

Abstract
Objectives: The aim of this study was to determine whether normal human embryonic stem cells (hESC) would secrete factors that arrest growth of human epithelial cancer cell lines.
Materials and methods: Cell proliferation was examined using the MTT assay then haemocytometer cell counts. Staining with propidium iodide followed by flow cytometry was used to detect cell cycle stages. Heat denaturation and molecular fractionation experiments were also performed.
Results: We found that hESC conditioned medium (hESC CM) inhibited SKOV‐3 and HEY cell proliferation. Similar results were also obtained when we used breast and prostate cancer cell lines, whereas little or no inhibitory effect was observed when human fibroblasts were tested. Moreover, a co‐culture model confirmed that inhibition of cancer cell proliferation is mediated by soluble factors produced by hESCs. We also determined that the proportion of cancer cells in G1 phase was increased by hESC CM treatment, accompanied by decrease in cells in S and G2/M phases, suggesting that the factors slow progression of cancer cells by cell cycle inhibition. Heat denaturation and molecular fractionation experiments indicated a low molecular weight thermostable factor was responsible for these properties.
Conclusions: Our findings provide evidence that the human embryonic microenvironment contains soluble factor(s) that are capable of inhibiting growth of cancer cells, and that exposure to such factors may represent a new cancer treatment strategy.
Introduction
Epithelial cancers account for the deaths of approximately 250 000 North Americans annually. These tumours are thought to arise from cancer stem cells (1, 2, 3, 4, 5), a concept developed to explain clonality of many tumours. Most malignant neoplasms consist of heterogeneous cell populations, including tumour stem cells (initiating cells) that are rare, and the rest being tumour non‐initiating cells (6, 7, 8). Tumour initiating cells or cancer stem cells (CSC) have the capability of unlimited self‐renewal, are resistant to drugs and retain markers that are expressed by normal stem cells. It has been suggested that CSCs originate from normal stem cells or from progenitor cells, as a consequence of genetic and epigenetic changes. CSCs can also originate from somatic tumour cells by dedifferentiation or reprogramming to a stem‐like cell. It is possible that both mechanisms are involved in the origin of CSCs. One of the requirements for development of cancer is dysregulation of stem cell self‐renewal (6, 7). Normally, through asymmetrical cell division, a single stem cell gives rise to one stem cell and one progenitor cell, which will give rise to a large number of cells all progressing towards a terminally differentiated state (9).
Despite human embryonic stem cells (hESC) and cancer cells sharing many similarities, hESCs do not form tumours following blastocyst implantation. This could be explained by the fact that hESCs are protected from tumorigenesis in vivo because they differentiate when present in an environment that otherwise would drive them to tumorigenesis (10). Thus, it is believed that this microenvironment termed ‘the stem cell niche’ is able to regulate stem cell fate (9, 11). In normal physiological conditions, the stem cell niche provides an interactive environment that helps maintain a balance between cell proliferation, differentiation and apoptosis, thereby preserving tissue homeostasis (12). In this tightly controlled situation, stem cells provide replacement cells throughout life but only as needed. In contrast, the CSC has unlimited proliferation potential, due to genetic abnormalities and a malignant (or reactive) stem cell niche (13). It remains possible that components of stem cells in the normal stem cell niche are lost in cancer and that CSCs retain the ability to be controlled if the niche is restored.
We hypothesized that stem cells may produce an autocrine or paracrine factor that limits their ability to proliferate. It is possible that cancer cells or CSCs have lost such factor(s); thus, these factors, as part of the hESC microenvironment, could be used to inhibit malignant cell growth. In support of this hypothesis, previous studies have demonstrated that embryonic microenvironments suppress the tumorigenicity of several cancer cell lines (14, 15, 16, 17, 18), for example, a tumour suppressive effect of embryonic zebrafish microenvironment on melanoma cells has been shown (16). Other studies have demonstrated that hESCs grown in a three‐dimensional microenvironment were able to reverse invasion of human metastatic melanoma cells into a three‐dimensional matrix (17).
The objective of the present study was to determine whether early‐passage hESCs would secrete soluble factors into the culture medium that arrest or slow population growth of human epithelial cancer cells; a variety of ovarian, prostate and breast cancer cell lines was studied. We also investigated the effect of hESC conditioned medium (hESC CM) on non‐malignant fibroblast cell population growth. We were able to demonstrate that a soluble factor (or factors) secreted by hESCs, inhibited population growth of all the cancer cell lines tested, whereas feeder‐cell conditioned medium had no effect. In addition, little or no inhibitory effect of hESC CM was observed for benign human fibroblast cells. Our data suggest that our factors act by slowing progression of cancer cells through the cell cycle.
Materials and Methods
Materials
The SKOV‐3 cell line obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) is a moderately well differentiated ovarian adenocarcinoma cell derived from ascites fluid of a 64‐year‐old Caucasian woman. HEY cells were provided by Dr Alexander Marks (Banting and Best Research Institute, University of Toronto, Toronto, Ontario, Canada) and were orthotopically established from a moderately well differentiated papillary (serous) cystadenocarcinoma of the ovary (19). PC‐3 and DU145 cells obtained from ATCC were derived from a grade IV prostate adenocarcinoma and a lesion of the brain of a patient with widespread metastatic prostate carcinoma, respectively. MCF‐7 and T47D breast adenocarcinoma cells obtained from ATCC were derived from pleural effusion from a 69‐year‐old and 54‐year‐old woman, respectively.
hESC lines (CA1 and CA2 hESCs) were derived from two separate human embryos at Mount Sinai Hospital, Samuel Lunenfeld Research Institute, Toronto, Ontario, Canada. The International Stem Cell Initiative has characterized both cell lines (20). Mouse primary embryonic fibroblasts (MEF) were obtained from the embryonic stem cell facility (Mount Sinai Hospital, Samuel Lunenfeld Research Institute). Human embryonic fibroblasts were obtained from human tissue derived at Mount Sinai Hospital, Samuel Lunenfeld Research Institute. Unless otherwise stated, all culture media and supplements were purchased from Gibco‐BRL (Gaithersburg, MD, USA). Cell culture inserts (0.4 μm high pore density) were obtained from Becton Dickinson (Franklin Lakes, NJ, USA) and MTT (3,[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Propidium iodide (PI), RNase type A and annexin V were purchased from Sigma Chemical Co. Amicon Ultra 50 K and Amicon Ultra 10 K centrifugal filter devices were purchased from Millipore (Bedford, MA, USA). Dimethyl sulphoxide (DMSO) was obtained from Fisher Scientific (Pittsburgh, PA, USA).
Cell culture conditions
Cancer cell lines were maintained in RPMI 1640 medium supplemented with 10% foetal bovine serum (FBS), l‐glutamine (2 mm), penicillin (100 U/ml), streptomycin (100 μg/ml), and fungizone (250 μg/ml). Medium for T47D was supplemented with 0.16 U/ml insulin. CA1 and CA2 hESCs were grown in 90‐mm dishes coated with a feeder monolayer of MEFs that had been mitotically inactivated with mitomycin. In this study, we used hESCs between passages 14 and 40. hESCs were cultured in knockout Dulbecco’s modified Eagle’s medium supplemented with 18% knockout serum replacement, 4.5% Plasmanate, 2 mm GlutaMAX 1, 50 U/μg/μl Penstrep, 55 μmβ‐mercaptoethanol, 0.1 mm non‐essential amino acids and 0.02 μg/ml FGF2 (Gibco‐BRL). All cells were grown at 37 °C in a humidified incubator in an atmosphere of 5% CO2 in air.
Conditioned medium culture
Control conditioned medium was produced by incubating MEFs with stem cell medium for 24, 48 or 72 h (day 1, day 2, day 3 MEF CM). hESC CM was obtained by overlaying MEF cells with hESCs in stem cell medium for 24, 48 or 72 h (day 1, day 2, day 3 MEF + hESC CM). hESCs were analysed for stem cell markers SSEA3 (anti‐stage‐specific embryonic antigen‐3), TRA‐1‐60 and Oct‐3/Oct‐4 and SSEA1 (anti‐stage‐specific embryonic antigen‐1) to verify that they did not differentiate over this time (Appendix S1, Supporting Information). Feeders and hESCs were grown on 90‐mm plates with 10 ml of stem cell medium. Feeders were plated at 800 000 cells per plate and hESCs were plated at 200 000 cells per plate. CM was harvested and passed through a 0.2‐μm syringe filter to remove any cellular debris.
Cell proliferation assay
Cell proliferation was determined by the MTT reduction method (21) and by direct cell counts. Cells were plated at a density of 5000 or 6000 cells/well in 200 μl of growth medium in 96‐well plates, at a density of 70 000 or 100 000 cells/well in 2 ml of growth medium in 6‐well plates. Cells were allowed to attach overnight. Twenty‐four hours later, the medium was removed and cells were then incubated with different conditioned media as indicated, for each experiment. For MTT assays, 30 μl of 5 mg/ml solution of MTT in phosphate‐buffered saline (PBS) was added to each well to be analysed. Cells were incubated for 4 h with MTT at 37 °C, after which the medium was aspirated and dimethyl sulphoxide was added to each well. Plates were placed in a microplate shaker for 5 min and absorbance at 570 nm was determined using an ELISA microplate reader (μQuant Microplate Spectrophotometer, Bio‐Tek, Instruments, Winooski, VT, USA). Direct cell counting was performed using a haemocytometer and trypan‐blue dye exclusion. Cells were harvested using trypsin‐EDTA (Gibco‐BRL) and counted using a light microscope. Stained cells were also counted to determine the level of dead cells present.
DNA cell cycle analysis
Cells were plated at a density of 40 000–100 000 cells/well in 2 ml of growth medium in 6‐well plates and were allowed to attach overnight. On reaching 50–60% confluence, cell cultures were synchronized by serum deprivation as follows. Medium was aspirated and cells were rinsed in PBS. They were then overlaid with 2 ml of 0.5% stripped serum in RPMI 1640 and incubated for 48 h. At the end of the synchronization process, medium was replaced by one of the conditioned media: day 3 MEF CM as control and day 3 MEF + hESC CM. Cells were harvested into 15 ml sterile conical tubes, centrifuged and washed in ice‐cold PBS. Cells (1 × 106 per test) were fixed in 70% cold ethanol for 30–60 min at −20 °C, washed in cold PBS and resuspended in PBS solution containing 50 μg/ml of PI and 100 units/ml of RNase type A. Cells were then left to incubate in the dark for 30–60 min at room temperature. Samples were filtered using a 50‐μm nylon membrane and flow cytometry analysis was performed after gating, to eliminate doublets and debris.
Annexin V assay
Cells were plated at a density of 100 000 cells/well in 2 ml of RPMI 1640 in 6‐well plates and were allowed to attach overnight. Twenty‐four hours later, medium was removed and cells were then incubated with different conditioned media as indicated previously. Cells were harvested into 15 ml sterile conical tubes, centrifuged and washed in PBS. Then they were resuspended in 1× binding buffer (10 mm HEPES/NAOH containing 0.14 m NaCl and 2.5 mm CaCl2) at a concentration of 1 × 106 cells/ml. Five microlitres of annexin V–FITC (Sigma Chemical Co.) and 10 μl of PI solution were added to cell suspensions. Cells were then left to incubate in the dark for 10 min at room temperature. Samples were filtered using a 50 μm nylon membrane, and flow cytometric analysis was performed.
Flow cytometry analysis
DNA cell cycle and annexin V were evaluated by flow cytometry using a FAC‐Scan flow cytometer (Becton Dickinson) equipped with a 488 nm argon laser as light source. Two fluorescence detectors were used according to which assay utilized, to measure fluorescence corresponding to green (FL‐1 detector 525 nm wavelength band) and to red (FL‐3 detector 670 nm wavelength band). In all, 10 000 events were measured for each sample at a flow rate of 100–200 events per second. Cellular debris was gated out by drawing a region on forward versus side scatter dot plots enclosing the population of cells of interest.
Cancer and hESCs co‐cultures
SKOV‐3 and HEY cells were plated at a density of 80 000 and 50 000 cells/well, respectively, in 2 ml RPMI 1640 medium supplemented with 10% FBS in 6‐well plates. Separately, hESCs on feeder layers, and feeders only, were plated at a density of 100 000 cells/insert. Cells were allowed to attach overnight. Twenty‐four hours later, RPMI 1640 was replaced with stem cell medium and inserts containing feeders only or hESCs on feeders were placed in wells containing the cancer cell lines. Co‐culture occurred for 72–120 h wherein medium was changed every 24 h. Cell number was assessed at each time point using the MTT dye‐reduction method and by direct cell counts.
Rescue experiment
SKOV‐3 and HEY cells were plated at a density of 80 000 and 50 000 cells/well, respectively, in 2 ml of RPMI 1640 containing 10% FBS in 6‐well plates. Twenty‐four hours later, RPMI 1640 was replaced with one of the following media: MEF CM, MEF + hESC CM or stem cell medium. After 72 h, media were aspirated and cells were rinsed in PBS. Cells were overlaid with fresh stem cell medium and were left to incubate for another 72 h. Cell number was assessed using the MTT dye‐reduction method.
Heat inactivation and size fractionation of MEF CM and MEF + hESC CM
Day 3 MEF CM and day 3 MEF + hES CM samples were heated at 55 °C, 65 °C or 90 °C for 5, 10, 20 or 30 min to study the impact of heat denaturation on growth inhibitory effect of MEF + hES CM. To determine molecular size of the inhibitory factor or factors, MEF CM and MEF + hESC CM were fractionated using Amicon Ultra 50 K and Amicon Ultra 10 K sieves (Millipore). Media were ultrafiltered through a 50 kDa membrane and centrifuged at 4000 g for 45 min. This step was repeated twice to recover concentrated > 50 kDa fraction. Eluates were then ultrafiltered through a 10 kDa membrane (concentrate = fraction > 10 kDa). The procedure was repeated as above. The final eluate represented the < 10 kDa fraction. Concentrates were then diluted in fresh stem cell medium. Cell number was assessed using the MTT dye‐reduction method.
Statistical analysis
Results are reported as mean ± standard error of the mean of four to eight replicates per group. Data were analysed using one‐way and two‐way analysis of variance followed by the Student–Newman–Keuls multiple range test using SPSS 11.0 for Windows (SPSS Inc., Chicago, IL, USA). A statistically significant difference was accepted when the P‐value was lower than 0.05.
Results
Effects of hESC CM on SKOV‐3 and HEY cell proliferation
To determine whether embryonic stem cells secrete factors that affect cancer cells, we tested the impact of day 1, day 2 and day 3 MEF + hESC CM on HEY and SKOV‐3 cell population growth in culture. CM was collected from either MEFs alone (control) or MEFs and hESC cells after 24, 48 or 72 h of incubation. The undifferentiated state of CA1 and CA2 hESCs was demonstrated by expression of three stem cell markers: SSEA3, TRA‐1‐60 and Oct‐3/Oct‐4 (Fig. S1, Supporting Information). Since CA1 hESC CM was able to inhibit cancer cell proliferation similar to that observed among the CA2 hESC CM (Fig. S2, Supporting Information), the study was carried out using the CA2 cell line only. Non‐conditioned embryonic stem cell medium and RPMI medium + 10% serum were used as additional controls. Since the cell lines consistently grew as well in stem cell medium conditioned by MEF cells alone as in stem cell medium or RPMI 1640, these controls were eventually discontinued. Media from SKOV‐3 and HEY cells seeded into 96‐ or 6‐well plates 24 h earlier was replaced with embryonic stem cell medium, RPMI 1640, MEF CM, or MEF + hESC CM, and MTT dye‐reduction assay or haemocytometer counting were performed at 0, 24, 48, or 72 h. Medium conditioned with MEF + hESC for only 24 h was sufficient to reduce proliferation of SKOV‐3 cells after 48 or 72 h of culture but not after 24 h. Use of 48 and 72 h CM resulted in stronger inhibition than was apparent with 24 h CM (Fig. 1a). Virtually identical results were obtained for HEY ovarian cancer cells (Fig. 1b). There was no difference between cells exposed to MEF CM and both ES and RPMI 1640 at 24–72 h. Here we show results obtained at 72 h with both MTT (Fig. 1c,d) and direct cell counts (Fig. 1e,f).
Figure 1.
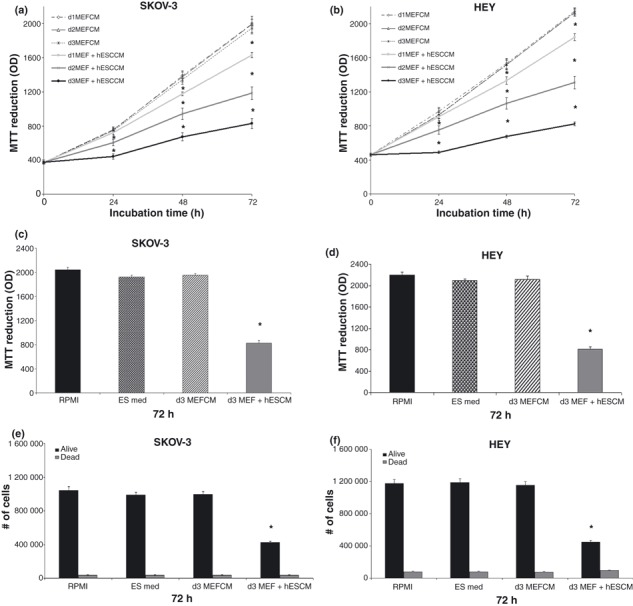
Human embryonic stem cell conditioned medium (hESC CM) inhibits SKOV‐3 and HEY cell proliferation. SKOV‐3 and HEY cells were treated with day 1, day 2, day 3 mouse primary embryonic fibroblast (MEF) CM or MEF + hESC CM, ES and RPMI media and incubated for 24–72 h. Cell number was determined by MTT assay (a–d) or by direct cell counting (e,f). Bars represent mean ± standard error of the mean of four to eight replicates. *Significantly different from control values (P < 0.05).
Effect of serial dilutions of day 3 hESC CM on ovarian cancer cell proliferation
In the next series of experiments, we ruled out the possibility that observed inhibitory effect could be due to depletion of essential nutrients from the CM, due to presence of hESC cells. First, dilutions of day 3 hESC CM with fresh medium were tested. Dilution of CM 1 : 1 with fresh medium (50%) would be sufficient to replenish nutrients in the medium and cell proliferation should not be inhibited if the effect was simply due to cell starvation. At 70%, 50% and 25% dilution, we observed a statistically significant inhibitory effect of MEF + hESC CM on SKOV‐3 and HEY cell proliferation as determined by both MTT (Fig. 2a,b) and direct cell counts (data not shown). Cell numbers decreased in a dose‐dependent manner with slight reductions at 10% and 5% CM dilution not attaining statistical significance. This dose–response relationship suggests that the inhibitory effect of hESC CM was not due to nutrient depletion.
Figure 2.
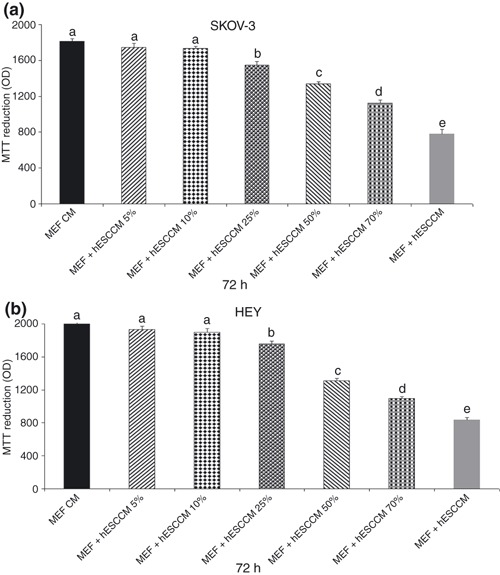
Effect of serial dilutions of day 3 mouse primary embryonic fibroblast (MEF) + human embryonic stem cell conditioned medium (hESC CM) on ovarian cancer cell proliferation. SKOV‐3 and HEY cells were treated with day 3 MEF + hESC CM at 70%, 50% and 25% dilution for 72 h. Cell number was determined by MTT assay (a,b). Bars represent mean ± standard error of the mean of eight replicates. Means with different letters are significantly different (a, b, c, d, e: P < 0.05).
Effects of day 1, day 2, and day 3 hESC CM on breast and prostate cancer cell proliferation
Breast cancer cell lines MCF‐7 and T47D were tested under the same conditions as the ovarian cell lines. Results were identical to those of the ovarian cancer lines tested (Fig. 3a,b). Forty‐eight‐ and 72‐h MEF + hESC CM were capable of reducing cell proliferation significantly at the 24–72 h culture time points. The most pronounced effect with the 48 and 72 h CM was observed within the first 24 h. 24 h MEF + hESC CM induced a significant inhibition of proliferation only after 48–72 h. PC‐3 and DU145 human prostate cancer cells responded similarly as ovarian and breast cancer cell lines (Fig. 3c,d). Medium conditioned with MEF + hESC for 48 or 72 h was able to reduce cell proliferation after 24–72 h of culture, with 72 h MEF + hES CM evoking the strongest response. 24 h MEF + hESC CM produced no effect on the cells within the first 24 h of culture, reducing cell proliferation after 48–72 h only.
Figure 3.
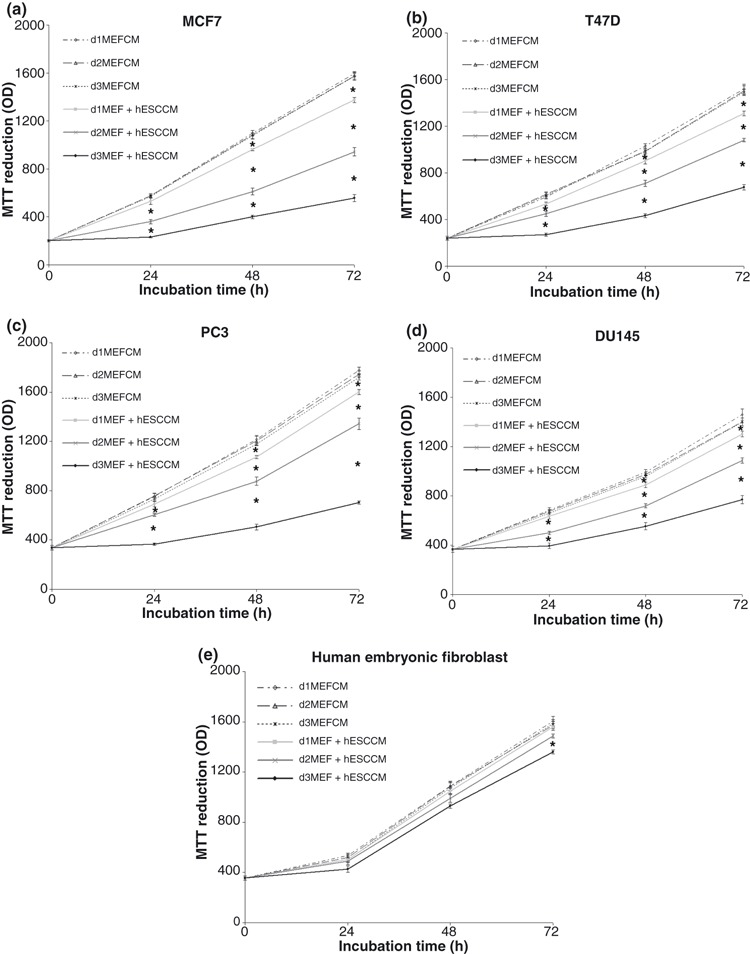
Effect of day 1, day 2, day 3 mouse primary embryonic fibroblast (MEF) + human embryonic stem cell conditioned medium (hESC CM) on cancer and human embryonic fibroblast cell proliferation. Breast cancer (MCF‐7 and T47D), prostate cancer (PC3 and DU145) and human embryonic fibroblasts were treated with day 1, day 2, day 3 MEF CM or MEF + hESC CM for 24–72 h. Cell number was determined by MTT assay (a–e). *Significantly different from control values (P < 0.05). Bars represent the mean ± standard error of the mean of eight replicates.
hESC CM did not alter population growth of fibroblasts
As a first step in determining the potential therapeutic index of the as yet unidentified growth suppressive factor or factors, we tested ability of hESC‐CM to inhibit population growth of normal human fibroblasts. The cells were tested under the same conditions described for the malignant cells. Of the various treatments, only 72 h MEF + hESC CM resulted in decreased fibroblast cell number; however, this was a slight effect and observed only after 3 days of culture (Fig. 3e). These findings provide a preliminary indication that the inhibitory effect was not generalizable to non‐malignant human cells.
Effect of MEF + hESC CM on cell death
We observed that 72 h MEF + hES CM had the strongest effect on cancer cell proliferation, within the first 24 h. On the basis of these data, we investigated whether cancer cells underwent early apoptosis. SKOV3 and HEY cells were treated with MEF + hECS CM or MEF CM for 6–24 h prior to harvesting. Cells were stained with annexin V–FITC and PI and were analysed by flow cytometry. Annexin V–FITC binds to phosphatidylserine, which is normally found on the inner plasma membrane, and becomes translocated to the external membrane surface at the onset of apoptosis. PI binds to DNA in cells where the cell membrane has been totally compromised. Cells that do not stain with either annexin V–FITC or PI are live cells (Annexin V− PI−); cells positive for annexin V–FITC and negative for PI uptake represent early apoptotic cells (Annexin 5+ PI−), while cells positive for annexin V–FITC and positive for PI uptake represent late apoptotic/necrotic cells (Annexin V+ PI+). We found no significant increase in the number of early apoptotic cells (4.24% compared to 4.05%) or in the number of late apoptotic/necrotic cells (2.4% compared to 1.91%) when SKOV‐3 cells were treated with MEF + hESC CM, compared to MEF CM control (data not shown). Similar results were obtained with HEY cells. We observed no significant increase in the number of early apoptotic cells (4% versus 3.8) or late apoptotic/necrotic cells (3.2% versus 2.9) when HEY cells were treated with MEF + hESC CM (data not shown). These data indicate that MEF + hESC CM does not mediate its growth inhibitory effects through increased apoptosis.
Effect of hESC CM on ovarian cancer cell cycle
We tested the effect of day 3 MEF + hESC CM on SKOV‐3 and HEY cell cycle progression. Cells were treated with day 3 MEF CM or MEF + hESC CM and harvested 24–72 h later and stained with PI for fluorescence‐activated cell sorting analysis. PI binds DNA and detects the percentage of cells in each phase of the cell cycle. We found an increase in SKOV‐3 cell number in G1 phase from 55% to 68%, decrease in cell number in S phase from 23% to 16% and in G2/M from 18% to 13%, when cell were treated with day 3 MEF + hESC CM (Fig. 4a,b).
Figure 4.
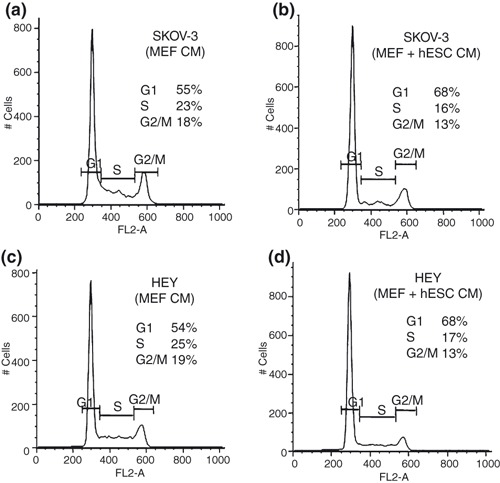
Effect of human embryonic stem cell conditioned medium (hESC CM) on ovarian cancer cell cycle. SKOV‐3 and HEY cells were treated with day 3 mouse primary embryonic fibroblast (MEF) CM or MEF + hESC CM for 24–72 h. Cells were harvested, fixed, stained with propidium iodide and analysed by flow cytometry. DNA histograms show percentage of cells in G1, G2/M and S phases (a–d). Results shown were representative of three independent experiments.
Similar results were obtained with HEY cells. Increased proportion of cells treated with day 3 MEF + hESC CM were in G1 phase as compared to MEF CM‐treated cells (68% versus 54%). Decreased proportion of cells in S (17% compared to 25%) and G2/M phases (13% compared to 19%) due to MEF + hESC CM was also observed (Fig. 4c,d).
Co‐culture of ovarian cancer cells with hESC results in decreased cell population growth
Co‐culture experiments were performed to confirm that soluble factors produced by hESCs rather than nutrient depletion affects SKOV‐3 and HEY cell proliferation. Feeder cells only and feeder + hESCs were plated in inserts placed over cancer cells, in culture. Inserts separated hESCs from cancer cells by a 0.4 μm pore high‐density membrane, thus allowing soluble factors secreted by hESCs to diffuse to the environment of the cancer cells, but preventing direct contact between stem cells and cancer cells. Co‐culture lasted for 72–120 h and MTT assays and cell counts were carried out at each time point. By 72 h, hESCs caused inhibition of SKOV‐3 cell proliferation, and the effect was observed also at 96 and 120 h (Fig. 5a,b). At 120 h, direct cell counts showed increase in dead cells compared to 72 h. There was no difference in the number of dead cells between MEF + SKOV‐3 only (control) and MEF + hESCs + SKOV‐3 treated cultures (Fig. 5b). MTT assay showed similar results with HEY cells (Fig. 5c). At 120 h, there was an increase of dead cells compared to 72 and 96 h (Fig. 5d).
Figure 5.
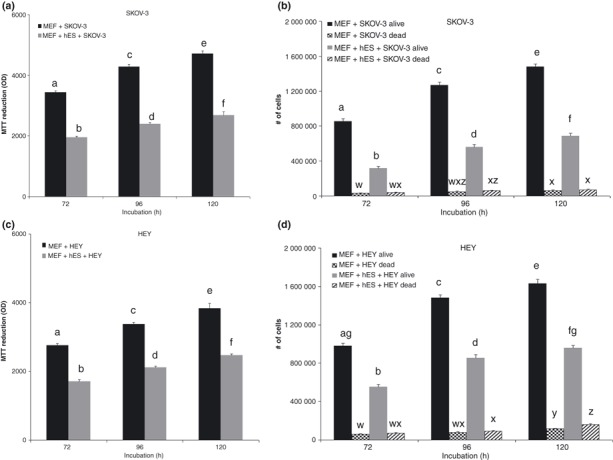
Effect of human embryonic stem cells (hESC) on ovarian cancer cells in a co‐culture model. Mouse primary embryonic fibroblasts (MEF) only and MEF + hESCs were plated in inserts placed over cancer cells in culture. Inserts separated hESCs from cancer by a 0.4 μm pore high‐density membrane. Co‐culture occurred for 72–120 h. Cell number was determined by MTT assay (a,c) or by direct cell counting (b,d). Bars represent mean ± standard error of the mean of four to six replicates. Means with different letters are significantly different. Alive cells: a, b, c, d, e, f, g: P < 0.05. Dead cells: w, x, y, z: P < 0.05.
Characterization of inhibitory factors: susceptibility to heat inactivation. To further characterize the secreted factors, heat inactivation studies were performed. When the MEF + hESC CM was heat inactivated at 65 °C for 5, 10 or 30 min, there was no difference in SKOV‐3 and HEY cell population growth, when compared to untreated MEF + hESC CM (data not shown). Heat inactivation at 90 °C for 5–30 min demonstrated loss of growth inhibition (as shown by direct cell count) as compared to untreated MEF + hESC CM, although the 90 °C treatment did not completely block the growth inhibitory effect (Fig. 6a).
Figure 6.
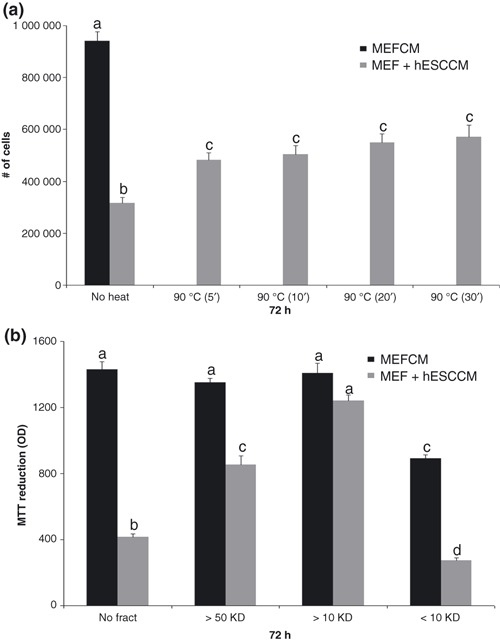
Heat inactivation and size fractionation of human embryonic stem cell conditioned medium (hESC CM). CM was heated at 90 °C for 5–30 min and then tested on SKOV‐3 and HEY cells for 72 h. Cancer cell number was determined by direct cell counting (a). CM was fractionated based on molecular size ranging from 0–10 kDa (< 10), between 10–50 kDa (> 10) and greater than 50 kDa (> 50). Fractions were tested on SKOV‐3 cells for 72 h. Cancer cell number was determined by MTT assay (b). Bars represent mean ± standard error of the mean of four to eight replicates. Means with different letters are significantly different (a, b, c, d: P < 0.05).
Characterization of the inhibitory factors: molecular size. To determine the molecular sizes of the growth inhibitory factor, components within the conditioned medium were fractionated, based on molecular size, ranging from 0–10 kDa, between 10–50 kDa and greater than 50 kDa. Fractions were tested on SKOV‐3 cells for 72 h. Unfractionated MEF + hESC CM showed the previously demonstrated significant reduction in proliferation of SKOV‐3 cells (Fig. 6b). MEF + hESC CM was then fractionated and compared to unfractionated MEF + hESC CM. Fractionated and unfractionated MEF CM was used as control in order to identify an effect on cell proliferation, resulting from the fractionation procedure itself or the unidentified soluble factor. The > 50 kDa fraction and the < 10 kDa fraction were both capable of maintaining the inhibition of cancer cell proliferation similar to the unfractionated MEF + hESC CM and statistically different from the controls (Fig. 6b). The > 10 kDa component showed no difference in cell proliferation when compared to control medium. Data were similar for HEY cancer cells (data not shown). Bovine serum albumin from the serum replacement found in medium segregates with the 50 kDa + fraction and made up the major protein content of the medium, as determined by two‐dimensional gel electrophoresis (data not shown). It is possible that the inhibitory factor is a protein complex or that the BSA (65 kDa) found in the > 50 kDa fraction acts as a carrier for the same small protein isolated in the < 10 kDa fraction, resulting in co‐segregation of some of the small protein with the BSA. We also tested the fractions by diluting them in fresh stem cell medium (data not shown). This demonstrated that the inhibition was not due to depletion of medium but due to presence of the active factor, capable of inhibiting cell proliferation.
Discussion
The potential use of hESCs as a cell source for tissue replacement is based on their capacity for self‐renewal in the undifferentiated state on the one hand and their ability to differentiate into the entire spectrum of specialized cell types of the body, on the other (22, 23). Control of the cell cycle is essential to maintain a proper balance between proliferation and differentiation in most cells. To prevent inappropriate cell proliferation, cyclin‐dependent kinase plays a role as a gatekeeper (24). The cell cycle phases have checkpoints that regulate cell cycle progression or arrest. At all the checkpoints, cells remain arrested until damage is repaired. Cancer cells arise when these checkpoints fail and damaged cells survive. Restoring the cell cycle checkpoints or blocking cancer cells in the G1 phase will prevent the growth of the cancer. In the present study, we determined that there was an increased percentage of the ovarian cancer cell lines SKOV‐ 3 and HEY in the G1 phase when the cells were treated with MEF + hESC CM. This increase was accompanied by the expected decrease of cells in the S and G2/M phases compared to cells cultured in control CM. Therefore, it appears that our factor blocked the progression of cancer cells through the cell cycle, resulting in growth inhibition.
Here, we have demonstrated that the strongest effect on inhibition of ovarian cancer cell proliferation was at 72 h when day 3 MEF + hESC CM was used. Moreover, we performed experiments using different dilutions of day 3 MEF + hESC CM. It was found that 25%, 50% and 70% dilution caused decrease in cell proliferation when compared to MEF CM. Thus, we confirmed that the effect of inhibiting cancer cell growth was not the result of depleting culture medium of crucial growth factors over the 3‐day period. In addition, to determine the effect of soluble factors produced by hESCs in mediating cancer cell proliferation, hESCs were plated in inserts placed over the cancer cells. The inserts allowed soluble factors produced by hESCs to diffuse to the cancer cells, but prevented direct contact between hESCs and the cancer cell lines. This experiment, demonstrated that the observed inhibitory effects were due to inhibitory factor (or factors) secreted by hESCs. Moreover, we observed that cell population growth inhibition of cells treated with MEF + hESC CM was reversed once the cells were grown in fresh stem cell medium. Together, these results confirmed that inhibition of cancer cell proliferation can be mediated by soluble factors produced by hESCs cells. Subsequently, this study was extended to several other cancer cell lines, in particular prostate and breast cancer cells. We found that 48 and 72 h MEF + ES CM reduced PC3 and DU145 prostate cancer cell line proliferation at each time point examined. Breast cancer cell lines MCF‐7 and T47D were tested under the same conditions as the prostate and ovarian cells. Results were similar to the population growth inhibition seen of the other cancer cells used. These findings suggest that the soluble factor (or factors) will likely be effective in inhibiting proliferation of many, if not all, epithelial tumour types.
In order to provide an initial assessment of the potential therapeutic index of the unidentified factor(s), we used human fibroblasts cultured under the same conditions described above for the other cells. Intriguing results were obtained showing only slight inhibition of human fibroblast proliferation when day 3 MEF + hESC CM was used after 72 h of culture. This finding demonstrates that the factor may not affect proliferation of non‐malignant human cells and suggests a favourable therapeutic index for the stem cell medium factor. This observation needs to be extended to other cell types, initially using immortalized non‐malignant cell lines.
Of interest in regard to the present study, a complex relationship has been shown to exist between stem cells and their microenvironment, that plays an important role in cell fate determination. Thus, it is important to study the hESC microenvironment and specially its potential, on several cell types. In this study, we showed that soluble signals produced by hESCs played an important role in cancer cell growth. Similarly, it has been shown that diffusible factors derived from skin of a developing mouse embryo were also able to inhibit growth of melanoma cells (15). In addition, there is evidence that human mesenchymal stem cells have an inhibitory effect on cancer cell proliferation (25, 26, 27, 28). For example, it has been demonstrated that human mesenchymal stem cells have intrinsic tumour‐inhibitory effects using an in vivo model of Kaposi’s sarcoma (27). Others have found that human mesenchymal stem cells are able to inhibit hepatoma cell growth in vitro and in vivo (28).
The apparent paracrine findings described by us and by previous studies may introduce a new dimension on the use of stem cells in regenerative medicine and cancer therapy. Instead of using cells themselves for therapy (29, 30, 31), it will likely be possible to identify and synthesize the specific tumour‐suppressing factors secreted by embryonic stem cells, thereby bypassing the practical and ethical issues currently associated with embryonic stem cell therapy.
In conclusion, in this report we demonstrate that hESCs produce soluble factors that are capable of blocking cancer cell proliferation by slowing progression of cancer cells through the cell cycle, without affecting cell death. Our challenge now is to identify the factor or factors involved and the mechanism of action. We have demonstrated heat resistance and a molecular weight of less than 10 kDa. Once the factor is identified, we will synthesize it for use in xenograph studies with cancer cell lines as well as in existing mouse models of ovarian, breast and prostate cancer, to test its ability to block cancer progression.
Supporting information
Appendix S1. Expression of SSEA3, TRA‐1‐60 and Oct 4 stem cells markers in CA1 and Ca2 hESCs and inhibition of SKOV‐3 and HEY cell proliferation by CA1 hESC CM.
Figure S1. Expression of stem cells markers in hESCs.
Figure S2. CA1 hESC CM inhibits SKOV‐3 and HEY cell proliferation.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
We thank Sherry Zhao and Leanne Jamieson at the SickKids‐UHN Flow Cytometry Facility for DNA cell cycle analysis. We also wish to thank Nobuko Yamanaka for helpful input. This work was supported by a Canadian Institutes of Health Research (CIHR) grant (# FRN 14058; Ottawa, ON, KIAOW9).
References
- 1. Al‐Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF (2004) Therapeutic implications of cancer stem cells. Curr. Opin. Genet. Dev. 14, 43–47. [DOI] [PubMed] [Google Scholar]
- 2. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T (2005) Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835. [DOI] [PubMed] [Google Scholar]
- 3. Bapat SA, Mali AM, Koppikar CB, Kurrey NK (2005) Stem and progenitor‐like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 65, 3025–3029. [DOI] [PubMed] [Google Scholar]
- 4. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ (2005) Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65, 10946–10951. [DOI] [PubMed] [Google Scholar]
- 5. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, Clarke MF (2007) Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 104, 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soltysova A, Altanerova V, Altaner C (2005) Cancer stem cells. Neoplasma 52, 435–440. [PubMed] [Google Scholar]
- 7. Wei G, Joseph L, Lasky III, Hong W (2006) Cancer stem cells. Pediatr. Res. 59, 59–64. [DOI] [PubMed] [Google Scholar]
- 8. Burkert J, Wright NA, Alison MR (2006) Stem cells and cancer: an intimate relationship. J. Pathol. 209, 287–297. [DOI] [PubMed] [Google Scholar]
- 9. Fuchs E, Tumbar T, Guasch G (2004) Socializing with the neighbors: stem cells and their niche. Cell 116, 769–778. [DOI] [PubMed] [Google Scholar]
- 10. Lotem J, Sachs L (2006) Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene 25, 7663–7672. [DOI] [PubMed] [Google Scholar]
- 11. Naveiras O, Daley GQ (2006) Stem cells and their niche: a matter of fate. Cell. Mol. Life Sci. 63, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He XC, Zhang J, Li L (2005) Cellular and molecular regulation of hematopoietic and intestinal stem cell behavior. Ann. N. Y. Acad. Sci. 1049, 28–38. [DOI] [PubMed] [Google Scholar]
- 13. Scadden DT (2006) The stem‐cell niche as an entity of action. Nature 441, 1075–1079. [DOI] [PubMed] [Google Scholar]
- 14. Dolberg DS, Bissell MJ (1984) Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature 309, 552–556. [DOI] [PubMed] [Google Scholar]
- 15. Gerschenson M, Graves K, Carson SD, Wells RS, Pierce GB (1986) Regulation of melanoma by the embryonic skin. Proc. Natl. Acad. Sci. USA 83, 7307–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee LMJ, Seftor EA, Bonde G, Cornell RA, Hendrix MJ (2005) The fate of human malignant melanoma cells transplanted into zebrafish embryos: Assessment of migration and cell division in the absence of tumor formation. Dev. Dyn. 233, 1560–1570. [DOI] [PubMed] [Google Scholar]
- 17. Postovit LM, Seftor EA, Seftor RE, Hendrix MJ (2006) A three‐dimensional model to study the epigenetic effects induced by the microenvironment of human embryonic stem cells. Stem. Cells 24, 501–505. [DOI] [PubMed] [Google Scholar]
- 18. Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW, Abbott DE, Seftor RE, Hendrix MJ (2008) Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl. Acad. Sci. USA 105, 4329–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buick RN, Pullano R, Trent JM (1985) Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Res. 45, 3668–3676. [PubMed] [Google Scholar]
- 20. International Stem Cell Initiative , Adewumi O, Aflatoonian B, Ahrlund‐Richter L, Amit M, Andrews PW, Beighton G, Bello PA, Benvenisty N, Berry LS, Bevan S, Blum B, Brooking J, Chen KG, Choo AB, Churchill GA, Corbel M, Damjanov I, Draper JS, Dvorak P, Emanuelsson K, Fleck RA, Ford A, Gertow K, Gertsenstein M, Gokhale PJ, Hamilton RS, Hampl A, Healy LE, Hovatta O, Hyllner J, Imreh MP, Itskovitz‐Eldor J, Jackson J, Johnson JL, Jones M, Kee K, King BL, Knowles BB, Lako M, Lebrin F, Mallon BS, Manning D, Mayshar Y, McKay RD, Michalska AE, Mikkola M, Mileikovsky M, Minger SL, Moore HD, Mummery CL, Nagy A, Nakatsuji N, O’Brien CM, Oh SK, Olsson C, Otonkoski T, Park KY, Passier R, Patel H, Patel M, Pedersen R, Pera MF, Piekarczyk MS, Pera RA, Reubinoff BE, Robins AJ, Rossant J, Rugg‐Gunn P, Schulz TC, Semb H, Sherrer ES, Siemen H, Stacey GN, Stojkovic M, Suemori H, Szatkiewicz J, Turetsky T, Tuuri T, Van Den Brink S, Vintersten K, Vuoristo S, Ward D, Weaver TA, Young LA, Zhang W (2007) Characterization of human embryonic stem cell lines by the international stem cell initiative. Nat. Biotechnol. 25, 803–816. [DOI] [PubMed] [Google Scholar]
- 21. Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. [DOI] [PubMed] [Google Scholar]
- 22. Carpenter MK, Rosler E, Rao MS (2003) Characterization and differentiation of human embryonic stem cells. Cloning Stem Cells 5, 79–88. [DOI] [PubMed] [Google Scholar]
- 23. Sato N, Sanjuan IM, Heke M, Uchida M, Naef F, Brivanlou AH (2003) Molecular signature of human embryonic stem cells and its comparison with the mouse. Dev. Biol. 260, 404–413. [DOI] [PubMed] [Google Scholar]
- 24. Morgan DO (1997) Cyclin‐dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291. [DOI] [PubMed] [Google Scholar]
- 25. Ohlsson LB, Varas L, Kjellman C, Edvardsen K, Lindvall M (2003) Mesenchymal progenitor cell‐mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp. Mol. Pathol. 75, 248–255. [DOI] [PubMed] [Google Scholar]
- 26. Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda H, Bizen A, Honmou O, Niitsu Y, Hamada H (2004) Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 11, 1155–1164. [DOI] [PubMed] [Google Scholar]
- 27. Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, Nguyen AT, Malide D, Combs CA, Hall G, Zhang J, Raffeld M, Rogers TB, Stetler‐Stevenson W, Frank JA, Reitz M, Finkel T (2006) Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J. Exp. Med. 203, 1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC, Ye L, Zhang X (2008) Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 18, 500–507. [DOI] [PubMed] [Google Scholar]
- 29. Arnhold S, Klein H, Semkova I, Addicks K, Schraermeyer U (2004) Neurally selected embryonic stem cells induce tumor formation after long‐term survival following engraftment into the subretinal space. Invest. Ophthalmol. Vis. Sci. 45, 4251–4255. [DOI] [PubMed] [Google Scholar]
- 30. Andrews PW, Matin MM, Bahrami AR, Damjanov I, Gokhale P, Draper JS (2005) Embryonic stem (ES) cells and embryonal carcinoma (EC) cells: opposite sides of the same coin. Biochem. Soc. Trans. 33, 1526–1530. [DOI] [PubMed] [Google Scholar]
- 31. Fujikawa T, Oh SH, Pi L, Hatch HM, Shupe T, Petersen BE (2005) Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell‐derived insulin‐producing cells. Am. J. Pathol. 166, 1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Expression of SSEA3, TRA‐1‐60 and Oct 4 stem cells markers in CA1 and Ca2 hESCs and inhibition of SKOV‐3 and HEY cell proliferation by CA1 hESC CM.
Figure S1. Expression of stem cells markers in hESCs.
Figure S2. CA1 hESC CM inhibits SKOV‐3 and HEY cell proliferation.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item