

Abstract
Objectives: Peroxisome proliferator‐activated receptors (PPARs) are implicated in epithelial cell proliferation and differentiation, but investigation has been confounded by potential off‐target effects of some synthetic PPAR ligands. Our aim was to determine mechanisms underlying the pro‐apoptotic effect of synthetic PPAR agonists in normal human bladder uro‐epithelial (urothelial) cells and to reconcile this with the role of PPARs in urothelial cytodifferentiation.
Materials and methods: Normal human urothelial (NHU) cells were grown as non‐immortal lines in vitro and exposed to structurally diverse agonists ciglitazone, troglitazone, rosiglitazone (PPARγ), ragaglitazar (PPARα/γ), fenofibrate (PPARα) and L165041 (PPARβ/δ).
Results: NHU cells underwent apoptosis following acute exposure to ciglitazone, troglitazone or ragaglitazar, but not fenofibrate, L165041 or rosiglitazone, and this was independent of ERK or p38 MAP‐kinase activation. Pro‐apoptotic agonists induced sustained increases in intracellular calcium, whereas removal of extracellular calcium altered the kinetics of ciglitazone‐mediated calcium release from sustained to transient. Cell death was accompanied by plasma‐membrane disruption, loss of mitochondrial membrane‐potential and caspase‐9/caspase‐3 activation. PPARγ‐mediated apoptosis was unaffected following pre‐treatment with PPARγ antagonist T0070907 and was strongly attenuated by store‐operated calcium channel (SOC) inhibitors 2‐APB and SKF‐96365.
Conclusions: Our results provide a mechanistic basis for the ability of some PPAR agonists to induce death in NHU cells and demonstrate that apoptosis is mediated via PPAR‐independent mechanisms, involving intracellular calcium changes, activation of SOCs and induction of the mitochondrial apoptotic pathway.
Introduction
Peroxisome proliferator‐activated receptors (PPARs) are members of the NR1C nuclear hormone receptor family, which heterodimerize with retinoid X receptors and bind specific DNA peroxisome proliferator response elements to activate gene transcription. The three PPAR isoforms, PPARα, PPARβ/δ and PPARγ, are products of distinct genes and vary in their tissue distribution and function. PPARs have important roles in regulating expression of genes involved in modulating lipid and carbohydrate metabolism. In particular, PPARα and PPARγ are important therapeutic targets for treatment of dyslipidaemia, insulin resistance and type II diabetes. This has led to development of specific agonists, including the hypolipidaemic fibrates that bind PPARα, antidiabetic thiazolidinediones that are high‐affinity PPARγ ligands, and to a growing range of other agonists with dual‐ or pan‐activity, such as the ‘‐glitazars’ of dual α/γ activity. Despite proven benefits of targeting PPARα and PPARγ in metabolic diseases, there remains concern about potential adverse effects of PPAR agonists in humans, including carcinogenicity, myopathy, weight gain, oedema and cardiac dysfunction (1). PPAR activators have pleiotropic effects, which not only modify expression of genes involved in metabolism, but also affect expression of genes regulating inflammation, proliferation and differentiation (2).
Involvement of PPARγ signalling is best characterized in adipogenesis, where induction, activation and downstream transcriptional activity of PPARγ are critical events in the cascade of gene expression changes accompanying differentiation (3). Activation of PPARγ has been shown to induce terminal differentiation of liposarcoma cells in vitro (3) and in vivo (4), and ectopic expression of PPARγ2 in fibroblasts has been shown to promote adipogenesis via activation of the adipocyte gene expression programme (5). A role for PPARγ in the urinary bladder has been implicated from high levels of expression found in both developing and mature uro‐epithelium from multiple species (6, 7, 8). We have previously demonstrated a role for PPARγ in induction of differentiation in normal human urothelial (NHU) cells in vitro, where specific activation of PPARγ induced expression of cytokeratin, claudin and uroplakin genes associated with late/terminal urothelial differentiation (9, 10, 11), mediated via PPARγ‐induced intermediary transcription factors (12).
Concern about the specificity of PPAR activators is due in part to the propensity for PPAR‐independent effects by some agonists. For example, PPARγ agonists have been reported to induce apoptosis in a wide range of tumour‐derived cell lines. This has led to interest in their potential anti‐cancer properties. PPARγ activators have also been shown to decrease lung tumour growth in mice (13) and mammary tumour growth in rats in vivo (14), although PPARγ activation did not induce a clear response in human breast cancer in vivo (15). There is emerging evidence that growth inhibitory and pro‐apoptotic effects of PPARγ agonists are mediated via receptor‐independent (non‐genomic) mechanisms (16). Such effects are seen at high agonist concentrations (typically > 10 µm) and have also been reported in the absence of PPAR expression (17). Non‐genomic effects of PPAR agonists have well‐established parallels in, for example, non‐genomic actions of steroid hormones, the mechanisms of which are only partly understood (18). The importance of understanding specific and non‐specific effects of PPAR agonists has been highlighted by emerging reports that PPAR agonists may play a role in inducing or promoting bladder carcinogenesis in rats (19, 20), although the mechanism and relevance of these findings for PPAR therapy in humans have yet to be determined (reviewed by Oleksiewicz et al. (21)).
In our studies of PPARγ‐mediated urothelial cytodifferentiation, we have previously observed that the thiazolidinedione troglitazone induced apoptosis at concentrations above those required to initiate differentiation (9, 10, 11). This agrees with a number of other studies of normal (22, 23) and carcinoma‐derived (24, 25) urothelial cells, which have documented growth inhibition and/or induction of apoptosis with PPAR agonists, although the majority of these studies have not distinguished between cytostasis and cell death, or determined the PPAR dependency of their observations.
Given the limited understanding of PPAR‐independent effects of thiazolidinediones and ‐glitazars in human urothelium and their potential role in adverse responses to therapy, we have investigated the mechanisms underlying the pro‐apoptotic effects of PPAR agonists in NHU cells.
Materials and methods
Cell culture
Collection of specimens was approved by the relevant local research ethics committees and had full, informed patient consent. Surgical specimens of normal ureteric urothelium were obtained from patients with no histological evidence of urothelial dysplasia or malignancy. Tissues were collected in Hank's balanced salt solution (HBSS) containing 10 mm HEPES (pH 7.6) and 20 KIU aprotinin (Trasylol, Bayer plc, Newbury, UK). Preparation and maintenance of finite NHU cell lines have been previously detailed (26, 27). Briefly, once established in culture, NHU cell lines were maintained in KSFMc consisting of keratinocyte‐serum free medium (KSFM) supplemented with bovine pituitary extract and epidermal growth factor, as recommended by the manufacturer (Invitrogen, Paisley, UK), and 30 ng/ml cholera toxin (Sigma Aldrich, Poole, UK). NHU cell lines were harvested for subculture or analysis in suspension by incubation for 5 min in phosphate‐buffered saline containing 0.1% (w/v) EDTA, followed by minimal incubation with 0.25% (w/v) trypsin in 0.02% (w/v) EDTA to detach cells for collection into medium containing 1 mg/ml trypsin inhibitor (Sigma Aldrich). Five independent NHU cell lines were used in this study, between passages 2 and 5. NHU cells were grown to sub‐confluence (80–90%) in T25 tissue culture Primaria® flasks (BD Biosciences, Oxford, UK). Cells were then exposed to PPAR agonists or vehicle control over a time course (5 min to 72 h).
Antibodies and reagents
Primary antibodies with the following antigenic specificities were used for Western blotting: pan‐ERK (100 ng/ml; rabbit polyclonal; BD Biosciences, catalogue no. 610123), phospho‐ERK (Thr202/Tyr204 100 ng/ml, mouse monoclonal; Cell Signaling, catalogue no. 9101), pan‐p38 (100 ng/ml; rabbit polyclonal; Cell Signaling, catalogue no. 9212), phospho‐p38 (Thr180/Tyr182 100 ng/ml, rabbit polyclonal; Cell Signaling, catalogue no. 9216), caspase‐9 (Asp315 100 ng/ml; rabbit polyclonal; Cell Signaling, catalogue no. 9505), and caspase‐3 (2 µg/ml, mouse monoclonal; Imgenex Corp. supplied by Cambridge Bioscience, Cambridge, UK, catalogue no. IMG‐144 A).
Troglitazone, rosiglitazone, ciglitazone and fenofibrate were obtained from Sigma Aldrich, L165041 was from Merck Biosciences (Nottingham, UK), and ragaglitazar was obtained from NovoNordisk A/S (Maalov, Denmark). Stock concentrations (100 mm) of these agonists were prepared in dimethyl sulphoxide (DMSO); maximum DMSO concentration (0.1%) was used in vehicle‐only controls. Store‐operated calcium channel (SOC) inhibitors, 2‐aminoethoxydiphenyl borate (2‐APB) and SKF‐96365 were obtained from Sigma Aldrich. Inhibitors of p38 (SB203580), PPARγ (T0070907), caspase‐9 (Z‐LEHD‐FMK) and caspase‐3 (Z‐DEVD‐FMK) were from Calbiochem (supplied by Merck Biosciences). All other chemicals and reagents were of either analytical or laboratory grade and obtained from either Sigma Aldrich or Fisher Scientific (Loughborough, UK).
Cell proliferation assays
Growth of NHU cell cultures in the presence of PPAR agonists was assessed using the thiazolyl blue tetrazolium (MTT; Sigma Aldrich) dye reduction assay. Briefly, NHU cells were seeded overnight in 96‐well flat‐bottom Primaria® tissue culture plates at a density of 2 × 104 cells/ml. PPAR agonists were added 24 h after seeding and tested over a range of concentrations. Growth medium and drugs were re‐applied every 3 days and cell proliferation was assessed over a period of 8 days, unless otherwise stated. At selected time points, MTT (500 µg/ml; 200 µl volume) was added to each well and incubated for 4 h at 37 °C. Following this period, MTT was aspirated from each well and MTT‐formazan crystals were solubilized by addition of DMSO (spectrophotometric grade; 200 µl). Absorbance at 570 nm of each sample well was measured using an automated plate reader. For clarity, results are presented at a single time point and full growth curves are presented as supplemental data.
Western blotting
Following treatment, cell culture experiments were terminated by in situ lysis in Tris‐HCl (125 mm pH 7.4) containing glycerol (20% v/v), sodium dodecyl sulphate (2% w/v), sodium fluoride (50 mm), sodium orthovanadate (2 mm), tetra‐sodium pyrophosphate (30 mm), dithiothreitol (0.2% v/v) and protease inhibitor cocktail (Protease Inhibitor Cocktail set III, Merck Biosciences). Protein lysates were sonicated prior to microcentrifugation for 30 min at 4 °C. Protein concentrations were determined using Coomassie Plus assay (Pierce, supplied by Perbio Science UK Ltd, Cheshire, UK). Cell extracts were resolved electrophoretically on NuPage® 4–12% Bis‐Tris acrylamide gels using 3‐(n‐morpholino)‐propanesulphonic acid buffer (Invitrogen) and transferred electrophoretically onto 0.45‐µm polyvinylidene fluoride membrane (GE Healthcare, UK) in 25 mm Tris base containing 192 mm glycine and methanol (10%, v/v) at 4 °C, 25 V for 90 min. Membranes were probed with primary antibodies overnight at 4 °C, and bound antibody detected with either goat anti‐rabbit immunoglobulin conjugated to IRDye® 800 (50 ng/ml; Rockland Immunochemicals; supplied by Tebu‐bio, Peterborough, UK) or anti‐mouse conjugated to Alexa Fluor® 680 (200 ng/ml; Invitrogen). Immunolabelled proteins were visualized using an Odyssey infrared imaging system (LiCor, Cambridge, UK).
Calcium imaging
NHU cell cultures were grown to 70–80% confluence and harvested by trypsinization. Collected cells were washed in KSFMc containing 1 mg/ml trypsin inhibitor (Invitrogen), and the final pellet was resuspended in HBSS and adjusted to 0.5 × 106 cells/ml. Cells were incubated with the fluorescence Ca2+ indicator fluo‐3‐AM (5 µm; Invitrogen) in HBSS containing Pluronic® F‐127 (0.2%; Invitrogen) for 30 min at 37 °C in 5% CO2, thereafter cells were washed three times in HBSS. NHU cells were transferred to quartz cuvettes containing a micro‐magnetic stirrer. A baseline fluorescence measurement (excitation wavelength 485 nm; emission wavelength 520 nm) was obtained using a FluoroMax‐2 spectrofluorometer (Jobin Yvon Horiba Ltd, Stanmore, UK). PPAR agonists were added manually and fluorescence was measured at 1 second intervals. Ionomycin (5 µm) was added at the end of each experiment to determine cell viability and maximum fluorescence of dye‐bound cytosolic calcium.
Assessment of cell death by flow cytometry
Mitochondrial‐mediated apoptosis was assessed using the MitoCapture™ apoptosis kit (Biovision Ltd, supplied by Merck Biosciences). Briefly, NHU cells were grown to 70–80% confluence and following treatment with ciglitazone (10 µm) for 3 or 6 h, cells were harvested as above and labelled with MitoCapture™ according to the manufacturer's instructions. Thereafter, NHU cells were analysed by flow cytometry using a CyAn™ instrument (DakoCytomation, Ely, UK) on the FL‐1 (FITC) channel. Time‐matched vehicle‐only‐treated cells were used as controls. In addition, apoptosis was quantified by flow cytometry following annexin V–FITC and propidium iodide (PI) labelling as described in detail previously (28). The cells were treated as above for 8 or 24 h with ciglitazone (10 µm) prior to harvesting and labelling with annexin V–FITC and PI, which were analysed on FL‐1 (FITC) and FL‐3 (PI) channels, respectively. For certain experiments, cells were pre‐incubated for 30 min in medium containing SOC inhibitors, 2‐APB and SKF‐96365 or PPARγ antagonist T0070907, at indicated concentrations. In all flow cytometry experiments, 106 cells/ml per sample were prepared and at least 104 cells were acquired and analysed using Summit® software (DakoCytomation).
Statistical analysis
Each experiment was performed in triplicate and replicated on three independent NHU cell lines, unless otherwise stated. All results are expressed as mean ± standard deviation. Statistical analysis was performed using the SPSS 11.0 software (Surrey, UK). Comparisons were analysed by one‐way analysis of variance. Differences were considered statistically significant if the P‐value was less than 0.05.
Results
PPAR agonist effect on growth and apoptosis of NHU cell cultures
NHU cell growth was assessed by MTT assays and even at the highest concentration tested (30 µm), the PPARα agonist fenofibrate had no significant effect on cell growth, compared to vehicle control (P = 0.20; Fig. 1a). By contrast, significant attenuation of growth was apparent with the PPARβ/δ agonist L165041 (≥ 1 µm; P < 0.05), PPARα/γ agonist ragaglitazar (≥ 10 µm; P < 0.01), and PPARγ agonists rosiglitazone (≥ 10 µm; P < 0.01), troglitazone (≥ 1 µm; P < 0.01) and ciglitazone (≥ 1 µm; P < 0.01), compared to vehicle‐treated controls (Fig. 1a–c).
Figure 1.
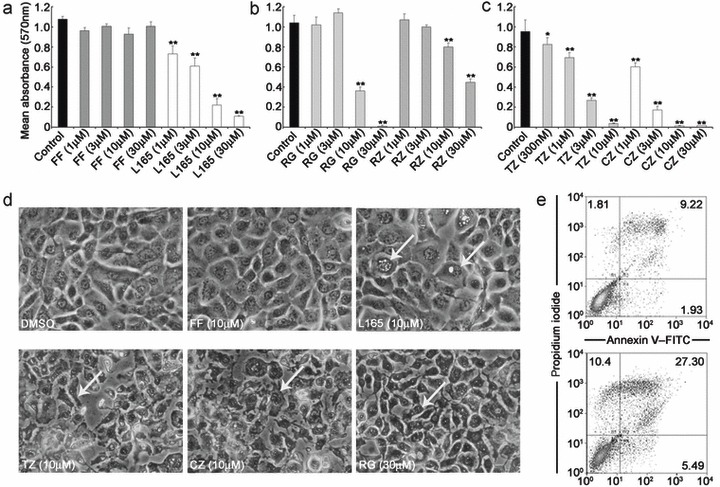
Grading of cytotoxic effects of peroxisome proliferator‐activated receptor (PPAR) agonists in normal human urothelial (NHU) cells. (a–c) NHU cells were treated with the indicated concentrations of PPAR agonist (or vehicle alone) and proliferation was assessed over 8 days by MTT assays. Bars represent mean absorbance from three independent experiments (± standard deviation) taken at day 6. Control, DMSO alone; FF, fenofibrate; L165, L165041; RG, ragaglitazar; RZ, rosiglitazone; TZ, troglitazone; CZ, ciglitazone. Statistical difference was determined using one‐way analysis of variance, *P < 0.05; **P < 0.01 compared to vehicle‐treated cells. (d) Following treatment of NHU cells for 72 h with the indicated concentrations of PPAR agonists or vehicle alone (DMSO), morphological changes associated with apoptosis were visualized by phase contrast microscopy and representative photomicrographs are shown; arrows indicate apoptotic changes. (e) Cells were treated with ciglitazone (10 µm) for 8 h and apoptosis was assessed by annexin V–FITC/PI labelling and flow cytometry. Representative results of cells treated with vehicle alone (upper panel) or ciglitazone (lower panel) are shown as dot plots of annexin V–FITC (x‐axis) vs. propidium iodide (PI, y‐axis) log10 fluorescence intensity. Quadrants were used for quantification of percentage positive cell populations as indicated in the dot plots.
To distinguish between cytostatic and apoptotic effects, cultured NHU cells were treated with ciglitazone, troglitazone, ragaglitazar, rosiglitazone, fenofibrate and L165041 (0.1–100 µm) and monitored by phase contrast microscopy over a period of 72 h (Fig. 1d). From 6 h onwards, ciglitazone (≥ 10 µm), troglitazone (≥ 10 µm) and ragaglitazar (≥ 30 µm) induced morphological changes associated with apoptosis, including membrane blebbing and cytoplasmic shrinkage (Fig. 1d). By contrast, fenofibrate, rosiglitazone (data not shown) and L165041 did not induce these changes at the concentrations tested (< 100 µm; Fig. 1d). PPARβ/δ agonist L165041 (10 µm) induced nuclear vacuolization in NHU cells from 24 h onwards, but this was not associated with cytoplasmic shrinkage nor membrane blebbing.
To confirm apoptosis, annexin V–FITC/PI assays were performed on the cells following treatment with ciglitazone (10 µm; Fig. 1e). By 8 h, there was an increase in the proportion of early (annexin V+ PI−) and late (annexin V+ PI+) apoptotic cells in NHU cell cultures treated with ciglitazone (10.4% early and 27.3% late) compared to vehicle‐only control (1.8% early and 9.2% late). This indicated that the growth inhibition and morphological changes seen with particular PPAR agonists were attributable to a rapid induction of apoptotic cell death.
Induction of apoptosis in NHU cells by ciglitazone
To determine whether PPAR agonist‐mediated apoptosis was due to a PPARγ receptor‐dependent mechanism, the effect of T0070907, a selective PPARγ antagonist (29), was assessed.
NHU cell cultures were pre‐treated with ciglitazone (10 µm) in the absence or presence of T0070907 (5 µm) for 24 h and apoptosis was assessed by annexin V–FITC/PI labelling and flow cytometry (Fig. 2a). Although treatment with ciglitazone alone induced high levels of apoptosis, there was no difference in the proportion of early (annexin V+ PI−) or late (annexin V+ PI+) apoptotic cells in the cell cultures treated with ciglitazone in the presence of T0070907 (5 µm). Percentage levels of early and late apoptotic cells in absence and presence of T0070907 was 6.48%/46.9% and 2.35%/48%, respectively.
Figure 2.
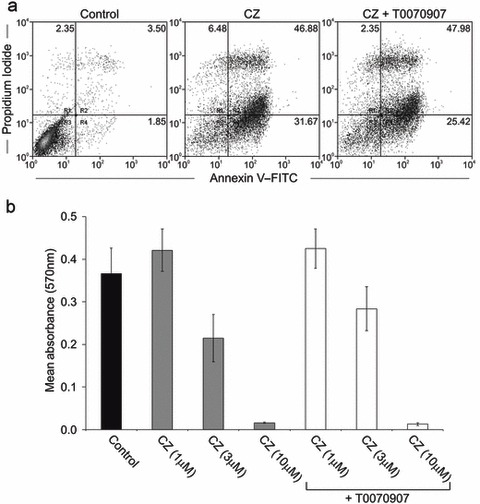
Induction of apoptosis by ciglitazone is an effect independent of peroxisome proliferator‐activated receptor (PPAR). (a) Normal human urothelial (NHU) cell cultures were treated with ciglitazone (10 µm), in the absence or presence of T0070907 (5 µm) for 24 h and apoptosis was assessed by annexin V–FITC/propidium iodide (PI) labelling and flow cytometry. Representative results of cells treated with vehicle alone (left panel), ciglitazone (10 µm; middle panel), or ciglitazone in the presence of T0070907 (5 µm; right panel) are shown as dot plots of annexin V–FITC (x‐axis) vs. PI (y‐axis) log10 fluorescence intensity. Quadrants were used for quantification of percentage of positive cell populations as indicated in the dot plots. (b) NHU cells were treated with the indicated concentration of ciglitazone (CZ) or vehicle control, in the absence or presence of the PPARγ antagonist, T0070907 (5 µm), as indicated. Cell proliferation was assessed over 3 days by MTT assay and results are shown from day 2. Bars represent mean absorbance (± standard deviation); data obtained from three independent NHU cell lines.
To confirm these findings, NHU cell viability was assessed using the MTT assay. Cell cultures were pre‐treated with T0070907 (5 µm) or vehicle (DMSO) for 1 h prior to exposure to ciglitazone (1–10 µm) and cultured for 48 h (Fig. 2b). Consistent with data obtained using annexin V–FITC/PI, there was no significant difference in growth of NHU cell cultures following treatment with ciglitazone in the absence or presence of T0070907 (P > 0.05). These findings demonstrate that PPARγ agonist‐mediated apoptosis of NHU cells is independent of direct receptor activation.
Effect of PPAR agonists on intracellular calcium in NHU cells
PPAR agonists were assessed for their ability to alter the concentration of intracellular calcium in cultured NHU cells loaded with the calcium indicator dye fluo‐3. In calcium‐containing HBSS, ciglitazone (30 µm), troglitazone (100 µm), ragaglitazar (100 µm) and L165041 (100 µm) evoked rapid increases of [Ca2+]i in the cells (Fig. 3a,b,d,f). These responses typically peaked within 1 min and were sustained. The mean ciglitazone (30 µm), troglitazone (100 µm), ragaglitazar (100 µm) and L165041 (100 µm) responses were 38.6 ± 3.6% (n = 3), 43.5 ± 19.4% (n = 3), 55 ± 8% (n = 3) and 11.3 ± 7% of the peak ionomycin (5 µm) response, respectively (Fig. 3a,b,d,f). By contrast, rosiglitazone and fenofibrate, at concentrations tested (1–100 µm), did not evoke changes in [Ca2+]i in NHU cells (Fig. 3c,e).
Figure 3.
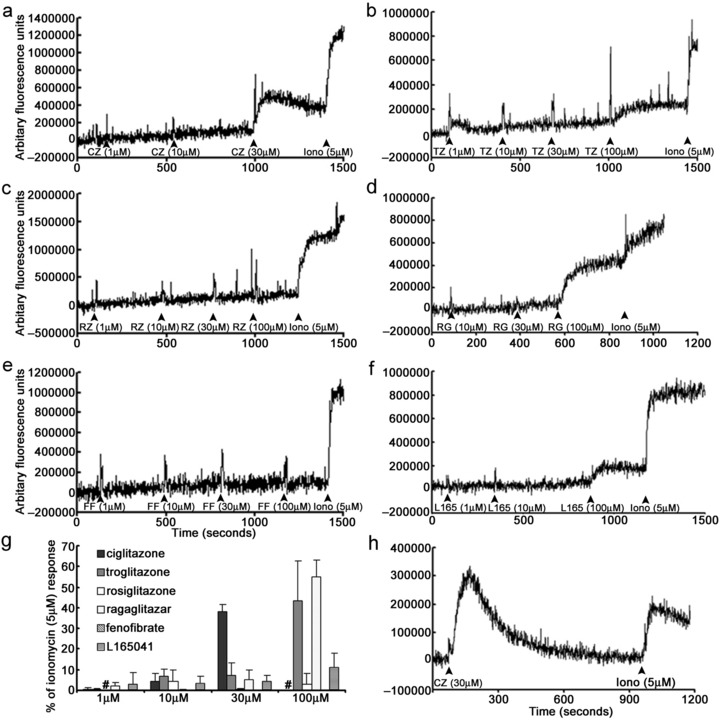
Grading of peroxisome proliferator‐activated receptor (PPAR) agonist effect on intracellular calcium levels in normal human urothelial (NHU) cells. (a–f) Representative continuous traces illustrating the changes in intracellular calcium in NHU cells loaded with the calcium indicator dye, fluo‐3‐AM, following addition of increasing concentrations of PPAR agonists, ciglitazone, troglitazone, rosiglitazone, ragaglitazar, fenofibrate and L165041. (g) Illustrates the mean amplitude of the maximum changes in intracellular calcium mediated by PPAR agonists expressed as a percentage of the maximum ionomycin (5 µm) response (± standard deviation) from three independent NHU cell lines; #, not done. (h) Representative continuous trace illustrating the changes in intracellular calcium in NHU cells following addition of ciglitazone (30 µm) in Ca2+‐free extracellular medium. FF, fenofibrate; L165, L165041; RG, ragaglitazar; RZ, rosiglitazone; TZ, troglitazone; CZ, ciglitazone; Iono, ionomycin.
Ciglitazone‐evoked calcium responses (30 µm) in NHU cells maintained in calcium‐free HBSS. However, these responses were transient and [Ca2+]i fully recovered to baseline levels between 10 and 15 min post‐application (Fig. 3h); this was in contrast to the sustained calcium responses evoked in calcium‐containing HBSS (Fig. 3a).
Induction of ERK and p38 phosphorylation by PPAR agonists in NHU cells
Relative to vehicle‐treated cultures, an increase in the ERKPhosphorylated/ERKTotal ratio was detected in NHU cells treated with troglitazone, ragaglitazar, ciglitazone and rosiglitazone, although there were differences in the kinetics between agonists. Troglitazone (10 µm) and ragaglitazar (30 µm) induced rapid ERK phosphorylation, peaking 5 min following exposure (Fig. 4a). ERK phosphorylation was also induced by ciglitazone (10 µm) and rosiglitazone (10 µm), peaking at 2–4 h. Neither fenofibrate (10 µm) nor L165041 (10 µm) induced ERK phosphorylation (Fig. 4a).
Figure 4.
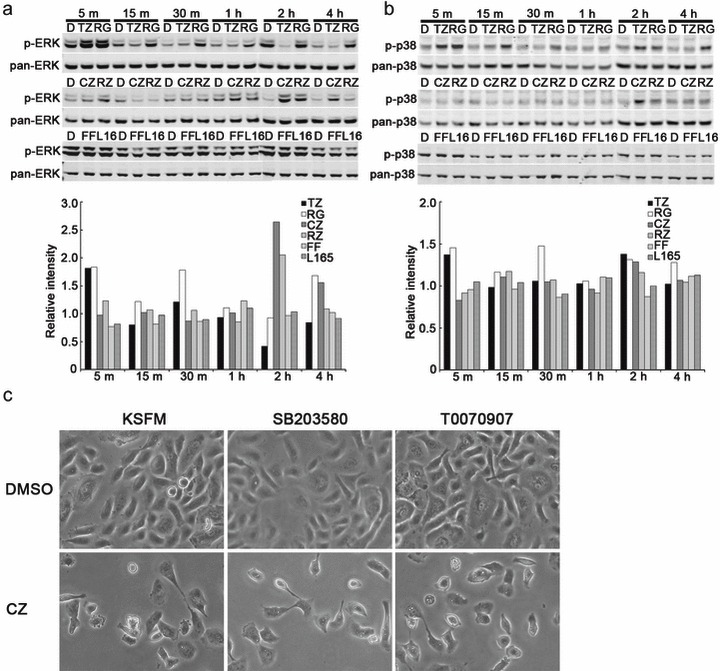
Pro‐apoptotic peroxisome proliferator‐activated receptor (PPAR) agonists induce phosphorylation of ERK and p38‐MAPK, but cell death is PPAR‐ and MAPK‐independent in cultured normal human urothelial (NHU) cells. Representative Western blots of the effects of PPAR agonists, troglitazone (TZ; 10 µm), ragaglitazar (RG; 30 µm), ciglitazone (CZ; 10 µm), rosiglitazone (RZ; 10 µm), fenofibrate (FF; 10 µm), and L165041 (10 µm) on ratios between (a) ERKPhosphorylated/ERKTotal and (b) p38Phosphorylated/p38Total were assessed in cultured NHU cells (three independent NHU cultures). Phosphorylation of Thr202/Tyr204 and Thr180/Tyr182 were examined for ERK and p38, respectively. (c) Illustrates representative photomicrographs of NHU cells that were pre‐treated with SB203580 (10 µm), T0070907(5 µm) or vehicle control (DMSO) alone for 1 h; cells (n = three independent cultures) were maintained in medium containing ciglitazone (10 µm) for 24 h and phenotype changes assessed using phase contrast microscopy.
Phosphorylation of p38 was observed in NHU cells following exposure to troglitazone (10 µm), ciglitazone (10 µm), ragaglitazar (30 µm) and, to a lesser extent, rosiglitazone (10 µm; Fig. 4b). Neither fenofibrate (10 µm) nor L165041 (10 µm) induced p38 phosphorylation in the cells (Fig. 4b). Thus, the PPAR agonists that induced ERK phosphorylation also induced p38 phosphorylation, but signal/noise ratios appeared lower in the p38 assay (Fig. 4, compare TZ, RG, CZ and RZ in 4a and 4b).
NHU cell death by pro‐apoptotic PPAR agonists is PPAR and MAPK independent
As phosphorylation of p38 MAPK has been implicated in induction of apoptosis, we examined the potential involvement of p38 in NHU cell death by functional inhibition. Using the same approach, we also sought to confirm that apoptosis was triggered by PPAR receptor‐independent mechanisms. As shown in Fig. 4c, treatment of the cells with ciglitazone (10 µm) resulted in extensive cell death in comparison to vehicle‐only controls. Pre‐treatment of the cells with inhibitors of either PPARγ (T0070907; 5 µm) or p38 (SB203580; 10 µm) pathways had no effect on ciglitazone‐induced cell death (Fig. 4c).
Change in mitochondrial membrane potential occurs early after ciglitazone exposure
Disruption of mitochondrial membrane potential (ΔΨm) is an early event following induction of apoptosis via the intrinsic pathway. We used the MitoCapture™ flow cytometric assay to assess changes mediated by ciglitazone (10 µm) on ΔΨm in cultured NHU cells. A time‐dependent increase in FITC fluorescence, indicating altered ΔΨm due to inability of the dye to form aggregates in mitochondria, was evident over 6 h compared to vehicle control (Fig. 5a, right panel).
Figure 5.
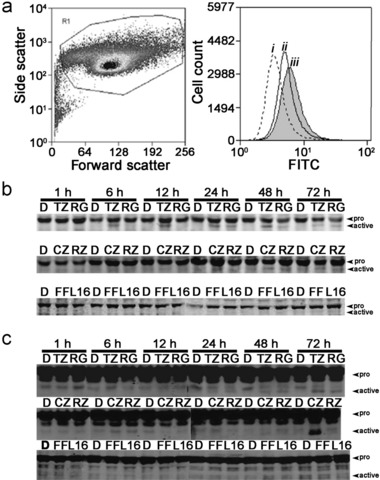
Peroxisome proliferator‐activated receptor (PPAR) agonist‐induced apoptosis is mediated by disruption of mitochondrial membrane potential with concomitant activation of caspase‐9 and caspase‐3. (a) Disruption of mitochondrial membrane potential (ΔΨm) was assessed using the MitoCaptureTM apoptosis detection kit following treatment with ciglitazone (10 µm) in cultured NHU cell populations (left panel) over a period of 6 h (i, vehicle control; ii–iii, 3 and 6 h post‐treatment, respectively). A time‐dependent increase in FITC‐fluorescence (right panel) was evident over 6 h, suggesting disruption of ΔΨm, by decreased ability of the mitochondria to form aggregates of the dye. (b and c) The effects of PPAR agonists troglitazone (TZ; 10 µm), ragagli‐ tazar (RG; 30 µm), ciglitazone (CZ; 10 µm), rosiglitazone (RZ; 10 µm), fenofibrate (FF; 10 µm) and L165041 (L165; 10 µm) on activation of caspase‐9 (b) and caspase‐3 (c) was assessed in cultured NHU cells over a period of 72 h using Western blotting. Full length and active fragments of caspase‐9 were detected at 45 kDa and 38 kDa, respectively. Full length and active fragments of caspase‐3 were detected at 32 and 18 kDa, respectively. All experiments were conducted using at least three independent NHU cultures.
Caspase‐3 and caspase‐9 activation in NHU cells by PPAR agonists
Caspase activation over the 72‐h period following exposure of NHU cells to PPAR agonists was assessed by immunoblotting using antibodies that recognized both pro‐ and active, cleaved caspase forms. Treatment of the cells with ciglitazone (10 µm), troglitazone (10 µm) and ragaglitazar (30 µm) all resulted in caspase‐9 activation (Fig. 5b). Effects of troglitazone (10 µm) were seen at 6 h and maintained throughout the 72‐h period assessed. Activation of caspase‐9 was observable at 24 h onwards with ragaglitazar (30 µm) and ciglitazone (10 µm). Little or no caspase‐9 activation was evident following exposure to rosiglitazone (10 µm), fenofibrate (10 µm) or L165041 (10 µm) over the time periods assessed (Fig. 5b).
In accordance with the caspase‐9 results, troglitazone (10 µm), ciglitazone (10 µm) and ragaglitazar (30 µm) all activated caspase‐3 after 72 h exposure (Fig. 5c). No caspase‐3 cleavage was evident following treatment with rosiglitazone or fenofibrate over the course of these experiments (Fig. 5c). No caspase‐3 activation was evident with L165041 over the 72‐h period assessed, but was shown to cleave caspase‐3 at later time points (data not shown).
Of note, pre‐treatment of NHU cells with specific caspase‐9 and caspase‐3 inhibitors, Z‐LEHD‐FMK (20 µm) and Z‐DEVD‐FMK (20 µm), did not prevent ciglitazone‐mediated (10 µm) cell death. In fact, caspase activation‐blockade accelerated the death process and altered morphology of dying cells from nuclear condensation and membrane blebbing to a more ‘swollen cytoplasm’ phenotype (data not shown).
Role of SOCs
There is evidence that sustained release of [Ca2+]i may lead to depletion of Ca2+ from intracellular stores, which causes influx of extracellular Ca2+ via opening of SOCs (30). To assess the effects of SOC inhibitors on ciglitazone‐mediated (10 µm) apoptosis, NHU cells were pre‐treated with 2‐APB (10–25 µm) and SKF‐96365 (25–50 µm) for 30 min followed by annexin V–FITC/PI assays. Although ciglitazone (10 µm) induced extensive apoptosis in the cells, pre‐treatment with 2‐APB (10 µm) significantly attenuated ciglitazone‐mediated apoptosis compared to vehicle‐treated controls (P < 0.01; Fig. 6a,c). Pre‐treatment of cells with SKF‐96365 (25 µm) reduced the pro‐apoptotic effects of ciglitazone (Fig. 6b,d), but this did not reach statistical significance (P > 0.05). Effects of 2‐APB (10 µm) and SKF‐96365 (25 µm) were also apparent at the cellular level by phase contrast microscopy, where ciglitazone‐mediated apoptotic changes, including membrane blebbing and cytoplasmic shrinkage, were reduced following pre‐treatment with 2‐APB (10 µm) and SKF‐96365 (25 µm; Fig. 6e). It should, however, be noted that at the higher concentrations both SOC inhibitors alone exhibited cytotoxicity in our cells (data not shown). This cytotoxicity was detected by annexin V–FITC/PI labelling and was evident from the reduced ability of 2‐APB (20 µm) to inhibit cell death (Fig. 6c), and particularly by the complete loss of any inhibitory effect of SKF‐96365 (50 µm; Fig. 6d).
Figure 6.
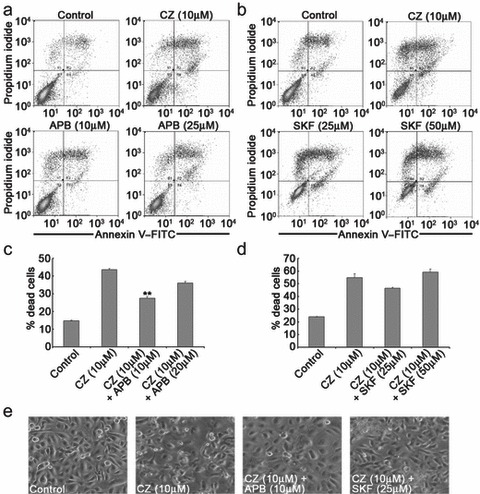
Peroxisome proliferator‐activated receptor (PPAR) agonist‐mediated apoptosis is mediated partially by activation of store‐operated calcium channels. (a,c) The increase in annexin V–FITC/PI staining in normal human urothelial (NHU) cells caused by ciglitazone (10 µm) was significantly attenuated following pre‐treatment with the SOC inhibitor, 2‐APB (10 µm). (b, d) Pre‐treatment with the SOC inhibitor, SKF‐96365 (25–50 µm for 30 min before ciglitazone) also reduced the ciglitazone‐induced increase in annexin V–FITC/PI staining, but was not statistically significant. (a) and (b) show representative experiments; data in (c) and (d) show mean percentage of apoptotic cells (annexin V and/or PI positive) from at least two independent NHU cell lines (± standard deviation). Statistical difference was determined using one‐way analysis of variance, **P < 0.01 compared to vehicle‐treated cells. (e) Illustrates macroscopic changes in the morphology of NHU cells associated with apoptosis following treatment with ciglitazone (10 µm for 8 h). These effects were partially inhibited following pre‐treatment with SOC inhibitors 2‐APB and SKF‐96365 (10 and 25 µm for 30 min before ciglitazone, respectively).
Discussion
We have demonstrated that troglitazone, ciglitazone or ragaglitazar, but not rosiglitazone, fenofibrate or L165041, may induce apoptosis in NHU cells, as indicated by morphological changes, annexin V/PI positivity and pro‐caspase cleavage. These effects were apparent within a pharmacologically relevant drug concentration range, although concentrations were 3‐ to 100‐fold higher than previously described to induce differentiation in NHU cells (9, 10, 11, 12). Induction of apoptosis was rapid (within 6 h), could not be inhibited using a specific PPARγ antagonist, T0070907, was restricted in response to some PPAR agonists only and occurred at concentrations above affinity constants for PPAR activation, thereby strongly suggesting that apoptosis was induced by PPAR‐independent (non‐genomic) mechanisms. While it has previously been shown that PPARα and PPARγ agonists may exhibit non‐genomic actions (2, 16, 31), our work extends this phenomenon also to PPARβ/δ and dual PPARα/γ agonists.
Previous studies have suggested that thiazolidinedione‐mediated elevation of intracellular calcium levels may contribute to their anti‐proliferative effects by inhibiting translation initiation and subsequent block of G1‐S transition (17). However, with the exception of fenofibrate, all PPAR agonists attenuated NHU cell growth, including rosiglitazone, which did not induce calcium transients, suggesting different mechanisms involved in cytostasis. Our study revealed a clear association between those ligands that induced an immediate increase in intracellular calcium and loss of viability in the medium term (hours‐days). Morphological changes strongly suggested that loss of viability was associated with induction of apoptosis. This was examined in further detail using ciglitazone (the most potent cytotoxic agent) as an example. Ciglitazone treatment caused surface exposure of phosphatidylserine that was not prevented by pre‐treatment with PPAR antagonist, T0070907, confirming the apoptotic mode of cell death induced by a PPAR‐independent mechanism. Large and sustained changes in [Ca2+]i are associated with signalling cascades regulating apoptosis (32). Elevated cytosolic calcium can, under certain pathological conditions, interact with cyclophilin D to open permeability transition pores in mitochondria, leading to dissipation of mitochondrial membrane potential (ΔΨm) and release of pro‐apoptotic mediators (33). Likewise, sustained rises in [Ca2+]i can lead to depletion of intracellular Ca2+ stores, such as the sarcoplasmic reticulum, and signal Ca2+ influx from the extracellular medium via opening of SOCs (30). Our data support this as the mechanism by which ciglitazone affected ΔΨm.
Removal of calcium from the extracellular medium significantly altered the kinetics of ciglitazone‐mediated calcium release in NHU cells from sustained to transient release, with full recovery to basal levels. Ciglitazone‐induced apoptosis was partially inhibited by SOC inhibitors SKF‐96365 and, particularly, 2‐APB. These findings demonstrate that cell death induced by ciglitazone involved disruption of mitochondrial ΔΨm, required elevated and sustained rise in intracellular calcium and functioning SOCs (summarized in Fig. 7). Similarly, inhibition of SOCs by lanthanum or SKF‐96365 has been reported to attenuate ciglitazone‐mediated anti‐proliferative effects in human uterine leiomyoma cells (34). Interestingly, a sustained increase in [Ca2+]i through SOCs has been reported as necessary for EGF receptor transactivation by ciglitazone (35).
Figure 7.
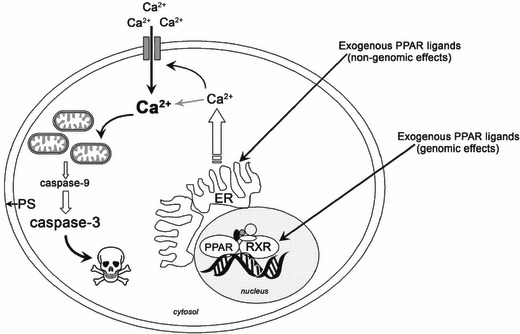
Proposed non‐genomic actions of pro‐apoptotic PPAR agonists independent of peroxisome proliferator‐activated receptor (PPAR). Acute exposure to PPARγ agonists ciglitazone, troglitazone and the dual PPARα/γ agonist, ragaglitazar rapidly induced apoptosis in normal human urothelial (NHU) cells. Pro‐apoptotic PPAR agonists induced rapid and sustained increases in intracellular calcium in NHU cells, leading to loss of mitochondrial membrane potential and activation of caspase‐9 and caspase‐3, suggesting activation of intrinsic apoptotic pathways. In addition to phenotypic changes, cell membrane disruption was observable as indicated by increased annexin V/PI positivity, which was partly attenuated by block of store‐operated calcium channels (SOC). These findings demonstrate that some PPARα/γ agonists activate the intrinsic apoptotic pathway in NHU cells in a PPAR‐independent manner, involving increases in [Ca2+]i and activation of SOCs. ER, endoplasmic reticulum; PS, phosphatidylserine.
Increased phosphorylation of ERK and p38 seen at high concentrations of troglitazone, ciglitazone and ragaglitazar may not have been unexpected, as activation of p38 is associated with induction of apoptosis in response to a variety of stimuli and pro‐survival pathways are activated in many cell death paradigms, including PI3 K/AKT and ERK (36). Moreover, although phosphorylation of the ERK pathway is generally considered a survival signal, a role for ERK activation in induction of apoptosis has also been described (37). By contrast, our study revealed clear independence between pro‐apoptotic effects of particular agonists and activation of p38/ERK. These included (i) rosiglitazone activated both ERK and p38, although it induced no increase in [Ca2+]i and was not pro‐apoptotic; (ii) there was disparity in concentrations of PPAR agonists used to evoke changes in [Ca2+]i, which were 0.5 log unit higher than concentrations that invoked changes in protein phosphorylation; (iii) p38 inhibition did not abrogate the apoptotic effect; and (iv) increases in p‐ERK occurred rapidly and transiently and were countered by marked attenuation at later time periods.
Ciglitazone, troglitazone and ragaglitazar, but not rosiglitazone or fenofibrate, induced cleavage of procaspase‐9, followed by later activation of caspase‐3. Activation of caspase‐9 is intimately associated with intrinsic apoptotic pathways leading to caspase‐3 activation (38). Intriguingly, pre‐treatment with caspase‐9 and caspase‐3 inhibitors did not prevent NHU cell death induced by ciglitazone, but accelerated the death process and altered the morphology of dying NHU cells from nuclear condensation and membrane blebbing to a ‘swollen cytoplasm’ phenotype, which is characteristic of a necrotic response (39). This suggests that although caspase activation occurred as a consequence of rapid mitochondrial membrane permeabilization, it is not obligatory for PPAR‐mediated death. Our observations are not anomalous, as it is well‐documented that caspase activation and subsequent biochemical and morphological phenotype of apoptotic death are not mutually interdependent (reviewed by Subramaniam & Unsicker (40) and Jones et al. (41)). In particular, there are reports of caspase‐independent, calcium‐mediated ‘paraptotic’ cell death (42) and other studies have also demonstrated that caspase inhibitors may not prevent cell death, but instead lead to necrosis, whereas certain stimuli can result in either apoptotic or necrotic death, depending on the availability of activated caspases (43).
Previous studies have demonstrated changes in [Ca2+]i by PPARγ agonists in a number of cell systems: human uterine myometrium/leiomyoma cells, PPAR−/– as well as PPAR+/+ mouse embryonic stem cells, human monocytes and rat hepatocytes (17, 34, 35). Thus, our study highlights the striking similarity of the non‐genomic effects of PPAR agonists across different cell systems and across structurally different PPAR agonists. Likewise, steroids and thyroid hormone, substances exhibiting even larger structural variability than the PPAR agonists examined in the present study, exhibit a similar spectrum of non‐genomic actions across a wide variety of cell types (18). Non‐genomic effects of steroids and PPAR agonists are strikingly similar: both are accompanied by rises in intracellular calcium, activation of p38 and ERK MAPK and ensuing cytotoxicity/apoptosis (2, 16, 18). For PPAR agonists, non‐genomic effects have been defined as (i) occurring very rapidly, or (ii) in cells not expressing PPARs, or (iii) despite pre‐treatment of cells with PPAR antagonists, or (iv) with structural PPAR agonist variants lacking agonist activity such as Δ2‐ciglitazone or Δ2‐troglitazone (2, 16). Similar methodology has been used to define non‐genomic effects of steroids, and importantly, these methods do not rule out the presence of non‐classical receptors mediating the non‐genomic effect (18). Determination of whether non‐classical receptors are involved in non‐genomic actions of PPAR agonists may help in developing compounds with improved toxicity profiles.
Conclusion
The results of the present study provide a mechanistic basis for the ability of some PPAR agonists to induce death in human urothelial cells, and demonstrate that apoptosis is mediated at non‐pharmacological concentrations via PPAR‐independent mechanisms that involve intracellular calcium changes, activation of SOCs and induction of the mitochondrial apoptotic pathway. These findings highlight paradoxical PPAR‐dependent and ‐independent agonist effects on urothelial cell differentiation and survival responses, which may play contributory roles in bladder pathophysiology. However, it remains as yet unknown whether PPAR‐induced bladder carcinogenesis in rodents is relevant in humans.
Acknowledgements
We are grateful to clinical colleagues for supplying tissue samples for our research. NTG was funded by the Engineering and Physical Sciences Research Council (EPSRC), Grant GR/S62338/01. This study was funded by Novo Nordisk. J.S. is supported by York Against Cancer.
References
- 1. El‐Hage G (2004) Preclinical and Clinical Safety Assessments for PPAR Agonists [WWW document]. http://www.fda.gov/cder/present/DIA2004/Elhage.ppt [accessed on 2 July 2009].
- 2. Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM (2006) The toxicology of ligands for peroxisome proliferator‐activated receptors (PPAR). Toxicol. Sci. 90, 269–295. [DOI] [PubMed] [Google Scholar]
- 3. Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM (1997) Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator‐activated receptor γ and the retinoid X receptor. Proc. Natl. Acad. Sci. USA 94, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, Spiegelman BM, Singer S (1999) Induction of solid tumor differentiation by the peroxisome proliferator‐activated receptor‐γ ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. USA 96, 3951–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM (1994) mPPARγ2: tissue‐specific regulator of an adipocyte enhancer. Genes Dev. 8, 1224–1234. [DOI] [PubMed] [Google Scholar]
- 6. Guan Y, Zhang Y, Davis L, Breyer MD (1997) Expression of peroxisome proliferator‐activated receptors in urinary tract of rabbits and humans. Am. J. Physiol. 273, F1013–F1022. [DOI] [PubMed] [Google Scholar]
- 7. Jain S, Pulikuri S, Zhu Y, Qi C, Kanwar YS, Yeldandi AV, Rao MS, Reddy JK (1998) Differential expression of the peroxisome proliferator‐activated receptor γ (PPARγ) and its coactivators steroid receptor coactivator‐1 and PPAR‐binding protein PBP in the brown fat, urinary bladder, colon, and breast of the mouse. Am. J. Pathol. 153, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chopra B, Hinley J, Oleksiewicz MB, Southgate J (2008) Trans‐species comparison of PPAR and RXR expression by rat and human urothelial tissues. Toxicol. Pathol. 36, 485–495. [DOI] [PubMed] [Google Scholar]
- 9. Varley CL, Garthwaite MA, Cross W, Hinley J, Trejdosiewicz LK, Southgate J (2006) PPARγ‐regulated tight junction development during human urothelial cytodifferentiation. J. Cell Physiol. 208, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varley CL, Stahlschmidt J, Lee WC, Holder J, Diggle C, Selby PJ, Trejdosiewicz LK, Southgate J (2004a) Role of PPARγ and EGFR signalling in the urothelial terminal differentiation programme. J. Cell Sci. 15, 2029–2036. [DOI] [PubMed] [Google Scholar]
- 11. Varley CL, Stahlschmidt J, Smith B, Stower M, Southgate J (2004b) Activation of peroxisome proliferator‐activated receptor‐γ reverses squamous metaplasia and induces transitional differentiation in normal human urothelial cells. Am. J. Pathol. 164, 1789–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Varley CL, Bacon EJ, Holder JC, Southgate J (2009) FOXA1 and IRF‐1 intermediary transcriptional regulators of PPARγ‐induced urothelial cytodifferentiation. Cell Death Differ. 16, 103–114. [DOI] [PubMed] [Google Scholar]
- 13. Shaik MS, Chatterjee A, Singh M (2004) Effect of a selective cyclooxygenase‐2 inhibitor, nimesulide, on the growth of lung tumors and their expression of cyclooxygenase‐2 and peroxisome proliferator‐ activated receptor‐γ. Clin. Cancer Res. 10, 1521–1529. [DOI] [PubMed] [Google Scholar]
- 14. Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, Sporn MB (1999) A new ligand for the peroxisome proliferator‐activated receptor‐γ (PPAR‐γ), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 59, 5671–5673. [PubMed] [Google Scholar]
- 15. Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP (2003) Use of the peroxisome proliferator‐activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res. Treat. 79, 391–397. [DOI] [PubMed] [Google Scholar]
- 16. Gardner OS, Dewar BJ, Graves LM (2005) Activation of mitogen‐activated protein kinases by peroxisome proliferator‐activated receptor ligands: an example of nongenomic signaling. Mol. Pharmacol. 68, 933–941. [DOI] [PubMed] [Google Scholar]
- 17. Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA (2001) Anticancer effects of thiazolidinediones are independent of peroxisome proliferator‐activated receptor γ and mediated by inhibition of translation initiation. Cancer Res. 61, 6213–6218. [PubMed] [Google Scholar]
- 18. Lösel R, Wehling M (2003) Nongenomic actions of steroid hormones. Nat. Rev. Mol Cell Biol. 42, 46–56. [DOI] [PubMed] [Google Scholar]
- 19. Lubet RA, Fischer SM, Steele VE, Juliana MM, Desmond R, Grubbs CJ (2008) Rosiglitazone, a PPAR gamma agonist: Potent promoter of hydroxybutyl (butyl) nitrosamine‐induced urinary bladder cancers. Int. J. Cancer. 123, 2254–2259. [DOI] [PubMed] [Google Scholar]
- 20. Long GG, Reynolds VL, Lopez‐Martinez A, Ryan TE, White SL, Eldridge SR (2008) Urothelial carcinogenesis in the urinary bladder of rats treated with naveglitazar, a γ‐dominant PPAR α/γ agonist: lack of evidence for urolithiasis as an inciting event. Toxicol. Pathol. 36, 218–231. [DOI] [PubMed] [Google Scholar]
- 21. Oleksiewicz MB, Southgate J, Iversen L, Egerod FL (in press) Rat urinary bladder carcinogenesis by dual‐acting PPAR α+γ agonists. PPAR Res. 2009 in press (PMID: 19197366) [DOI] [PMC free article] [PubMed]
- 22. Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD (1999) Expression of peroxisome proliferator‐activated receptor γ (PPARγ) in human transitional bladder cancer and its role in inducing cell death. Neoplasia 1, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawakami S, Arai G, Hayashi T, Fujii Y, Xia G, Kageyama Y, Kihara K (2002) PPARγ ligands suppress proliferation of human urothelial basal cells in vitro . J. Cell. Physiol. 191, 310–319. [DOI] [PubMed] [Google Scholar]
- 24. Chaffer CL, Thomas DM, Thompson EW, Williams ED (2006) PPARγ‐independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakashiro KI, Hayashi Y, Kita A, Tamatani T, Chlenski A, Usuda N, Hattori K, Reddy JK, Oyasu R (2001) Role of peroxisome proliferator‐activated receptor γ and its ligands in non‐neoplastic and neoplastic human urothelial cells. Am. J. Pathol. 159, 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Southgate J, Hutton KA, Thomas DF, Trejdosiewicz LK (1994) Normal human urothelial cells in vitro: proliferation and induction of stratification. Lab. Invest. 71, 583–594. [PubMed] [Google Scholar]
- 27. Southgate J, Masters JR, Trejdosiewicz LK (2002) Culture of human urothelium In: Freshney RI, Freshney MG, eds. Culture of Epithelial Cells, pp. 381–400. New York: John Wiley & Sons. [Google Scholar]
- 28. Steele LP, Georgopoulos NT, Southgate J, Selby PJ, Trejdosiewicz LK (2006) Differential susceptibility to TRAIL of normal versus malignant human urothelial cells. Cell Death Differ. 13, 1564–1576. [DOI] [PubMed] [Google Scholar]
- 29. Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y (2002) T0070907, a selective ligand for peroxisome proliferator‐activated receptor γ, functions as an antagonist of biochemical and cellular activities. J. Biol. Chem. 277, 19649–19657. [DOI] [PubMed] [Google Scholar]
- 30. Lewis RS (2007) The molecular choreography of a store‐operated calcium channel. Nature 446, 284–287. [DOI] [PubMed] [Google Scholar]
- 31. Jain A, Agus DB (2004) PPARgamma signaling: one size fits all? Cell Cycle 3, 1352–1354. [DOI] [PubMed] [Google Scholar]
- 32. Scorrano L (2003) Divide et impera: Ca2+ signals, mitochondrial fission and sensitization to apoptosis. Cell Death Differ. 10, 1287–1289. [DOI] [PubMed] [Google Scholar]
- 33. Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P (2005) Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 280, 18558–18561. [DOI] [PubMed] [Google Scholar]
- 34. Kim BY, Cho CH, Song DK, Mun KC, Suh SI, Kim SP, Shin DH, Jang BC, Kwon TK, Cha SD, Bae I, Bae JH (2005) Ciglitizone inhibits cell proliferation in human uterine leiomyoma via activation of store‐operated Ca2+ channels. Am. J. Physiol. Cell Physiol. 288, C389–C395. [DOI] [PubMed] [Google Scholar]
- 35. Dewar BJ, Gardner OS, Chen CS, Earp HS, Samet JM, Graves LM (2007) Capacitative calcium entry contributes to the differential transactivation of the epidermal growth factor receptor in response to thiazolidinediones. Mol. Pharmacol. 72, 1146–1156. [DOI] [PubMed] [Google Scholar]
- 36. Zarubin T, Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18. [DOI] [PubMed] [Google Scholar]
- 37. Chang F, Steelman LS, Shelton JG, Lee JT, Navolanic PM, Blalock WL, Franklin R, McCubrey JA (2003) Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway. Int. J. Oncol. 22, 469–480. [PubMed] [Google Scholar]
- 38. Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281, 1312–1316. [DOI] [PubMed] [Google Scholar]
- 39. Denecker G, Vercammen D, Steemans M, Vanden Berghe T, Brouckaert G, Van Loo G, Zhivotovsky B, Fiers W, Grooten J, Declercq W, Vandenabeele P (2001) Death receptor‐induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ. 8, 829–840. [DOI] [PubMed] [Google Scholar]
- 40. Subramaniam S, Unsicker K (2006) Extracellular signal‐regulated kinase as an inducer of non‐apoptotic neuronal death. Neuroscience 138, 1055–1065. [DOI] [PubMed] [Google Scholar]
- 41. Melino G, Knight RA, Nicotera P (2005) How many ways to die? How many different models of cell death? Cell Death Differ. 12(Suppl. 2), 1457–1462. [DOI] [PubMed] [Google Scholar]
- 42. Jambrina E, Alonso R, Alcalde M, Del Carmen Rodríguez M, Serrano A, Martínez AC, García‐Sancho J, Izquierdo M (2003) Calcium influx through receptor‐operated channel induces mitochondria‐triggered paraptotic cell death. J. Biol. Chem. 278, 14134–14145. [DOI] [PubMed] [Google Scholar]
- 43. Jones BE, Lo CR, Liu H, Srinivasan A, Streetz K, Valentino KL, Czaja MJ (2000) Hepatocytes sensitized to tumor necrosis factor‐α cytotoxicity undergo apoptosis through caspase‐dependent and caspase‐independent pathways. J. Biol. Chem. 275, 705–712. [DOI] [PubMed] [Google Scholar]