliferation and migration.44 Meanwhile, through bioinformatics analysis such as JASPAR and TargetScan, we found the 3′UTR regions of KLF3 and Sp1 contained the putative binding sites of miR‐326, suggesting Sp1 and KLF3 may be potential downstream targets of miR‐326. To verify the prediction, we firstly examined the level of miR‐326 in lung cancer tissues and cell lines. As shown in Figure 7A, compared with the normal tissues, miR‐326 was significantly downregulated in lung cancer tissues. We further analysed the relations between Sp1, KLF3 and miR‐326 and found that the level of miR‐326 was significantly negatively correlated with Sp1 expression (R = −0.483, P < 0.01) but positively with KLF3 level (R = 0.503, P < 0.001) (Figure 7B, C). Based on this finding, we then detected the miR‐326 level in distinct types of cells and the data indicated that the expressions in lung cancer cell lines were statistically reduced that in HBECs (Figure 7D). Furthermore, to investigate whether miR‐326 could regulate Sp1 and KLF3 expression, we transfected miR‐326 mimics into lung cancer cells and real‐time PCR results revealed that overexpression of miR‐326 suppressed the expression of Sp1 and KLF3 in both A549 (Figure 7E) and 95D (Figure 7F) cells. Similarly, Western blot analysis also showed that the expression of Sp1 and KLF3 was markedly deterred due to miR‐326 upregulation (Figure 7G). We then employed dual‐luciferase reporter assays to verify the direct relationship. The results showed that transfection of miR‐326 mimics significantly decreased the luciferase activity of Sp1 3′UTR compared with cells transfected with NC, whereas the effect was abolished when the binding sites of miR‐326 were mutant (Figure 7H). However, the no significant change regarding luciferase activity of KLF3 3′UTR was found by using miR‐326 mimics, suggesting KLF3 was not a direct gene of miR‐326 (Figure 7I). In order to further validate the miR‐326/Sp1/KLF3 regulatory axis, we impeded the expression of Sp1. Notably, the KLF3 level was remarkably inhibited on account for the overexpression of miR‐326 (Figure 7E‐G). The downregulation of miR‐326 thus induced the level of KLF3, but this can be reversed by suppression of Sp1, which suggests that miR‐326 downregulation increases the KLF3 overexpression through the upregulation of Sp1 (Figure 7J, K).
Figure 7.
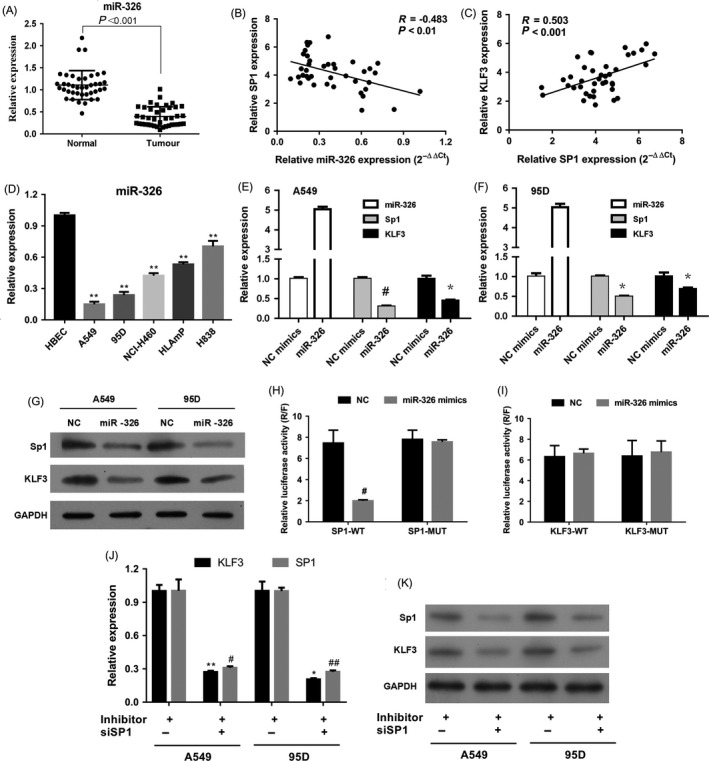
Sp1 is a direct downstream target of miR‐326. A, Relative expression of miR‐326 in lung cancer tissues was confirmed by real‐time PCR. N = 40. P < 0.001. B, Correlation analysis between miR‐326 and Sp1 levels. P < 0.01. C, Correlation analysis between Sp1 and KLF3 expressions. P < 0.001. D, Real‐time PCR results revealed that miR‐326 was downregulated in lung cancer cell lines. **P < 0.001 vs HBECs. E, Overexpression of miR‐326 downregulated the expression of Sp1 and KLF3 in A549 cells. Mean ± SD. *P < 0.05; #P < 0.05. F, Overexpression of miR‐326 reduced the expression of Sp1 and KLF3 in 95D cells. Mean ± SD. *P < 0.05; #P < 0.05. G, Western blot analysis demonstrated that miR‐326 inhibited Sp1 and KLF3 expression in A549 and 95D cells. H, Luciferase activity of Sp1 wild type (wt) was inhibited by using miR‐326 mimics, compared to Sp1 mutant (mut). #P < 0.05. I, No significant difference of luciferase activity of KLF3 wild type (wt) and KLF3 mutant (mut) was found by using miR‐326 mimics. #P > 0.05. J, The levels of Sp1 and KLF3 mRNAs were significantly reduced in group of miR‐326 inhibitor+siSP1 than those of miR‐326 inhibitor+siRNA‐NC in both A549 and 95D cells. miR‐326 antagomir was used as inhibitor. *P < 0.05; #P < 0.05; **P < 0.01; ##P < 0.01. K, The expressions of Sp1 and KLF3 proteins were obviously decreased in group of miR‐326 inhibitor+siSP1 than those of miR‐326 inhibitor+siRNA‐NC in both A549 and 95D cells. miR‐326 antagomir was used as inhibitor
3.8. miR‐326 and KLF3 are associated with overall survival in patients with lung cancer
Based on the above findings, we further evaluated whether miR‐326 and KLF3 expression are associated with clinical prognosis. Through analysis of a dataset of lung cancer that includes 40 patients, we found that decreased miR‐326 expression was associated with poor prognosis among patients with lung cancer (Figure 8A, P = 0.003). Of note, the patients with high level of KLF3 were associated with poor overall survival (OS) caused by lung cancer (Figure 8B, P = 0.001). Clinicopathologic features were compared between these groups (Table 4), and significantly higher recurrence as well as death rate was observed in lung cancer patients with low miR‐326 and high KLF3 levels, when compared to the high‐miR‐326 group and the low‐KLF3 group (P < 0.05). However, the miR‐326 and KLF3 expression levels were not associated with age (P = 0.093 and P = 0.069, respectively), gender (P = 0.05) and TNM tumour stage (P = 0.278 and P = 0.207, respectively). These findings indicate that miR‐326/Sp1/KLF3 regulatory axis participated in lung cancer progression.
Figure 8.
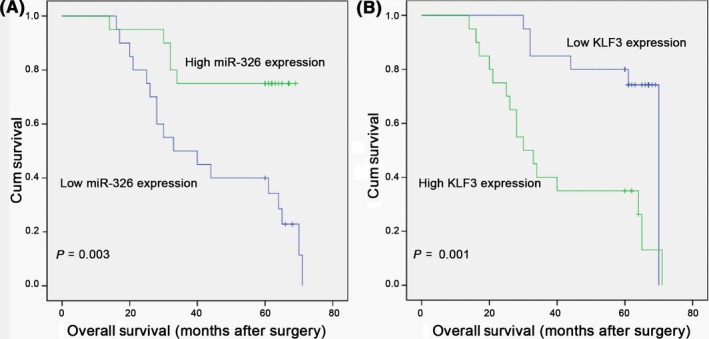
miR‐326 and KLF3 are associated with overall survival (OS) in patients with lung cancer. A, Kaplan–Meier plots of OS in patients with lung cancer, stratified by miR‐326 expression. The P value was calculated by a log‐rank test. B, Kaplan–Meier plots of OS stratified by KLF3 expression. The results showed that high levels of KLF3 were associated with significantly poorer OS in patients with lung cancer, P = 0.001
Table 4.
Correlation between miR‐326 and KLF3 mRNA expression and clinicopathological characteristics with lung cancer
| Parameter Clinical | Overall | Expression of miR‐326 | Expression of KLF3 | ||||
|---|---|---|---|---|---|---|---|
| Low | High | P value | Low | High | P value | ||
| In Total | 40 | 20 (50.00) | 20 (50.00) | ‐ | 20 (50.00) | 20 (50.00) | ‐ |
| Age (y, mean ± SD) | 63.90 ± 9.98 | 61.25 ± 9.59 | 66.55 ± 9.88 | 0.093 | 67.55 ± 9.71 | 60.25 ± 9.05 | 0.069 |
| Male (n, %) | 33 (82.50) | 17 | 16 | 0.5 | 16 | 17 | 0.5 |
| Follow‐up duration (months, mean ± SD) | 48.80 ± 19.33 | 42.65 ± 20.47 | 54.95 ± 16.37 | 0.043 | 58.60 ± 12.98 | 39.00 ± 19.90 | 0.001 |
| TNM (n, %) | |||||||
| IB | 17 (42.50) | 6 | 11 | 0.278 | 11 | 6 | 0.207 |
| IIA | 10 (25.00) | 6 | 4 | 3 | 7 | ||
| IIB | 13 (32.50) | 8 | 5 | 6 | 7 | ||
| Recurrence (n, %) | |||||||
| Overall | 23 (57.50) | 18 | 5 | <0.001 | 7 | 16 | 0.01 |
| 1‐y recurrence | 11 (27.50) | 9 | 2 | 0.031 | 1 | 10 | 0.003 |
| 5‐y recurrence | 12 (30.00) | 9 | 3 | 0.082 | 6 | 6 | 1 |
| Death | 22 (55.00) | 17 | 5 | <0.001 | 6 | 16 | 0.004 |
| 5‐y actual survival | 23 (57.50) | 5 | 12 | 0.054 | 13 | 4 | 0.01 |
3.9. The decrease of KLF3 inhibited the growth of xenograft tumour in nude mice
To further investigate the effect of KLF3 on tumour growth in vivo, both A549 and 95D cells were transfected with NC or si‐KLF3 and then hypodermically injected into the BALB/c nude mice. The reduction in KLF3 significantly depressed the tumour volumes in a time‐dependent manner (P < 0.05) and also remarkably lowered the tumour weights in both A549 and 95D cells (Figure 9A‐E, P < 0.05). Consistent with the results in vitro, Western blot and immunohistochemical assays revealed that the phosphorylation levels of JAK2, STAT3, PI3K and AKT proteins also declined in BALB/c nude mice injecting A549 and 95D cells with decreasing level of KLF3 (Figure 10A and B). Together, these results suggest that KLF3 is involved in the tumour growth of A549 and 95D cells through regulating JAK2/STAT3 and PI3K/AKT signalling pathways in vivo.
Figure 9.
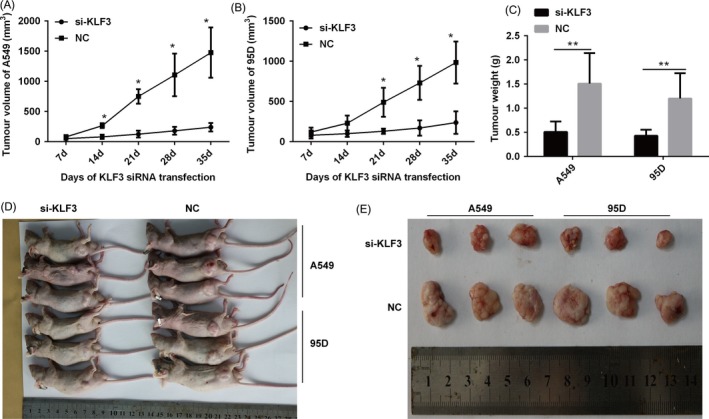
Knockdown of KLF3 inhibited the growth of xenograft tumour in nude mice. (A and B) Line graphs showed that knockdown the expression of KLF3 significantly inhibited the tumour volume in nude mice injecting A549 (A) and 95D cells (B). *indicates that P < 0.05. (C) Bar graph demonstrated that knockdown the expression of KLF3 remarkably suppressed the tumour weight in nude mice injecting A549 and 95D cells. **indicates that P < 0.01. (D and E) Gross observation of xenograft tumour revealed that the tumour volume in nude mice injecting KLF3‐silenced A549 and 95D cells was much smaller
Figure 10.
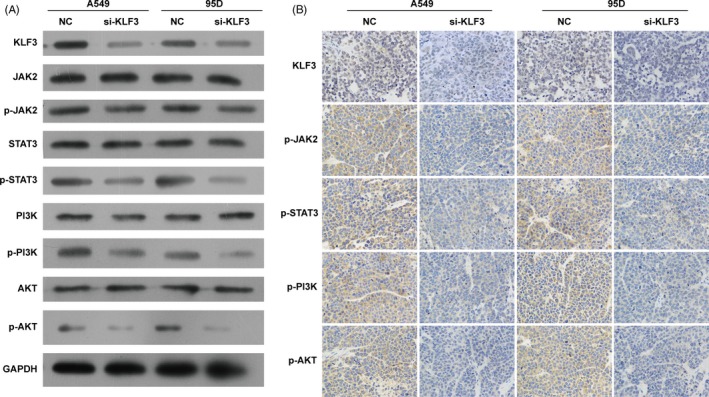
Knockdown of KLF3 suppressed the activation of JAK2/STAT3 and PI3K/AKT signalling pathways in vivo. A and B, Western blot and immunohistochemical analysis (×400) revealed that knockdown the expression of KLF3 obviously downregulated the phosphorylation levels of JAK2, STAT3, PI3K, and AKT proteins in nude mice injecting KLF3‐silenced A549 and 95D cells in vivo
4. DISCUSSION
KLF3 was originally discovered as a CACCC box‐binding transcription factor in erythroid cells.48 In fact, similar to other KLF members, KLF3 exhibits both transcriptional activation and repression via interaction with other factors.27, 49 Although the role of KLF3 has been extensively studied in the development of hematopoietic system, little is known about KLF3 in cancer. In 2011, Magali et al reported that the expression of KLF3 was downregulated in patients with acute myeloid leukaemia compared with healthy donors.50 In addition, Sachdeva et al found that KLF3 expression was downregulated in both primary mouse and human metastatic sarcomas due to the promoter hypermethylation.51 Intriguingly, increased Krüppel‐like factor 6 splice variant 1 (KLF6‐SV1) expression is associated with poor prognosis in prostate, lung and ovarian cancer.52 By contrast, the mRNA expression level of Krüppel‐like factor 6 splice variant 2 (KLF6‐SV2) in colorectal cancer tissues was decreased than in corresponding normal tissues.53 KLF6 expression is also decreased in gastric cancers. It has been implicated that the difference of KLF3 expression is owing to the loss of heterozygosity, mutations and alternative splicing. 54, 55 Identically, the difference in expressional changes exists among various types of cancers. For instance, in our study, KLF3 expression was elevated in lung cancer tissues, and abnormal high level of KLF3 was associated with poor overall survival in patients with lung cancer, which was consistent with previous finding.56 The decrease in KLF3 reduced cell proliferation, migration and invasion in vitro, and also inhibited tumour growth in BALB/c nude mice, suggesting that KLF3 may function as an oncogene during lung cancer progression. Nevertheless, KLF3 was downregulated in acute myeloid leukaemia and sarcomas,50, 51 and loss of KLF3 was correlated with aggressive phenotypes and poor survival outcomes in colorectal cancer.57 Additionally, downregulation of KLF3 blocked JAK2/STAT3 and PI3K/AKT signalling pathways. It is generally known that aberrant JAK2/STAT3 and PI3K/AKT signalling pathways are associated with the occurrence and development of cancers including tumour cell proliferation, migration and apoptosis.58, 59, 60 Thus, we propose KLF3 may mediate JAK2/STAT3 and PI3K/AKT signalling pathways during lung cancer development. The distinct splicing, differential enrichment of KLF3 and leading pathways in diverse organs may result in contrasting level of KLF3 in the development of tumours.
As a transcription factor, KLF3 was already known to exert as a strong transcriptional repressor. Some studies have identified the potential regulatory mechanisms of KLF3 in different tissues. Dewi et al found that phosphorylation of KLF3 and CtBP2 by HIPK2 strengthens the interaction between these two factors and increases transcriptional repression by KLF3.29 KLF3 can also be modified by small ubiquitin‐like modification (SUMOylation).61 Lack of SUMOylation and disconnection with CtBP result in a loss of transcription repression and gain of transcription activation potential.61 Through bioinformatics analysis, certain putative binding sites of Sp1 were found related to the promoter of KLF3, which suggests that Sp1 presents as a positive regulator of KLF3. Next, we examined the expression of KLF3 after impeding Sp1 level and found that KLF3 was downregulated in A549 and 95D cells. Finally, the luciferase reporter assay, EMSA and ChIP assays validated that KLF3 was a novel direct target gene of Sp1.
Sp1 is an ubiquitously expressed transcription factor and is considered to be a general transcription factor required for transcription of a large number of housekeeping genes.40, 62 However, accumulating evidence indicates that Sp1 is upregulated in various types of human cancer, including lung cancer.63 Consistent with these reports, our study also revealed that Sp1 was increased in lung cancer tissues compared with that in adjacent normal tissues.
Usually, miRNAs modulate cancer cell proliferation and migration by restricting expression of downstream targets. The interaction between lncRNA PDIA3P and miR‐185‐5p affects oral squamous cell carcinoma progression by targeting cyclin D2.64 Sp1 contributes to tumour progression and is regulated by many miRNAs. Mao et al demonstrated that miR‐330 inhibited prostate cancer cell migration by targeting Sp1.65 Wang et al also revealed that miR‐375 could bind to the 3′UTR of Sp1. In lung cancer, Sp1 could be repressed by miR‐29c, miR‐27b, miR‐335 and miR‐429 in translational level.66, 67, 68 Similar to these findings, our previous study revealed that miR‐326 was deterred in lung cancer cells, and transfection of miR‐326 significantly decreased cell proliferation and migration.44 Recently, the increasing studies have focused on the target genes of miR‐326 that could interact with miR‐326 to impede tumour development and progression, such as NOB1,11 phox2a,44 CCND1,69 FSCN115 and NSBP1.70 In the present study, we demonstrated that Sp1 is a downstream target of miR‐326. We note that non‐coding RNAs, including lncRNA, miRNA, circRNA, as well as their interactions, play a pivotal role in regulation of various types of cancer and other diseases such as acute myocardial infarction, spinal muscular atrophy and cardiac dysfunction. 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81 Particularly, long non‐coding RNA XIST acts as an oncogene in non–small‐cell lung cancer by epigenetically repressing KLF2 expression, indicating that lncRNAs also exert mediating function by targeting KLF protein.75 Our further study may focus the regulatory axis of lncRNA‐miRNA in the progression of lung cancer, as well as to elucidate the possible mechanisms.
In summary, our study provides evidence that miR‐326/Sp1/KLF3 regulatory axis involves in lung cancer cell proliferation, migration and invasion, which provide leads for the further therapy to patients with lung cancer.
CONFLICT OF INTERESTS
None.
AUTHORS’ CONTRIBUTION
RW, YQS and WD conceived the study and performed the whole experimental work in its design and coordination. WZ, JLX, TZQ, JL, MLZ, QQW and TPX participated in parts of the experiments and analysis of the data. JX, LJZ, RHG, KHL, YMY, YHG, PWH, PL and LKL contributed to provide clinical samples and parameters. The manuscript was written by RW. All authors read and approved the final manuscript.
Supporting information
ACKNOWLEDGEMENT
This work was supported by the National Natural Science Foundation of China (Grant No: 81502513), The Jiangsu Province Natural Science Foundation of China (Grant No: BK2015‐1028) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801).
Wang R, Xu J, Xu J, et al. MiR‐326/Sp1/KLF3: A novel regulatory axis in lung cancer progression. Cell Prolif. 2019;52:e12551 10.1111/cpr.12551
REFERENCES
- 1. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1‐19. [DOI] [PubMed] [Google Scholar]
- 2. Dela Cruz CS, Tanoue LT, Matthay RA. Lung cancer: epidemiology, etiology, and prevention. Clin Chest Med Dec. 2011;32(4):605‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steliga MA, Dresler CM. Epidemiology of lung cancer: smoking, secondhand smoke, and genetics. Surg Oncol Clin N Am. 2011;20(4):605‐618. [DOI] [PubMed] [Google Scholar]
- 4. Yano T, Haro A, Shikada Y, Maruyama R, Maehara Y. Non‐small cell lung cancer in never smokers as a representative ‘non‐smoking‐associated lung cancer’: epidemiology and clinical features. Int J Clin Oncol. 2011;16(4):287‐293. [DOI] [PubMed] [Google Scholar]
- 5. Wen J, Fu J‐H, Zhang W, Guo M. Lung carcinoma signaling pathways activated by smoking. Chin J Cancer. 2011;30(8):551‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sax SN, Zu K, Goodman JE. Air pollution and lung cancer in Europe. Lancet Oncol. 2013;14(11):e439‐e440. [DOI] [PubMed] [Google Scholar]
- 7. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271‐289. [DOI] [PubMed] [Google Scholar]
- 8. Kim J, Krichevsky A, Grad Y, et al. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci U S A. 2004;101(1):360‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Liu D, Zhang X, et al. miR‐326 regulates HbF synthesis by targeting EKLF in human erythroid cells. Exp Hematol. 2018;63:33‐40.e2. [DOI] [PubMed] [Google Scholar]
- 10. Liu W, Zhang B, Xu N, Wang MJ, Liu Q. miR‐326 regulates EMT and metastasis of endometrial cancer through targeting TWIST1. Eur Rev Med Pharmacol Sci. 2017;21(17):3787‐3793. [PubMed] [Google Scholar]
- 11. Ji S, Zhang B, Kong Y, Ma F, Hua Y. miR‐326 inhibits gastric cancer cell growth through downregulating NOB1. Oncol Res. 2017;25(6):853‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J, Li S, Chen Z, et al. miR‐326 reverses chemoresistance in human lung adenocarcinoma cells by targeting specificity protein 1. Tumour Biol. 2016;37(10):13287‐13294. [DOI] [PubMed] [Google Scholar]
- 13. Cao L, Wang J, Wang PQ. MiR‐326 is a diagnostic biomarker and regulates cell survival and apoptosis by targeting Bcl‐2 in osteosarcoma. Biomed Pharmacother. 2016;84:828‐835. [DOI] [PubMed] [Google Scholar]
- 14. Yu S, Huang H, Deng G, et al. miR‐326 targets antiapoptotic Bcl‐xL and mediates apoptosis in human platelets. PLoS One. 2015;10(4):e0122784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Y, Gao Y, Xu Y, Ma H, Yang M. Down‐regulation of miR‐326 is associated with poor prognosis and promotes growth and metastasis by targeting FSCN1 in gastric cancer. Growth Factors. 2015;33(4):267‐274. [DOI] [PubMed] [Google Scholar]
- 16. Cai M, Wang Z, Zhang J, et al. Adam17, a target of Mir‐326, promotes emt‐induced cells invasion in lung adenocarcinoma. Cell Physiol Biochem. 2015;36(3):1175‐1185. [DOI] [PubMed] [Google Scholar]
- 17. Zahednasab H, Balood M. The role of miR‐326 and miR‐26a in MS disease activity. Gene. 2014;548(1):158. [DOI] [PubMed] [Google Scholar]
- 18. Jiang Z, Cushing L, Ai X, Lü J. miR‐326 is downstream of sonic hedgehog signaling and regulates the expression of Gli2 and smoothened. Am J Respir Cell Mol Biol. 2014;51(2):273‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Valencia K, Martin‐Fernandez M, Zandueta C, et al. miR‐326 associates with biochemical markers of bone turnover in lung cancer bone metastasis. Bone. 2013;52(1):532‐539. [DOI] [PubMed] [Google Scholar]
- 20. Liang Z, Wu H, Xia J, et al. Involvement of miR‐326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance‐associated protein 1. Biochem Pharmacol. 2010;79(6):817‐824. [DOI] [PubMed] [Google Scholar]
- 21. Du C, Liu C, Kang J, et al. MicroRNA miR‐326 regulates TH‐17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10(12):1252‐1259. [DOI] [PubMed] [Google Scholar]
- 22. Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin SH. Isolation and characterization of the cDNA encoding BKLF/TEF‐2, a major CACCC‐box‐binding protein in erythroid cells and selected other cells. Mol Cell Biol. 1996;16(4):1695‐1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S. Kruppel‐like transcription factors: a functional family. Int J Biochem Cell Biol. 2008;40(10):1996‐2001. [DOI] [PubMed] [Google Scholar]
- 24. Ma Y, Wang B, Jiang F, et al. A feedback loop consisting of microRNA 23a/27a and the beta‐like globin suppressors KLF3 and SP1 regulates globin gene expression. Mol Cell Biol. 2013;33(20):3994‐4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Funnell AP, Norton LJ, Mak KS, et al. The CACCC‐binding protein KLF3/BKLF represses a subset of KLF1/EKLF target genes and is required for proper erythroid maturation in vivo. Mol Cell Biol. 2012;32(16):3281‐3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang W, Kadam S, Emerson BM, Bieker JJ. Site‐Specific Acetylation by p300 or CREB binding protein regulates erythroid Kruppel‐like factor transcriptional activity via its interaction with the SWI‐SNF complex. Mol Cell Biol. 2001;21(7):2413‐2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turner J, Crossley M. Cloning and characterization of mCtBP2, a co‐repressor that associates with basic Kruppel‐like factor and other mammalian transcriptional regulators. EMBO J. 1998;17(17):5129‐5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi Y, Sawada J, Sui G, et al. Coordinated histone modifications mediated by a CtBP co‐repressor complex. Nature. 2003;422(6933):735‐738. [DOI] [PubMed] [Google Scholar]
- 29. Dewi V, Kwok A, Lee S, et al. Phosphorylation of Kruppel‐like factor 3 (KLF3/BKLF) and C‐terminal binding protein 2 (CtBP2) by homeodomain‐interacting protein kinase 2 (HIPK2) modulates KLF3 DNA binding and activity. J Biol Chem. 2015;290(13):8591‐8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Funnell AP, Mak KS, Twine NA, et al. Generation of mice deficient in both KLF3/BKLF and KLF8 reveals a genetic interaction and a role for these factors in embryonic globin gene silencing. Mol Cell Biol. 2013;33(15):2976‐2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sue N, Jack BH, Eaton SA, et al. Targeted disruption of the basic Kruppel‐like factor gene (Klf3) reveals a role in adipogenesis. Mol Cell Biol. 2008;28(12):3967‐3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vu TT, Gatto D, Turner V, et al. Impaired B cell development in the absence of Kruppel‐like factor 3. J Immunol. 2011;187(10):5032‐5042. [DOI] [PubMed] [Google Scholar]
- 33. Turchinovich G, Vu TT, Frommer F, et al. Programming of marginal zone B‐cell fate by basic Kruppel‐like factor (BKLF/KLF3). Blood. 2011;117(14):3780‐3792. [DOI] [PubMed] [Google Scholar]
- 34. Bell‐Anderson KS, Funnell AP, Williams H, et al. Loss of Kruppel‐like factor 3 (KLF3/BKLF) leads to upregulation of the insulin‐sensitizing factor adipolin (FAM132A/CTRP12/C1qdc2). Diabetes. 2013;62(8):2728‐2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Himeda CL, Ranish JA, Pearson RC, Crossley M, Hauschka SD. KLF3 regulates muscle‐specific gene expression and synergizes with serum response factor on KLF binding sites. Mol Cell Biol. 2010;30(14):3430‐3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Briggs MR, Kadonaga JT, Bell SP, Tjian R. Purification and biochemical characterization of the promoter‐specific transcription factor, Sp1. Science. 1986;234(4772):47‐52. [DOI] [PubMed] [Google Scholar]
- 37. Yao JC, Wang L, Wei D, et al. Association between expression of transcription factor Sp1 and increased vascular endothelial growth factor expression, advanced stage, and poor survival in patients with resected gastric cancer. Clin Cancer Res. 2004;10(12 Pt 1):4109‐4117. [DOI] [PubMed] [Google Scholar]
- 38. Jiang NY, Woda BA, Banner BF, Whalen GF, Dresser KA, Lu D. Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2008;17(7):1648‐1652. [DOI] [PubMed] [Google Scholar]
- 39. Yen WH, Ke WS, Hung JJ, Chen TM, Chen JS, Sun HS. Sp1‐mediated ectopic expression of T‐cell lymphoma invasion and metastasis 2 in hepatocellular carcinoma. Cancer Med. 2016;5(3):465‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Black AR, Black JD, Azizkhan‐Clifford J. Sp1 and kruppel‐like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188(2):143‐160. [DOI] [PubMed] [Google Scholar]
- 41. Safe S, Imanirad P, Sreevalsan S, Nair V, Jutooru I. Transcription factor Sp1, also known as specificity protein 1 as a therapeutic target. Expert Opin Ther Targets. 2014;18(7):759‐769. [DOI] [PubMed] [Google Scholar]
- 42. Guan H, Cai J, Zhang N, et al. Sp1 is upregulated in human glioma, promotes MMP‐2‐mediated cell invasion and predicts poor clinical outcome. Int J Cancer. 2012;130(3):593‐601. [DOI] [PubMed] [Google Scholar]
- 43. Choi J, Park SY, Joo CK. Transforming growth factor‐beta1 represses E‐cadherin production via slug expression in lens epithelial cells. Invest Ophthalmol Vis Sci. 2007;48(6):2708‐2718. [DOI] [PubMed] [Google Scholar]
- 44. Wang R, Chen X, Xu T, et al. MiR‐326 regulates cell proliferation and migration in lung cancer by targeting phox2a and is regulated by HOTAIR. Am J Cancer Res. 2016;6(2):173‐186. [PMC free article] [PubMed] [Google Scholar]
- 45. Zohre S, Kazem NK, Abolfazl A, et al. Trichostatin A‐induced apoptosis is mediated by Kruppel‐like factor 4 in ovarian and lung cancer. Asian Pac J Cancer Prev. 2014;15(16):6581‐6586. [DOI] [PubMed] [Google Scholar]
- 46. Yang JH, Li XY, Wang X, et al. Long‐term persistent infection of HPV 16 E6 up‐regulate SP1 and hTERT by inhibiting LKB1 in lung cancer cells. PLoS One. 2017;12(8):e0182775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li M, Ling B, Xiao T, et al. Sp1 transcriptionally regulates BRK1 expression in non‐small cell lung cancer cells. Gene. 2014;542(2):134‐140. [DOI] [PubMed] [Google Scholar]
- 48. Miller IJ, Bieker JJ. A novel, erythroid cell‐specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol Cell Biol. 1993;13(5):2776‐2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Turner J, Nicholas H, Bishop D, Matthews JM, Crossley M. The LIM protein FHL3 binds basic Kruppel‐like factor/Kruppel‐like factor 3 and its co‐repressor C‐terminal‐binding protein 2. J Biol Chem. 2003;278(15):12786‐12795. [DOI] [PubMed] [Google Scholar]
- 50. Humbert M, Halter V, Shan D, et al. Deregulated expression of Kruppel‐like factors in acute myeloid leukemia. Leuk Res. 2011;35(7):909‐913. [DOI] [PubMed] [Google Scholar]
- 51. Sachdeva M, Dodd RD, Huang Z, et al. Epigenetic silencing of Kruppel like factor‐3 increases expression of pro‐metastatic miR‐182. Cancer Lett. 2015;369(1):202‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. DiFeo A, Martignetti JA, Narla G. The role of KLF6 and its splice variants in cancer therapy. Drug Resist Update. 2009;12(1–2):1‐7. [DOI] [PubMed] [Google Scholar]
- 53. Zhang B, Guo DD, Zheng JY, Wu YA. Expression of KLF6‐SV2 in colorectal cancer and its impact on proliferation and apoptosis. Eur J Cancer Prev. 2018;27(1):20‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cho YG, Kim CJ, Park CH, et al. Genetic alterations of the KLF6 gene in gastric cancer. Oncogene. 2005;24(28):4588‐4590. [DOI] [PubMed] [Google Scholar]
- 55. Sangodkar J, Shi J, DiFeo A, et al. Functional role of the KLF6 tumour suppressor gene in gastric cancer. Eur J Cancer. 2009;45(4):666‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gao L, Qiu H, Liu J, et al. KLF15 promotes the proliferation and metastasis of lung adenocarcinoma cells and has potential as a cancer prognostic marker. Oncotarget. 2017;8(66):109952‐109961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang X, Jiang Z, Zhang Y, Wang X, Liu L, Fan Z. RNA sequencing analysis reveals protective role of kruppel‐like factor 3 in colorectal cancer. Oncotarget. 2017;8(13):21984‐21993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(Pt 8):1281‐1283. [DOI] [PubMed] [Google Scholar]
- 59. Lin HL, Yang MH, Wu CW, et al. 2‐Methoxyestradiol attenuates phosphatidylinositol 3‐kinase/Akt pathway‐mediated metastasis of gastric cancer. Int J Cancer. 2007;121(11):2547‐2555. [DOI] [PubMed] [Google Scholar]
- 60. Ke F, Wang Z, Song X, et al. Cryptotanshinone induces cell cycle arrest and apoptosis through the JAK2/STAT3 and PI3K/Akt/NFkappaB pathways in cholangiocarcinoma cells. Drug Des Devel Ther. 2017;11:1753‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perdomo J, Verger A, Turner J, Crossley M. Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol Cell Biol. 2005;25(4):1549‐1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dynan WS, Tjian R. The promoter‐specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell. 1983;35(1):79‐87. [DOI] [PubMed] [Google Scholar]
- 63. Hsu TI, Wang MC, Chen SY, et al. Sp1 expression regulates lung tumor progression. Oncogene. 2012;31(35):3973‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun CC, Zhang L, Li G, et al. The lncRNA PDIA3P interacts with miR‐185‐5p to modulate oral squamous cell carcinoma progression by targeting cyclin D2. Mol Ther Nucleic Acids. 2017;9:100‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mao Y, Chen H, Lin Y, et al. microRNA‐330 inhibits cell motility by downregulating Sp1 in prostate cancer cells. Oncol Rep. 2013;30(1):327‐333. [DOI] [PubMed] [Google Scholar]
- 66. Zhang HW, Wang EW, Li LX, et al. A regulatory loop involving miR‐29c and Sp1 elevates the TGF‐beta1 mediated epithelial‐to‐mesenchymal transition in lung cancer. Oncotarget. 2016;7(52):85905‐85916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu D, Niu X, Pan H, et al. Tumor‐suppressing effects of microRNA‐429 in human renal cell carcinoma via the downregulation of Sp1. Oncol Lett. 2016;12(4):2906‐2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang H, Li M, Zhang R, et al. Effect of miR‐335 upregulation on the apoptosis and invasion of lung cancer cell A549 and H1299. Tumor Biol. 2013;34(5):3101‐3109. [DOI] [PubMed] [Google Scholar]
- 69. Sun C, Huang C, Li S, et al. Hsa‐miR‐326 targets CCND1 and inhibits non‐small cell lung cancer development. Oncotarget. 2016;7(7):8341‐8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li D, Du X, Liu A, Li P. Suppression of nucleosome‐binding protein 1 by miR‐326 impedes cell proliferation and invasion in non‐small cell lung cancer cells. Oncol Rep. 2016;35(2):1117‐1124. [DOI] [PubMed] [Google Scholar]
- 71. Sun CC, Li SJ, Li G, Hua RX, Zhou XH, Li DJ. Long Intergenic Noncoding RNA 00511 Acts as an Oncogene in Non‐small‐cell Lung Cancer by Binding to EZH2 and Suppressing p57. Mol Ther Nucleic Acids. 2016;5(11):e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sun CC, Li SJ, Zhang F, et al. The novel miR‐9600 suppresses tumor progression and promotes paclitaxel sensitivity in non‐small‐cell lung cancer through altering STAT3 expression. Mol Ther Nucleic Acids. 2016;5(11):e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang ZW, Chen JJ, Xia SH, et al. Long intergenic non‐protein coding RNA 319 aggravates lung adenocarcinoma carcinogenesis by modulating miR‐450b‐5p/EZH2. Gene. 2018;650:60‐67. [DOI] [PubMed] [Google Scholar]
- 74. Wang H, Bei Y, Huang P, et al. Inhibition of miR‐155 protects against LPS‐induced Cardiac dysfunction and apoptosis in mice. Mol Ther Nucleic Acids. 2016;5:e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fang J, Sun CC, Gong C. Long noncoding RNA XIST acts as an oncogene in non‐small cell lung cancer by epigenetically repressing KLF2 expression. Biochem Biophys Res Commun. 2016;478(2):811‐817. [DOI] [PubMed] [Google Scholar]
- 76. Sun C, Li S, Zhang F, et al. Long non‐coding RNA NEAT1 promotes non‐small cell lung cancer progression through regulation of miR‐377‐3p‐E2F3 pathway. Oncotarget. 2016;7(32):51784‐51814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sun CC, Li SJ, Yuan ZP, Li DJ. MicroRNA‐346 facilitates cell growth and metastasis, and suppresses cell apoptosis in human non‐small cell lung cancer by regulation of XPC/ERK/Snail/E‐cadherin pathway. Aging. 2016;8(10):2509‐2524. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78. Sun C, Li S, Yang C, et al. MicroRNA‐187‐3p mitigates non‐small cell lung cancer (NSCLC) development through down‐regulation of BCL6. Biochem Biophys Res Commun. 2016;471(1):82‐88. [DOI] [PubMed] [Google Scholar]
- 79. Catapano F, Zaharieva I, Scoto M, et al. Altered levels of microRNA‐9, ‐206, and ‐132 in spinal muscular atrophy and their response to antisense oligonucleotide therapy. Mol Ther Nucleic Acids. 2016;5(7):e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Huang W, Tian SS, Hang PZ, Sun C, Guo J, Du ZM. Combination of microRNA‐21 and microRNA‐146a attenuates cardiac dysfunction and apoptosis during acute myocardial infarction in mice. Mol Ther Nucleic Acids. 2016;5:e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hu W, Bi ZY, Chen ZL, et al. Emerging landscape of circular RNAs in lung cancer. Cancer Lett. 2018;427:18‐27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials