

Abstract
Objectives: Non‐steroidal anti‐inflammatory drugs have been shown to induce apoptosis in primary B‐cell chronic lymphocytic leukaemia (CLL) cells, but the molecular mechanisms that underpin this observation have not been fully elucidated. Here, we have analysed the effect two novel aspirin analogues, 2‐hydroxy benzoate zinc (2HBZ) and 4‐hydroxy benzoate zinc (4HBZ), on primary CLL samples.
Materials and methods: Cytotoxic effects of 2HBZ and 4HBZ were analysed in primary CLL cells derived from 52 patients, and normal B‐ and T‐lymphocytes. Mechanisms of action of these agents were also elucidated.
Results: Both analogues induced apoptosis in a dose‐dependent and time‐dependent manner. Apoptosis was associated with activation of caspase‐3 that could be partially abrogated by the caspase‐9 inhibitor (Z‐LEHD.fmk). Importantly, both agents demonstrated preferential cytotoxicity in CLL cells when compared to normal B‐ and T‐lymphocytes. In terms of their molecular mechanisms of action, 4HBZ and 2HBZ inhibited COX‐2 transcription and protein expression and this was associated with upstream inhibition of transcription factor Rel A. Co‐culture of CLL cells with CD40 ligand‐expressing mouse fibroblasts significantly increased COX‐2 expression and inhibited spontaneous apoptosis. Importantly, the most potent analogue, 4HBZ, overcame pro‐survival effects of the co‐culture system and significantly repressed COX‐2. Finally, elevated COX‐2 expression was associated with poor prognostic subsets and increased sensitivity to 4HBZ.
Conclusions: Our results demonstrate therapeutic potential of 4HBZ and are consistent with a mechanism involving suppression of Rel A nuclear translocation and inhibition of COX‐2 transcription.
Introduction
B‐cell chronic lymphocytic leukaemia (CLL) is the most common form of leukaemia in the Western world. It is characterized by clonal expansion of CD5+, CD19+, CD23+ and CD20+ B cells (1, 2, 3), and molecular pathogenesis of CLL appears to be due, at least in part, to defects in apoptosis (4, 5, 6, 7). Most CLL patients show initial responsiveness to frontline chemotherapeutic drugs and monoclonal antibodies, and introduction of agents such as purine nucleoside analogue fludarabine, and rituximab, has resulted in higher rates of remission (8, 9, 10, 11). However, treated patients almost invariably develop drug resistance and, as a result, there has been little improvement in overall survival for patients with this condition (12, 13). Consequently, there is much interest in identifying novel therapies that will add to the current therapeutic arsenal and reduce toxicities associated with combination chemotherapy/monoclonal antibody regimens.
Aspirin and other non‐steroidal anti‐inflammatory drugs (NSAIDs) have been shown to exhibit a broad spectrum of biological activities including anti‐proliferative, anti‐cancer, anti‐oxidant and anti‐inflammatory properties (14, 15, 16, 17, 18, 19). Epidemiological studies have shown close correlation between use of these compounds and reduced risk of developing certain human cancers (20, 21, 22, 23). The anti‐cancer effects of NSAIDs are clearly multi‐factorial, but inhibition of cyclo‐oxygenase 2 (COX‐2) appears to be an important molecular target. COX‐2 has a number of carcinogenic properties including ability to stimulate angiogenesis by promoting prostaglandin biosynthesis and by increasing vascular endothelial growth factor (VEGF) expression (24, 25). Increased COX‐2 may also contribute to elevated matrix metalloproteinases and increase Bcl‐2 expression (26, 27). Unlike normal B cells, CLL cells exhibit constitutive expression of COX‐2 and high expression is associated with resistance to apoptosis and poor prognosis in this disease (28, 29). Reasons for over expression of COX‐2 are not yet fully resolved, but transcription of COX‐2 is regulated by NF‐κB (30). As NF‐κB is also constitutively activated in CLL (31), it provides a potential rationale for over expression of COX‐2. Over the last decade, a number of studies have shown that NSAIDs may have efficacy in treatment of B‐cell malignancies including CLL (14, 32, 33, 34). In this study, we report on pre‐clinical evaluation of two novel aspirin analogues, 2‐hydroxy benzoate zinc (2HBZ) and 4‐hydroxy benzoate zinc (4HBZ). We compared their cytotoxic profiles with aspirin in 52 primary CLL patient samples as well to normal B‐ and T‐lymphocytes from 10 age‐matched controls. In addition, we investigated primary molecular mechanism(s) of action and found that inhibition of COX‐2 and NF‐κB subunit Rel A preceded induction of apoptosis.
Materials and methods
Patients’ cells and clinical details
Peripheral blood samples from 52 patients with CLL and 10 age‐matched normal controls were obtained with patients’ informed consent. CLL was defined by clinical criteria as well as cell morphology and co‐expression of CD19 and CD5 in lymphocytes, simultaneously displaying restriction of light‐chain rearrangement. Staging was based on the Binet classification system (35) and IGHV gene mutational status, CD38 expression, and ZAP‐70 expression was determined for all 52 patients using methods described previously (36). Clinical characteristics of the patient cohort are summarized in Table 1.
Table 1.
Clinical characteristics of the CLL patients in this study
| No. of patients | 52 |
| Mean age (years) | 67 |
| Gender (male/female) | 33/19 |
| Binet stage (A/B/C) | 29/10/13 |
| Previous treatment (untreated/treated) | 17/35 |
| IGHV gene mutation (mutated/unmutated) | 37/15 |
| CD38 expression (<20%/≥20%) | 29/23 |
| ZAP‐70 expression (<20%/≥20%) | 26/26 |
Preparation of novel aspirin analogues
The two novel aspirin analogues, 2HBZ and 4HBZ, were synthesized from 2‐hydroxy or 4‐hydroxybenzoic acid and zinc sulphate and liquid chromatographic analysis showed that both analogues were more than 97% pure (data not shown); structure of each analogue is given in Fig. 1a. Aspirin was purchased from Sigma Chemicals Co (Poole, UK).
Figure 1.
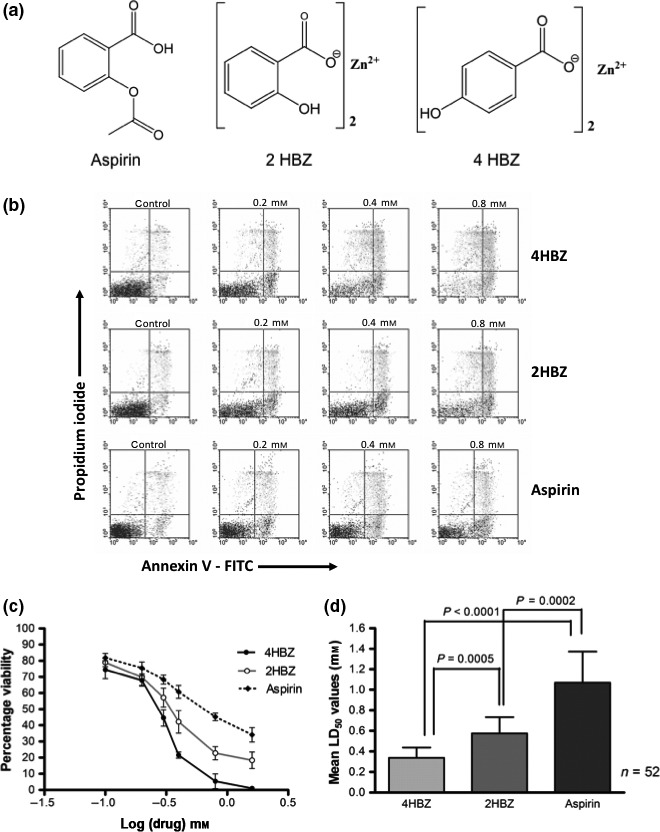
Comparison of in vitro cytotoxicity of aspirin, 2HBZ and 4HBZ on CLL cells. (a) Chemical structure of aspirin, 4HBZ and 2HBZ (b) annexin V/propidium iodide (PI) dotplots demonstrating dose‐dependent increase in apoptosis induced by aspirin, 4HBZ and 2HBZ following in vitro culture for 48 h. Apoptosis was quantified by summation of the lower right quadrant and upper right quadrants. (c) Representative example of dose–response curves for 4HBZ, 2HBZ and aspirin (d) 4HBZ was significantly more cytotoxic than either 2HBZ (P = 0.0005) or aspirin (P < 0.0001) as demonstrated by lower mean LD50 values in the cohort of 52 patients.
Primary CLL cell culture conditions
Freshly isolated peripheral blood lymphocytes (1 × 106/ml) were cultured in RPMI medium (Invitrogen, Paisley, UK) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 10% foetal calf serum and 5 ng/ml IL‐4. Normal B cells were purified by negative selection using CD3+ Dyanbeads, and normal T cells were purified by negative selection using CD19+ Dynabeads (Invitrogen); purity was assessed by flow cytometry and only samples with >95% target cells were used in subsequent experiments. Lymphocytes were incubated at 37 °C in humidified 5% carbon dioxide atmosphere in the presence of aspirin and the novel analogues (1 × 10−4–2 × 10−3 m) for up to 48 h. In addition, control cultures were set up in which no drug was added to normal and leukaemic lymphocytes. Cells were subsequently harvested by centrifugation and were analysed by flow cytometry using methods outlined below. Experiments were performed either in duplicate or in triplicate.
Co‐culture with mouse fibroblast L‐cells transfected with human CD40 ligand
Untransfected L‐cells (NTL) and CD40L‐expressing L‐cells were irradiated with 75 Gray and then seeded into six‐well plates (5 × 105 cells/well). Plates were then incubated for minimum of 2 h, to allow cells to adhere to the plastic, prior to addition of magnetically purified (CD19+) CLL cells (2 × 106 cells/well). Briefly, CLL cells were positively isolated using CD19+ Dynabeads (Invitrogen) and beads were then detached using DETACHaBEAD CD19 reagent (Invitrogen). Purity of CD5+/CD19+ CLL cells was >96% as assessed by flow cytometry. All cultures were maintained in RPMI media supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 10% foetal calf serum and 5 ng/ml IL‐4 (R&D Systems, Abingdon, UK). CLL cells were harvested from L‐cell co‐culture system by gently pipetting contents of the well.
Measurement of apoptosis in vitro
Cells were harvested and labelled with CD19‐allophycocyanin (APC) (Caltag, Buckingham, UK) and then resuspended in 200 μl of binding buffer containing 4 μl of annexin V labelled with fluorescein isothiocyanate (FITC) (Bender Medsystems, Vienna, Austria). Apoptosis was quantified in the CD19+ CLL cells using FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). At least 10 000 events were acquired and data were subsequently analysed using Summit 4.0 software (Dako, Ely, UK). All LD50 values (concentration of aspirin analogue required to kill 50% of cells) were derived from the dose–response curves.
Measurement of caspase‐3 activation
CLL cells were incubated at 37 °C in humidified 5% carbon dioxide atmosphere in the presence of phenolic analogues (1 × 10−4–2 × 10−3 m) for 12, 24 and 48 h. Cells were then harvested by centrifugation and incubated for 1 h at 37 °C in presence of PhiPhiLuxTM G1D2 substrate (Calbiochem, Nottingham, UK). This substrate contains two fluorophores separated by a quenching linker sequence that is cleaved by active caspase‐3. Once cleaved, the resulting product fluoresces green and can be quantified using flow cytometry. In additional experiments, pan‐caspase inhibitor, Z‐VAD.fmk, the caspase‐8 inhibitor, Z‐IETD.fmk, or the caspase‐9 inhibitor, Z‐LEHD.fmk (Cambridge Bioscience, Cambridge, UK) were added to treated cell cultures (final concentration 50 μm), to determine whether either of these inhibitors was able to abrogate apoptotic effects of the aspirin analogues in CLL cells.
Measurement of Cox‐2 protein expression
Samples were processed for triple immunofluorescent staining using Cox‐2 with CD5 and CD19 antibodies, within 4 h of sample collection. Briefly, 1 × 106 cells were incubated with anti‐CD5 FITC conjugated and anti‐CD19 APC conjugated antibodies or isotype‐matched controls (Dako). Cells were fixed and permeabilized using a commercially available kit (Dako) and then labelled with anti‐Cox‐2 phycoerythrin conjugated antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or an appropriate isotype‐matched control (Dako). At least 10 000 cells were analysed using an Accuri C6 flow cytometer (Accuri Cytometers Inc., Ann Arbor, MI, USA), and non‐specific binding was excluded by gating, using isotype control antibodies. Gating of CD5+/CD19+ cells was performed to quantify Cox‐2 protein expression in the leukaemic cells.
Rel A detection by enzyme‐linked immunosorbent assay
Nuclear proteins were assayed for Rel A DNA binding using TransAM NF‐κB kit according to the manufacturer’s instructions (Active Motif. Carlsbad, CA, USA); consensus oligonucleotide used for NF‐κB binding was 5′‐GGGACTTTCC‐3′. Wild‐type and mutated consensus oligonucleotides were used to monitor specificity of the assay. Optical density reading at 450 nm (OD450) was noted using a microtitre plate reader (Bio‐Rad, Hemel Hempstead, UK). OD450 values were converted into ng Rel A NF‐κB per μg of nuclear protein for each sample tested, from a standard curve constructed using known quantities of recombinant Rel A.
Real‐time reverse transcription‐PCR
Amounts of CFLAR, BIRC5, BCL2, MCL1, VEGF (5 NF‐κB regulated pro‐survival genes), COX‐1 and COX‐2 mRNA were assessed using real‐time reverse transcription‐PCR (RT‐PCR) using the Light Cycler System (Roche Diagnostics, Burgess Hill, UK). Total RNA was extracted, reverse‐transcribed with random hexamers and amplified using the following primers:
CFLAR: 5′‐AGAGTGAGGCGATTTGACCTG‐3′ (forward)
5′‐AAGGTG‐AGGGTTCCTGAGCA‐3′ (reverse)
BIRC5: 5′‐TGTTGGGAATCTGGAGATGA‐3′ (forward)
5′‐CGGATGAACTCCTGTCCTTT‐3′ (reverse)
BCL2: 5′‐GGTCATGTGTGTGGAGAGCG‐3′ (forward)
5′‐GGTGCCGGTTCAGGTACTCA‐3′ (reverse)
MCL1: 5′‐ AAAAGCAAGTGGCAAGAGGA ‐3′ (forward)
5′‐ TTAATGAATTCGGCGGGTAA ‐3′ (reverse)
VEGF: 5′‐AGCCTTGCCTTGCTGCTCTA‐3′ (forward)
5′‐GTGCTGGCCTTGGTGAGG‐3′ (reverse)
COX‐1: 5′‐TGGCTGGGCGTGCTAGAGGTT‐3′ (forward)
5′‐CAGCCTGCGTGAGGTGTGTCACT‐3′ (reverse)
COX‐2: 5′‐TCACAGGCTTCCATTGACCAG‐3′ (forward)
5′‐CCGAGGCTTTTCTACCAGA‐3′ (reverse)
ABL: 5′‐TTCAGCGGCCAGTAGCATCTGACTT‐3′ (forward)
5′‐CTGTTGACTGGCGTGATGTAGTTGCTT‐3′ (reverse)
The amount of ABL mRNA was quantified in all samples as internal housekeeping control and SYBRGreen was used as the detection method. All primers were purchased from Eurogentec Ltd (Southampton, UK). Results of real‐time RT‐PCR were expressed as normalized target gene values, for example, ratio between CFLAR and ABL transcripts calculated from crossing points of each gene. All experiments were performed in duplicate.
Statistical analysis
Data obtained in these experiments were evaluated using equal variance and paired Student’s t‐test, and correlation coefficients were calculated from least squares linear regression plots. LD50 values were calculated from line of best‐fit analysis of dose–response curves. All statistical analyses were performed using Graphpad Prism 3.0 software (Graphpad Software Inc., San Diego, CA, USA).
Results
Cytotoxic effect of novel aspirin analogues
Cytotoxic effects of the two novel aspirin analogues, 2HBZ and 4HBZ, were compared with aspirin, in primary cultures of CLL cells derived from 52 patients. Figure 1a shows the chemical structure of aspirin and the two novel analogues, and Fig. 1b shows an example of annexin V/propidium iodide plots derived for each agent following in vitro culture for 48 h. A clear dose response was observed for aspirin and both analogues (Fig. 1c) and mean LD50 values (±SD) for 4HBZ, 2HBZ and aspirin were 0.33 ± 0.12 mm, 0.54 ± 0.14 mm and 1.17 ± 0.33 mm, respectively (Fig. 1d). Both 2HBZ and 4HBZ were significantly more potent than aspirin (P = 0.0002 and P < 0.0001 respectively) and 4HBZ was more cytotoxic than 2HBZ (P = 0.0005). These results are in agreement with our recent findings that have demonstrated that 2HBZ is more cytotoxic than aspirin to the human HT‐1080 fibrosarcoma cell line (37). It should be noted that equimolar concentrations of zinc ions had negligible effect on CLL cell viability under the same experimental conditions (data not shown).
2HBZ, 4HBZ and aspirin induce caspase‐3 activation
We next investigated whether these compounds mediated their cell killing effects through activation of caspase‐3 in CLL cells. Experiments were conducted in which active form of caspase‐3 was specifically measured using an immunofluorescence‐labelled flow cytometric assay. Exposure to all three agents induced time‐dependent (Fig. 2a) and concentration‐dependent activation of caspase‐3 (data not shown). Given that 4HBZ was the most potent aspirin analogue, a series of experiments was performed using caspase inhibitors to evaluate their ability to block 4HBZ‐induced apoptosis. Pan caspase inhibitor Z‐VAD.fmk at final concentration of 50 μm, significantly inhibited cytotoxic effects of 4HBZ (P = 0.02) and reduced numbers of apoptotic cells towards those seen in control cultures incubated without 4HBZ. Caspase‐9 inhibitor (Z‐LEHD.fmk) also caused reduction in 4HBZ‐induced apoptosis at final concentration of 50 μm (P = 0.04), but caspase‐8 inhibitor (Z‐IETD.fmk) had no significant cytoprotective effect (P = 0.78) as shown in Fig. 2b. These results confirm that cytotoxic effects of 4HBZ are caspase‐dependent and that apoptosis is induced predominantly via the intrinsic apoptotic pathway.
Figure 2.
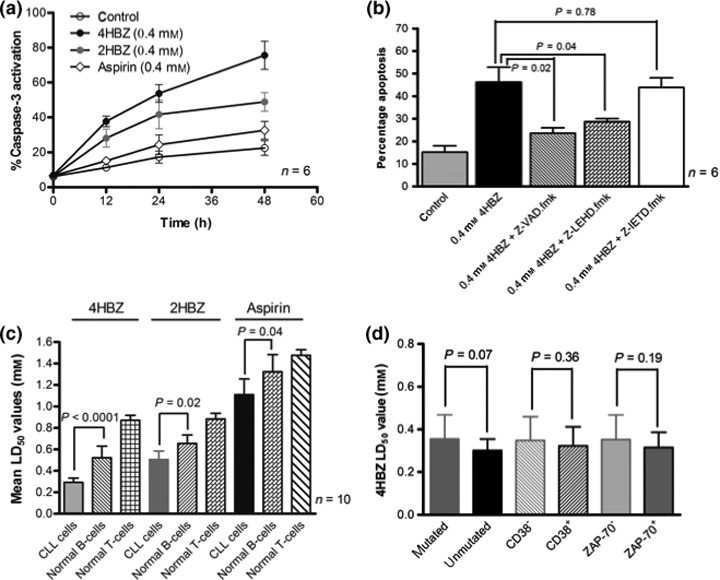
Caspase activation and relative sensitivity of normal lymphocytes and CLL subsets. (a) CLL cells were incubated with 0.4 mm 2HBZ, 0.4 mm 4HBZ or 0.4 mm aspirin for up to 48 h followed by addition of fluorogenic caspase‐3 substrate. All three agents induced a time‐dependent increase in caspase‐3 activation. (b) CLL cells were cultured alone and with 0.4 mm 4HBZ in presence and absence of a caspase‐8 inhibitor (50 μm), a caspase‐9 inhibitor (50 μm), or a pan caspase inhibitor (50 μm). The caspase‐9 inhibitor and the pan caspase inhibitor were able to abrogate effects of 4HBZ, whereas caspase‐8 inhibitor had little effect on its cytotoxic effects. (c) All three agents were significantly more cytotoxic to CLL cells when compared to normal B‐ and T‐lymphocytes from 10 age‐matched controls. (d) The most potent agent, 4HBZ, was equipotent in different prognostic subsets with a trend towards increased potency in poor prognostic groups.
Normal B‐ and T‐lymphocytes are less susceptible to cytotoxic effects of aspirin analogues
B‐lymphocytes from 10 age‐matched normal controls were assessed for their sensitivity to aspirin analogue‐induced apoptosis. Normal age‐matched control B‐lymphocytes demonstrated higher LD50 values for 4HBZ, 2HBZ and aspirin when compared to CLL cells. These differences were statistically significant for 4HBZ (P < 0.0001), 2HBZ (P = 0.02) and aspirin (P = 0.04); in addition, normal T‐lymphocytes were significantly more resistant to effects of all three agents. Relative sensitivities of normal lymphocytes and CLL cells to these compounds are illustrated in Fig. 2c. Interestingly, when we evaluated relative sensitivity of different prognostic subsets to apoptotic effects of 4HBZ, there was a trend towards increased sensitivity in the poor prognostic groups (unmutated IGHV, CD38+ and ZAP‐70+), although this did not reach significance (Fig. 2d). It is also worthy of note that two of the samples tested in this cohort had a 17p deletion and 4HBZ retained its potency in this important minority group.
Aspirin analogues selectively inhibit COX‐2, but not COX‐1 mRNA levels
It has recently been reported that COX‐2 expression promotes survival in CLL cells and selective inhibition of COX‐2 results in increased apoptosis (28). We therefore set out to establish whether our novel aspirin analogues were able to inhibit transcription of COX‐1 and COX‐2 in primary CLL cells. Real time RT‐PCR revealed that the analogues were potent and selective inhibitors of COX‐2 after only 4 h exposure (Fig. 3a). Aspirin and the two novel analogues significantly inhibited COX‐2 mRNA (P < 0.0001), but had no effect on COX‐1 mRNA.
Figure 3.
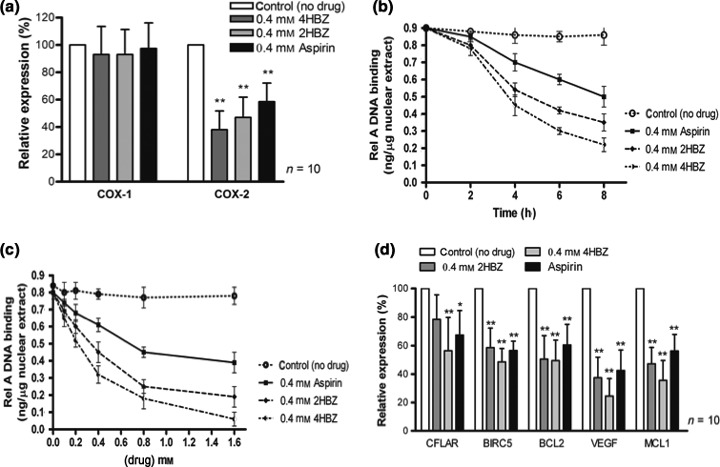
2HBZ, 4HBZ and aspirin selectively inhibit COX‐2 mRNA levels and suppress Rel A DNA binding. (a) CLL cells were treated with 0.4 mm of 2HBZ, 4HBZ or aspirin for 4 h. They were then harvested and RNA extracted. Real‐time PCR analysis revealed that COX‐2 mRNA levels were markedly reduced in cells treated with the aspirin analogues when compared to untreated controls (**P < 0.0001). In contrast, COX‐1 mRNA levels were not markedly altered under these conditions. Panels (b) and (c) show representative examples of nuclear Rel A DNA binding following culture with and without 0.4 mm 4HBZ. (B) CLL samples (n = 10) were treated with 0.4 mm of 2HBZ, 4HBZ or aspirin for 2, 4 and 8 h before apoptosis assay and nuclear protein extraction. ELISA analysis revealed a time‐dependent decrease in Rel A DNA binding. (c) CLL cells were treated with 2HBZ, 4HBZ or aspirin (0–1.6 mm) for 4 h before apoptosis assay and nuclear protein extraction. ELISA analysis revealed dose‐dependent decrease in Rel A DNA binding. (d) We also examined transcription of downstream NF‐κB target genes by real‐time PCR. CLL cells were cultured with or without 0.4 mm of 2HBZ, 4HBZ or aspirin for 4 h. Expression of CFLAR, BIRC5, BCL2, MCL1 and VEGF were quantified using real‐time PCR and were normalized to housekeeping gene ABL. All three agents induced similar, significant, reductions in these genes over the same time period (*P < 0.05, **P < 0.0001), suggesting a common mechanism of action. All experiments were carried out in duplicate in samples derived from 10 CLL patients.
Aspirin analogues inhibit Rel A DNA binding
5′‐UTR of the COX‐2 gene contains binding sites for numerous regulatory transcription factors, including two NF‐κB motifs, two AP‐1 (activator protein 1) sites and two CREs (cAMP‐response elements) (38). We therefore evaluated DNA binding of NF‐kB subunit, Rel A in response to short‐term exposure (up to 8 h) to aspirin and our novel aspirin analogues. We found that all three inhibited Rel A DNA binding in a time‐dependent and concentration‐dependent manner (Fig. 3b,c). Furthermore, this inhibition preceded induction of apoptosis (data not shown). We next examined whether the aspirin analogues had an effect on other downstream NF‐κB‐regulated genes using real time RT‐PCR. We selected genes that have been shown to be important in regulation of CLL cell viability including BCL2 and MCL1 (36, 39). Gene transcription was compared in paired samples from individual patients (n = 10) with and without addition of 0.4 mm of each analogue for 4 h. Figure 3d demonstrates significant reductions in transcription of all 4 NF‐κB regulated genes (CFLAR, BCL2, BIRC5, MCL1 and VEGF) following exposure to aspirin and the two novel analogues.
CLL cells induce COX‐2 mRNA and protein following co‐culture with CD40L‐expressing L‐cells
A recent study demonstrated that COX‐2 mRNA can be induced by pro‐survival factor CD40 ligand (CD40L) (28). Here we used a mouse fibroblast cell line transfected with human CD40L to assess whether COX‐2 mRNA and protein levels were altered under these co‐culture conditions. Consistent with the previous report, here COX‐2 mRNA expression levels were higher following co‐culture with CD40L‐expressing L‐cells. In contrast, COX‐2 mRNA expression levels were not significantly altered by co‐culture with untransfected L‐cells (P = 0.42) indicating that CD40L is critical for this effect. Importantly, addition of 0.4 mm 4HBZ to CD40L co‐culture system significantly suppressed COX‐2 mRNA levels in primary CLL cells (Fig. 4a; P = 0.002). COX‐1 mRNA levels were not modulated over the same 4 h timeframe under any of the conditions tested. Effects on COX‐2 transcription were mirrored at the protein level following 24 h in culture. We measured Cox‐2 protein levels by flow cytometry (Fig. 4b,c) and showed a consistent increase in Cox‐2 when CLL cells were co‐cultured with CD40L‐expressing L‐cells (n = 8, P = 0.002). This effect was consistently inhibited by addition of 0.4 mm 4HBZ and caused significant reduction in Cox‐2 protein levels (P = 0.014). We next evaluated the impact of CD40L‐expressing L‐cell co‐culture system on ability of 4HBZ to induce apoptosis (Fig. 4d). CLL cell spontaneous apoptosis was significantly inhibited in this co‐culture system when compared to standard liquid culture conditions (P < 0.0001). However, 4HBZ was able to induce apoptosis in presence of CD40L‐expressing L‐cells (P < 0.0001) and was equipotent when compared to liquid culture conditions (P = 0.11).
Figure 4.
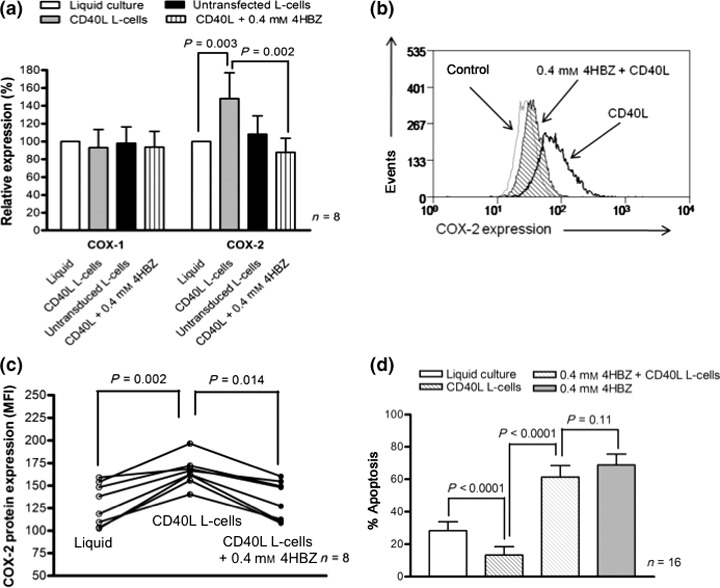
CLL cells co‐cultured on CD40L‐expressing mouse fibroblasts induce COX‐2 mRNA and protein, but retain their sensitivity to 4HBZ. (a) CLL cells were cultured under four different conditions; standard liquid culture, with untransfected L‐cells, L‐cells transfected with human CD40L and L‐cells transfected with human CD40L in the presence of 0.4 mm 4HBZ. All cultures were supplemented with IL‐4. COX‐2 mRNA levels were markedly increased in CLL cells co‐cultured with CD40L‐expressing L‐cells when compared to liquid culture conditions after 24 h in culture (P = 0.0003). Importantly, 4HBZ reversed the CD40L‐induced increase in COX‐2 mRNA expression (P = 0.002). In contrast, COX‐1 mRNA levels remained largely unaltered. (b and c) Co‐culture with CD40L‐expressing L‐cells also induced Cox‐2 protein expression and this could be reversed by addition of 4HBZ. (d) Co‐culture of CLL cells with CD40L‐expressing L‐cells caused significant reduction in spontaneous apoptosis (P < 0.0001). However, 4HBZ was able to induce apoptosis under these conditions (P < 0.0001) and showed potency similar to that under liquid culture conditions (P = 0.11).
COX‐2 mRNA expression is higher in poor prognostic subsets and is inversely correlated with resistance to 4HBZ
Given that there was considerable variation in COX‐2 mRNA levels within our CLL cohort, we assessed whether this was associated with any prognostic subset. Consistent with a previous study, we showed that COX‐2 mRNA expression was higher in samples derived from patients who had unmutated IGHV genes, high CD38 expression and high ZAP‐70 expression (Fig. 5a–c). It is of particular interest that there was inverse correlation between COX‐2 mRNA expression and resistance to 4HBZ (Fig. 5d). Given that poor prognostic subsets showed significantly higher COX‐2 expression, 4HBZ might be particularly effective in these poor risk groups.
Figure 5.
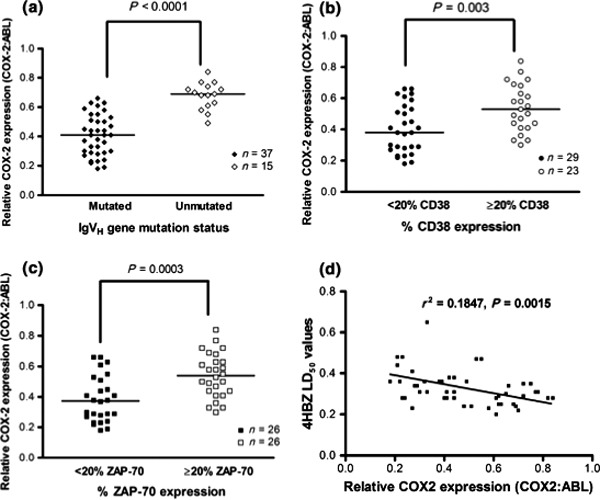
COX‐2 mRNA expression is significantly higher in poor prognostic subsets and is inversely correlated with resistance to 4HBZ. (a–c) show relative COX‐2 mRNA levels in different prognostic subsets within the cohort of 52 patients. COX‐2 mRNA levels were significantly higher in samples with unmutated IGHV genes (P < 0.0001), high CD38 expression (P = 0.003) and high ZAP‐70 expression (P = 0.0003). (d) There was a significant inverse correlation between COX‐2 mRNA expression and resistance to 4HBZ.
Discussion
Results presented in this study demonstrate that aspirin and two novel analogues, 2HBZ and 4HBZ, induce apoptosis in CLL cells in a dose‐dependent and time‐dependent manner. Our data confirm and extend the previous study of Bellosillo et al. (14). who demonstrated that aspirin and its metabolite, salicylate, induced apoptosis in CLL cells. Here, we show that apoptosis was predominantly triggered via the intrinsic apoptotic pathway as evidenced by blocking effects of caspase‐9 inhibitor Z‐LEHD.fmk. In contrast, caspase‐8 inhibitor Z‐IETD.fmk had little inhibitory effect on 2HBZ and 4HBZ‐induced apoptosis. Importantly, 2HBZ and 4HBZ were significantly more potent apoptosis‐inducing agents than aspirin (approximately 2‐fold and 3‐fold more potent respectively). These results are in keeping with those recently described for HT‐1080 fibrosarcoma cell line in which 2HBZ was shown to be approximately 7‐fold more potent than aspirin (37). Therefore, these novel phenolic agents represent promising lead molecules for development of clinically relevant chemotherapeutic agents. This assertion was further endorsed by the finding that 2HBZ and 4HBZ were preferentially cytotoxic to CLL cells when compared to normal B‐ and T‐lymphocytes, indicating that these agents may have a positive therapeutic index for treatment of CLL. In this regard, it is worthy of note that although LD50 values for 4HBZ required to kill CLL cells are in the sub‐millimolar range, and hence are significantly higher than conventional chemotherapeutic drugs, concentrations of 4HBZ used in this study equate to plasma concentrations that can be achieved with low‐dose aspirin (325 mg dose). Therefore, if 4HBZ shows a similar toxicity profile to aspirin in vivo, then these concentrations may be well tolerated and clinically useful.
A recent study has reported that a majority of CLL cells constitutively express both COX‐1 and COX‐2 enzymes (28). Here, we confirmed that COX‐1 and COX‐2 mRNA were present in all the primary CLL samples tested and that constitutive COX‐2 expression was higher in samples derived from patients with unmutated IGHV genes – an observation that was also true for CD38+ and ZAP‐70+ samples. Importantly, we demonstrated that the novel aspirin analogues under investigation were capable of selectively inhibiting COX‐2 mRNA expression levels after only 4 h in in vitro culture, whereas COX‐1 mRNA levels remained unaltered. After 24 h in culture, inhibition of COX‐2 transcription resulted in significant reductions in level of Cox‐2 protein. It is of considerable interest that samples derived from poor prognosis patients showed significantly higher COX‐2 expression, but showed a trend towards increased sensitivity to 4HBZ. This suggests that poor prognostic subsets may be particularly dependent on COX‐2‐mediated pro‐survival signals and that 4HBZ might be useful in treatment of these patients.
As the 5′‐UTR of the COX‐2 gene contains two putative binding sites for NF‐κB (40), and NF‐κB is constitutively active in CLL (31), we examined whether NF‐κB DNA binding was altered by exposure to the novel aspirin analogues. Consistent with a study of the novel NSAID SDX‐308 in multiple myeloma (38), we demonstrated inhibition of Rel A DNA binding that preceded induction of apoptosis. 4HBZ caused the most marked repression of Rel A DNA binding and this agent was also the most cytotoxic suggesting that these two phenomena may be related. In addition, 4HBZ also caused reduction in transcription of NF‐κB target genes including CFLAR, BIRC5, VEGF and BCL2. Consistent with the previous work of Iglesias‐Serret et al. (41). MCL1 transcription was also suppressed by aspirin and the novel analogues tested in this study. However, it remains uncertain whether MCL1 transcription is directly inhibited through suppression of NF‐κB or whether this is an indirect consequence of NF‐κB inhibition. In this regard, it is intriguing to note that other NF‐κB inhibitors do not appear to inhibit Bcl‐2 or Mcl‐1 protein expression in CLL (42). Furthermore, other Bcl‐2 family proteins, including Noxa, have been shown to be important in determining response to aspirin analogues in some cell types (41). Clearly, further work is necessary to untangle precise molecular mechanisms that contribute to aspirin analogue cytotoxicity in CLL cells and we are currently undertaking extensive temporal studies to evaluate exact relationship between pro‐ and anti‐apoptotic Bcl‐2 family transcription and protein expression following aspirin analogue treatment. Consistent with a complex mechanism of aspirin analogue‐induced cell killing, we have previously shown that there was no association between Rel A DNA binding and poor prognostic subgroups in CLL (43) and hence it would appear that COX‐2 is not solely regulated by Rel A in primary CLL cells. Therefore, that 4HBZ appears to inhibit both COX‐2 transcription and Rel A DNA binding may be therapeutically advantageous.
There is growing evidence that CLL cells are critically dependent on survival and proliferation signals derived from lymphoid tissues (44, 45, 46, 47) and that many of these pro‐survival signals are delivered when CLL cells come into direct contact with stromal cells, T cells and follicular dendritic cells (48, 49, 50). We therefore used a CD40L‐expressing mouse fibroblast co‐culture system to mimic tumour cell microenvironment to provide more representative in vitro drug testing for CLL cells to the aspirin analogue 4HBZ. In accordance with previously published data, this culture system inhibited CLL cell spontaneous apoptosis, but addition of 4HBZ largely abrogated this pro‐survival effect. Furthermore, we were able to demonstrate that the co‐culture system selectively increased COX‐2 mRNA and protein levels, but addition of 4HBZ under these conditions inhibited COX‐2 expression levels.
In conclusion, this study demonstrates that these novel aspirin analogues have significant therapeutic potential for CLL. Data presented here are consistent with a mechanism involving aspirin‐induced suppression of Rel A nuclear translocation and transcriptional inhibition of COX‐2. Both Rel A and COX‐2 have been shown to be important for CLL cell survival and therefore represent excellent therapeutic targets. The encouraging cytotoxic profile of these agents in samples derived from poor prognostic subsets raises the hope that they could improve the clinical management of this currently incurable disease.
Author contributions
C.P.: Designed research, performed research, analysed data and wrote the manuscript. J.G.M.: Designed research, contributed vital new reagents, analysed data and revised the manuscript. A.G.S.B. and T.T.L.: Performed research, analysed data and revised the manuscript. S.H.: Designed research, Performed research, contributed vital new reagents, analysed data and revised the manuscript. E.W. and A.H.: Performed research and analysed data. E.M. and A.J.M.: Contributed vital new reagents, analysed data and revised the manuscript. L.P. and L.M.: Performed research and revised the manuscript. I.D.B.: Designed research and revised the manuscript. P.B.: Designed research, analysed data and revised the manuscript. C.F.: Designed research, contributed vital new reagents, analysed data and wrote the manuscript.
Conflict of interests
The authors have no conflict of interests to declare.
Acknowledgements
This work was supported in part by grants from Leukaemia Lymphoma Research and the Leukaemia Research Appeal for Wales.
References
- 1. Bannerji R, Byrd JC (2000) Update on the biology of chronic lymphocytic leukemia. Curr. Opin. Oncol. 12, 22–29. [DOI] [PubMed] [Google Scholar]
- 2. Byrd JC, Stilgenbauer S, Flinn IW (2004) Chronic lymphocytic leukemia. Hematology, Am. Soc. Hematol. Ed. Program, 163–183. [DOI] [PubMed] [Google Scholar]
- 3. Keating MJ (1999) Chronic lymphocytic leukemia. Semin. Oncol. 26, 107–114. [PubMed] [Google Scholar]
- 4. Kolb JP, Kern C, Quiney C, Roman V, Billard C (2003) Re‐establishment of a normal apoptotic process as a therapeutic approach in B‐CLL. Curr. Drug Targets Cardiovasc. Haematol. Disord. 3, 261–286. [DOI] [PubMed] [Google Scholar]
- 5. Pepper C, Bentley P, Hoy T (1996) Regulation of clinical chemoresistance by bcl‐2 and bax oncoproteins in B‐cell chronic lymphocytic leukemia. Br. J. Haematol. 95, 513–517. [DOI] [PubMed] [Google Scholar]
- 6. Sanz L, Garcia‐Marco JA, Casanova B, de La Fuente MT, García‐Gila M, Garcia‐Pardo A, et al. (2004) Bcl‐2 family gene modulation during spontaneous apoptosis of B‐chronic lymphocytic leukemia cells. Biochem. Biophys. Res. Commun. 315, 562–567. [DOI] [PubMed] [Google Scholar]
- 7. Buggins AGS, Pepper CJ (2010) The role of Bcl‐2 family proteins in chronic lymphocytic leukaemia. Leuk. Res. 34, 837–842. [DOI] [PubMed] [Google Scholar]
- 8. Kay NE (2006) Purine analogue‐based chemotherapy regimens for patients with previously untreated B‐chronic lymphocytic leukemia. Semin. Hematol. 43, S50–S54. [DOI] [PubMed] [Google Scholar]
- 9. Lam CC, Ma ES, Kwong YL (2005) Therapy‐related acute myeloid leukemia after single‐agent treatment with fludarabine for chronic lymphocytic leukemia. Am. J. Hematol. 79, 288–290. [DOI] [PubMed] [Google Scholar]
- 10. Lamanna N, Weiss MA (2006) Purine analogue‐based chemotherapy regimens for second‐line therapy in patients with chronic lymphocytic leukemia. Semin. Hematol. 43, S44–S49. [DOI] [PubMed] [Google Scholar]
- 11. Tam CS, O’Brien S, Wierda W, Kantarjian H, Wen S, Do KA et al. (2008) Long‐term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 112, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robak T (2005) Therapy of chronic lymphocytic leukaemia with purine nucleoside analogues: facts and controversies. Drugs Aging 22, 983–1012. [DOI] [PubMed] [Google Scholar]
- 13. Yee KW, O’Brien SM (2006) Emerging drugs for chronic lymphocytic leukaemia. Expert Opin. Emerg. Drugs 11, 167–189. [DOI] [PubMed] [Google Scholar]
- 14. Bellosillo B, Piqué M, Barragán M, Castaño E, Villamor N, Colomer D et al. (1998) Aspirin and salicylate induce apoptosis and activation of caspases in B‐cell chronic lymphocytic leukemia cells. Blood 92, 1406–1414. [PubMed] [Google Scholar]
- 15. Hall JG, Cory AH, Hickerson DH, Cory JG (2001) Increased sensitivity to sodium salicylate‐induced apoptosis in drug‐resistant leukemia L1210 cells. Anticancer Res. 21, 173–180. [PubMed] [Google Scholar]
- 16. Nagaoka T, Banskota AH, Tezuka Y, Midorikawa K, Matsushige K, Kadota S (2003) Caffeic acid phenethyl ester (CAPE) analogues: potent nitric oxide inhibitors from the Netherlands propolis. Biol. Pharm. Bull. 26, 487–491. [DOI] [PubMed] [Google Scholar]
- 17. Kellner C, Zunino SJ (2004) Nitric oxide is synthesized in acute leukemia cells after exposure to phenolic antioxidants and initially protects against mitochondrial membrane depolarization. Cancer Lett. 215, 43–52. [DOI] [PubMed] [Google Scholar]
- 18. Cory AH, Cory JG (2005) Phenolic compounds, sodium salicylate and related compounds, as inhibitors of tumor cell growth and inducers of apoptosis in mouse leukemia L1210 cells. In Vivo 19, 31–35. [PubMed] [Google Scholar]
- 19. Mahdi JG, Alkarrawi MA, Mahdi AJ, Humam D, Bowen ID (2006) Calcium salicylate mediated apoptosis in human HT‐1080 fibrosarcoma cells. Cell Prolif. 39, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kasum CM, Blair CK, Folsom AR, Ross JA (2003) Non‐steroidal anti‐inflammatory drug use and risk of adult leukemia. Cancer Epidemiol. Biomarkers Prev. 12, 534–537. [PubMed] [Google Scholar]
- 21. Riboli E, Norat T (2003) Epidemiologic evidence of the protective effect of fruit and vegetables on cancer risk. Am. J. Clin. Nutr. 78, 559S–569S. [DOI] [PubMed] [Google Scholar]
- 22. Meyers KJ, Watkins CB, Pritts MP, Liu RH (2003) Antioxidant and antiproliferative activities of strawberries. J. Agric. Food Chem. 51, 6887–6892. [DOI] [PubMed] [Google Scholar]
- 23. Weiss JR, Baker JA, Baer MR, Menezes RJ, Nowell S, Moysich KB (2006) Opposing effects of aspirin and acetaminophen use on risk of adult acute leukemia. Leuk. Res. 30, 164–169. [DOI] [PubMed] [Google Scholar]
- 24. Nakao S, Kuwano T, Tsutsumi‐Miyahara C, Ueda S, Kimura YN, Hamano S et al. (2005) Infiltration of COX‐2‐expressing macrophages is a prerequisite for IL‐1beta‐induced neovascularization and tumor growth. J. Clin. Invest. 115, 2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhi YH, Liu RS, Song MM, Tian Y, Long J, Tu W et al. (2005) Cyclooxygenase‐2 promotes angiogenesis by increasing vascular endothelial growth factor and predicts prognosis in gallbladder carcinoma. World J. Gastroenterol. 11, 3724–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ito H, Duxbury M, Benoit E, Clancy TE, Zinner MJ, Ashley SW et al. (2004) Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets‐1‐dependent induction of matrix metalloproteinase‐2. Cancer Res. 64, 7439–7446. [DOI] [PubMed] [Google Scholar]
- 27. Park JH, Kang KH, Kim SH, Lee JH, Cho CM, Kweon YO et al. (2005) Expression of cyclooxygenase‐2 and Bcl‐2 in human gastric adenomas. Korean J. Intern. Med. 20, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ryan EP, Pollock SJ, Kaur K, Felgar RE, Bernstein SH, Chiorazzi N et al. (2006) Constitutive and activation‐inducible cyclooxygenase‐2 expression enhances survival of chronic lymphocytic leukaemia B cells. Clin Immunol. 120, 76–90. [DOI] [PubMed] [Google Scholar]
- 29. Secchiero P, Barbarotto E, Gonelli A, Tiribelli M, Zerbinati C, Celeghini C et al. (2005) Potential pathogenetic implications of cyclooxygenase‐2 overexpression in B chronic lymphoid leukemia cells. Am. J. Pathol. 167, 1599–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. D’Acquisto F, May MJ, Ghosh S (2002) Inhibition of nuclear factor kappa B (NF‐kB): an emerging theme in anti‐inflammatory therapies. Mol. Interv. 2, 22–35. [DOI] [PubMed] [Google Scholar]
- 31. Hewamana S, Alghazal S, Lin TT, Clement M, Jenkins C, Guzman ML et al. (2008) The NF‐kB subunit Rel A is associated with in vitro survival and clinical disease progression in chronic lymphocytic leukemia and represents a promising therapeutic target. Blood 111, 4681–4689. [DOI] [PubMed] [Google Scholar]
- 32. Lindhagen E, Nissle S, Leoni L, Elliott G, Chao Q, Larsson R et al. (2007) R‐etodolac (SDX‐101) and the related indole‐pyran analogues SDX‐308 and SDX‐309 potentiate the antileukemic activity of standard cytotoxic agents in primary chronic lymphocytic leukaemia cells. Cancer Chemother. Pharmacol. 60, 545–553. [DOI] [PubMed] [Google Scholar]
- 33. Lentzsch S, Elliott G, Roodman GD (2007) SDX‐308 and SDX‐101, non‐steroidal anti‐inflammatory drugs, as therapeutic candidates for treating hematologic malignancies including myeloma. Arch. Pharm. (Weinheim) 340, 511–516. [DOI] [PubMed] [Google Scholar]
- 34. Robak P, Linke A, Cebula B, Robak T, Smolewski P (2006) Cytotoxic effect of R‐etodolac (SDX‐101) in combination with purine analogs or monoclonal antibodies on ex vivo B‐cell chronic lymphocytic leukemia cells. Leuk. Lymphoma 47, 2625–2634. [DOI] [PubMed] [Google Scholar]
- 35. Binet JL, Auquier A, Dighiero G, Chastang C, Piguet H, Goasguen J et al. (1981) A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48, 198–206. [DOI] [PubMed] [Google Scholar]
- 36. Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L et al. (2008) Mcl‐1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood 112, 3807–3817. [DOI] [PubMed] [Google Scholar]
- 37. Mahdi JG, Pepper CJ, Alkarrawi MA, Mahdi AJ, Bowen ID (2010) Sub‐millimolar concentration of the novel phenolic‐based compound, JMC‐18, induce apoptosis in human HT‐1080 fibrosarcoma cells. Cell Prolif. 43, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feng R, Anderson G, Xiao G, Elliott G, Leoni L, Mapara MY et al. (2007) SDX‐308, a nonsteroidal anti‐inflammatory agent, inhibits NF‐kappaB activity, resulting in strong inhibition of osteoclast formation/activity and multiple myeloma cell growth. Blood 109, 2130–2138. [DOI] [PubMed] [Google Scholar]
- 39. Buggins AG, Pepper C, Patten PE, Hewamana S, Gohil S, Moorhead J et al. (2010) Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF‐kappaB activation and de novo gene transcription. Cancer Res. 70, 7523–7533. [DOI] [PubMed] [Google Scholar]
- 40. Appleby S, Ristimäki A, Neilson K, Narko K, Hla T (1994) Structure of the human cyclo‐oxygenase‐2 gene. Biochem. J. 302, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iglesias‐Serret D, Piqué M, Barragán M, Cosialls AM, Santidrián AF, González‐Gironès DM et al. (2010) Aspirin induces apoptosis in human leukaemia cells independently of NF‐kB and MAPKs through alteration of the Mcl‐1/Noxa balance. Apoptosis 15, 219–229. [DOI] [PubMed] [Google Scholar]
- 42. Pickering BM, de Mel S, Lee M, Howell M, Habens F, Dallman CL et al. (2007) Pharmacological inhibitors of NF‐kappaB accelerate apoptosis in chronic lymphocytic leukaemia cells. Oncogene 26, 1166–1177. [DOI] [PubMed] [Google Scholar]
- 43. Hewamana S, Lin TT, Rowntree C, Karunanithi K, Pratt G, Hills R et al. (2009) Rel A is an independent biomarker of clinical outcome in chronic lymphocytic leukemia. J. Clin. Oncol. 27, 763–769. [DOI] [PubMed] [Google Scholar]
- 44. Burger JA, Burger M, Kipps TJ (1999) Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood 94, 3658–3667. [PubMed] [Google Scholar]
- 45. Jurlander J (1998) The cellular biology of B‐cell chronic lymphocytic leukemia. Crit. Rev. Oncol. Hematol. 27, 29–52. [DOI] [PubMed] [Google Scholar]
- 46. Patten PE, Buggins AG, Richards J, Wotherspoon A, Salisbury J, Mufti GJ et al. (2008) CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood 111, 5173–5181. [DOI] [PubMed] [Google Scholar]
- 47. Caligaris‐Cappio F, Ghia P (2008) Novel insights in chronic lymphocytic leukemia: are we getting closer to understanding the pathogenesis of the disease? J. Clin. Oncol. 26, 4497–4503. [DOI] [PubMed] [Google Scholar]
- 48. Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A (2007) Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B‐cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch”. Leuk. Res. 31, 899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Edelmann J, Klein‐Hitpass L, Carpinteiro A, Führer A, Sellmann L, Stilgenbauer S et al. (2008) Bone marrow fibroblasts induce expression of PI3K/NF‐kappaB pathway genes and a pro‐angiogenic phenotype in CLL cells. Leuk. Res. 32, 1565–1572. [DOI] [PubMed] [Google Scholar]
- 50. Ghia P, Caligaris‐Cappio F (2000) The indispensable role of microenvironment in the natural history of low‐grade cell neoplasms. Adv. Cancer Res. 79, 157–173. [DOI] [PubMed] [Google Scholar]