

Abstract
Objectives
Tumour re‐population during radiotherapy was identified as an important reason for treatment failure in head and neck cancers. The process of re‐population is suggested to be caused by various mechanisms, one of the most plausible one being accelerated division of stem‐cells (i.e. drastic shortening of cell cycle duration). However, the literature lacks quantitative data regarding the length of tumour stem‐cell cycle time during irradiation.
Materials and methods
The presented work suggests that if accelerated stem‐cell division is indeed a key mechanism behind tumour re‐population, the stem‐cell cycle time can drop below 10 h during radiotherapy. To illustrate the possible implications, the mechanism of accelerated division was implemented into a Monte Carlo model of tumour growth and response to radiotherapy. Tumour response to radiotherapy was simulated with different stem‐cell cycle times (between 2 and 10 h) after the initiation of radiotherapy.
Results
It was found that very short stem‐cell cycle times lead to tumour re‐population during treatment, which cannot be overcome by radiation‐induced cell kill. Increasing the number of radiation dose fractions per week might be effective, but only for longer cell cycle times.
Conclusion
It is of crucial importance to quantitatively assess the mechanisms responsible for tumour re‐population, given that conventional treatment regimens are not efficient in delivering lethal doses to advanced head and neck tumours.
Introduction
Management of advanced head and neck cancer represents a multi‐disciplinary challenge. Despite considerable progress in surgery, radiotherapy and chemotherapy over the last few decades, long‐term survival has not improved significantly among these patients. One of the major challenges in advanced head and neck cancer is tumour re‐population during treatment. Protracted radiotherapy in squamous cell carcinoma in head and neck cases has been reported to result in poor clinical outcome, this being attributed to accelerated immigration of tumour cells during treatment 1. Due to the proliferative potential of head and neck cancer cells during radiotherapy, it has been advised that it is preferable to delay initiation of treatment rather than to interrupt treatment once started.
Several pre‐clinical studies have indicated that responses of normal and malignant squamous cells to cytotoxic injury (irrespective of its cause) include greatly increased mitotic rate. Accelerated re‐population is noted by a sharp increase in tumour growth rate (15–20 times faster) after the initiation of radiotherapy 2. Experimental data have shown that this response of squamous epithelia is analogous to the acute response of normal tissue after injury 3. It has been suggested 4 that squamous cell carcinomas retain some of their homeostatic control mechanisms specific to their tissue of origin. Similarly, other studies support evidence that normal and malignant squamous epithelia share the same behaviour in response to injury 5, 6. Consequently, mechanisms responsible for normal tissue re‐population may be considered relevant in tumour re‐population.
Furthermore, cell proliferation studies undertaken by Dörr et al. have shown that accelerated re‐population of human squamous mucosa begins within 1 week after the start of treatment 7. Histological analysis of human mucosa from head and neck cancer patients during a course of radiotherapy have indicated considerable reduction in cell density from approximately 1000 to 500 cells/mm by the end of the first week of treatment. This drop in cell density radically slowed in subsequent weeks reaching around 400 cells/mm by the end of treatment. This observation suggests that cell loss would be overcome by accelerated proliferation initiated within the first week.
As a response to cell destruction caused by irradiation, tumour tissue may accelerate a rate of stem cell production (considering that nomenclature of tumour stem cell should be accepted) by changing cell division patterns 3. One way to achieve such changes is through loss of asymmetrical division of stem‐cells, which results in stem‐cells dividing symmetrically into two stem‐cells instead of one stem cell and one differentiated cell. A further suggested mechanism is accelerated stem cell division (Fig. 1), a process in which stem cells shorten the duration of their cell cycle, resulting in higher mitotic rate 2, 3.
Figure 1.
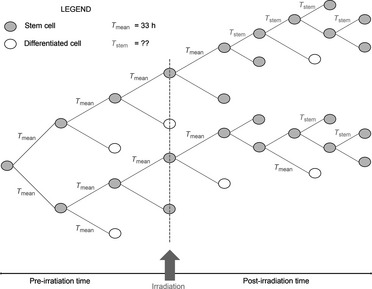
Schematic illustration of radiation‐induced accelerated stem cell division. Cells divide into two daughter cells, whereby one of the daughters retains stem cell‐like properties of the parent cell. The other daughter cell fate is to be either a differentiated cell or another stem cell.
Often being highly proliferative tumours, head and neck cancers cannot be successfully managed with conventionally fractionated radiotherapy which delivers overall dose of 70 Gy over 7 weeks, in a fractionated regimen with 2 Gy/fraction 5 days a week. As an aggressive response to an aggressive tumour, altered fractionation schedules, such as accelerated radiotherapy, have been developed and trialled with more success than conventional radiotherapy 8. Accelerated radiotherapy usually delivers smaller overall doses over shorter treatment courses (5–6 weeks), 6 days a week. Despite clinical implementation of more aggressive treatment, rate of locoregional recurrence is still high with 5‐year survival rate below 50% 9.
Knowing the impact of tumour re‐population, during therapy, on treatment outcome, it is of crucial importance to quantitatively determine duration of the stem‐cell cycle time here, during radiotherapy. Yet, there are no experimental data reporting values that reduced stem‐cell cycle times are likely to have.
The aim of this work was to use a computational modelling approach to tumour cell kinetics and behaviour during radiation therapy and to simulate the effect of various tumour stem‐cell cycle times on cell survival.
Materials and methods
Basic model of tumour growth
The present study has been based on the previously developed Monte Carlo tumour growth model 10 which showed that combination of probability functions for cell generation, duration of the four phases of the cell cycle, mean cell cycle time and cell loss factor creates a virtual tumour, with tumour growth parameters consistent with those found in the literature: volume doubling time of 50 days; cell loss factor of 85% and thymidine labelling index of 5%. Mean cell cycle time of such a virtual head and neck tumour was 33 h 11, standard deviation 13.7 h. Here, the main cell types comprising the tumour would be stem cells (unlimited division capacity and representing 2% of the pre‐treatment cell population), finitely proliferating cells (limited extent of cell division) and non‐proliferating (quiescent) cells. Cells followed exponential distribution along the G1/G0, S, G2 and M phases with the following proportions of cell cycle stages: G1/G0 – 40%, S – 30%, G2 – 23% and M – 7%.
The course of cell creation starting from a single stem cell, and cell proliferation was temporally controlled, the first stem cell being defined as entering mitosis at time zero. Each subsequent daughter cell was attributed a start and an end‐time, with each newly created cell having assigned a cell cycle time by random sampling from a truncated Gaussian distribution, with mean value of around 33 h. Start‐time was defined as sum of duration of cell cycle times since time zero, for all preceding generations. End‐time consisted of start‐time plus cell cycle time of the current cell. At each interval of a master clock, each cell was surveyed to check if its end‐time fell within the clock interval, and was processed accordingly. The results module kept track of overall number of cells, cell types, and also cell distribution along the four phases of the cycle, as well as population of quiescent cells. All these parameters were listed every 100 h of biological growth time. A growth rate factor (GRF) was defined as the ratio of cell counts between two consecutive 100‐h intervals. Kinetic parameters of the virtual tumour as well as distribution of cells along the cell cycle and quantitative tumour composition, were in accordance with basic biological properties of a head and neck carcinoma.
Model of radiotherapy and response to treatment
The virtual tumour was exposed to radiation, simulating real‐time conventional radiotherapy for squamous cell carcinomas of the head and neck, with a standard fractionation schedule: 2 Gy/fraction, 1 fraction/day, 5 days/week over 7 weeks. The surviving fraction after clinical dose of 2 Gy (SF2) for squamous cell carcinomas of the head and neck indicated by the literature is around 54% 12 and this was implemented into the model as such. Furthermore, the model has also taken into account differences in cell radiosensitivity along the various phases of the cell cycle. Therefore, supported by literature data 13, the following relationship between phase‐specific surviving fractions has been implemented into the model:
where SF2() relates to the surviving fraction of cells after radiotherapy in various phases of the cell cycle. As G1 is usually the longest and most variable phase of the cell cycle, for simplicity, SF2 for G1 was considered to be the average SF2 (i.e. 54%), linked to the other SF parameters via the relationship shown below:
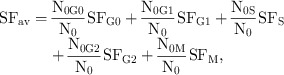 |
where N0 represents initial (pre‐irradiation) number of cells in the cycle and N0i represents initial number of cells in phase “i”. This formalism leads to non‐uniform radiation‐induced cell kill along the mitotic cycle, a fact that takes the model closer to the biological setting.
Tumour re‐population mechanisms were modelled according to experimental observations in which tumour stem cells reduce the length of their cell cycle as a response to cell destruction caused by radiation. Thus, mean cell cycle time of 33 h is shortened after initiation of radiotherapy. Possible mechanisms responsible for tumour re‐population during treatment have been analysed in a previous study 14 both as single mechanisms and as combined effects on tumour cell population. As a response to radiation‐induced cell loss, stem‐cell cycle time has been reduced in the aforementioned work to a fixed value of 3 h to simplify the modelling process; however, without initiation of a sensitivity study of other possible values and their likely effects on tumour behaviour. Consequently, the aim of the current model is (i) to simulate the effects of a wide range (from 2 to 10 h) of short stem‐cell cycle durations as a response to radiation‐induced cell killing and (ii) to assess tumour behaviour during treatment from the perspective of cell survival.
As there is experimental evidence on early onset of tumour re‐population 7, the current work simulates the mechanism of accelerated stem cell division from the first week of treatment. Accordingly, cycle time of stem cells is reduced from pre‐treatment value (33 h) to a value ranging between 2 and 10 h. Given that the literature lacks quantitative evaluation of duration of stem‐cell cycle time, the qualitative observation of ‘accelerated stem division’ has been translated into the model through a sensitivity study investigating the impact of stem‐cell cycle times reducing after initiation of treatment.
Results and discussion
Influence of stem‐cell cycle time on pre‐treatment tumour growth
As described in the Materials and methods section, the average value of stem‐cell cycle time during free growth has been considered to be 33 h. However, to study effects of various (extreme) stem‐cell cycle times on pre‐treatment tumour growth, besides the standard 33 h, one short (5 h) and one long (45 h) stem‐cell cycle times have also been considered and the results are presented in Figs 2 and 3. Growth Rate Factor, as defined in the Materials and methods, was determined for all three scenarios and plotted against tumour growth time (Fig. 2).
Figure 2.
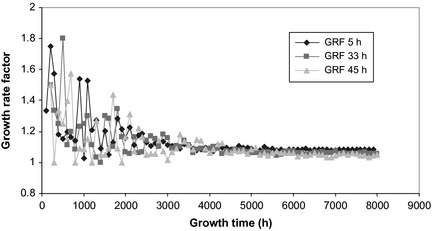
Tumour growth rate factor as a function of stem‐cell cycle time during free growth. Over time, growth of the tumour converges towards a stable growth rate factor, irrespective of cell cycle time.
Figure 3.
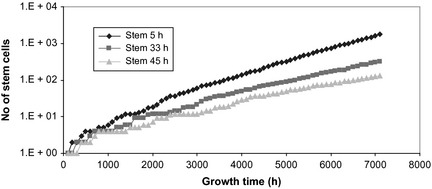
Tumour growth curves as a function of stem‐cell cycle time. Slope of the tumour growth curve is determined by initial variations among the different cell populations having varied cell cycle times.
Despite initial statistical fluctuation among GRF (due to low number of cells at the beginning of tumour development), in time, tumour growth converged towards a more stable growth rate factor, independent of cell cycle time (average GRF = 1.052 for the tumour with initial stem‐cell cycle time of 45 h and GRF = 1.084 for the tumour with initial stem‐cell cycle time of 5 h). Slope of the tumour growth curve (Fig. 3) is dictated by initial variations among the three cell populations having different cell cycle times. Clearly, shorter the cycle duration, steeper the slope of the tumour growth curve, and faster the development into a clinically detectable tumour.
Influence of stem‐cell cycle time on tumour re‐growth during conventional radiotherapy
Onset time for re‐population during radiotherapy greatly influences overall cell survival. Knowledge of early re‐population can determine the correct treatment choice to overcome uncontrollable tumour growth.
Looking closely into the stem cell population (Fig. 4), it is noted that cell killing increases with later onset re‐population. As each fraction of radiotherapy contributes to cell destruction, thus decreasing the stem cell population, the later re‐population activated, the lower is the number of affected stem cells. To consider the worst‐case scenario, and also to be in line with experimental data supporting early onset of accelerated stem division, this work has modelled re‐population being triggered by cell loss from the first radiotherapy fraction.
Figure 4.
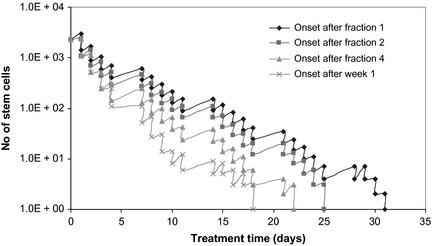
Onset time for re‐population and effect on stem population survival. The later the onset of re‐population, the higher the cell killing, as less stem cells are affected.
Tumour re‐growth is strongly dependent on duration of stem‐cell cycle time. As illustrated in Figs 5 and 6, very short cell cycle times can lead to uncontrollable tumour growth caused by conventional radiotherapy, even if the initial stem cell population affected by accelerated cell division is as low as 2%.
Figure 5.
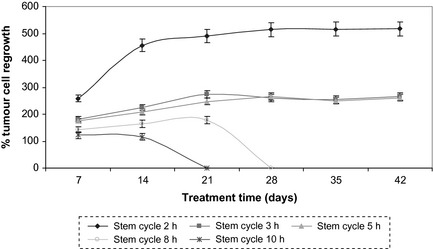
Percentage tumour re‐growth during the course of conventional radiotherapy, with different lengths of stem‐cell cycle times after initiation of treatment. In the first 2 weeks of therapy, radiation induced cell killing is overcome by re‐growth of tumour cell population in all situations.
Figure 6.
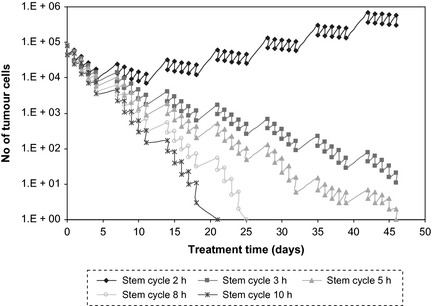
Tumour cell survival curves after conventional radiotherapy with different lengths of stem‐cell cycle times after initiation of treatment. For very short cell cycle times re‐population via tumour stem cells overcomes radiation‐induced cell kill.
Percentage tumour cell re‐growth during the course of radiotherapy is shown in Fig. 5. Each data point represents the percentage of malignant cell re‐growth over a weekend (representing the 6 weekends during the 7 weeks treatment course), when no treatment was delivered, allowing surviving tumour cells to proliferate and re‐populate the tumour.
If onset of re‐population is triggered during the first week of treatment, as suggested by experimental data, Fig. 5 shows that in the first 2 weeks of therapy, radiation‐induced cell killing is overcome by re‐emergence of the tumour population in all cases (illustrated by the increasing slope). After 3 weeks treatment, radiation starts to take effect and cell killing leads to successful tumour control in cases where stem‐cell cycle duration is considered to be greater than 8 h.
Results of simulating the course of conventional radiotherapy for various lengths of tumour stem‐cell cycle during irradiation are presented in Fig. 6. Graphs show that for very short cell cycle times, re‐population via tumour stem cells overcomes cell death due to radiation, and radiation doses administered with conventional radiotherapy cannot compensate for tumour re‐growth. Thus, altered fractionation regimens are recommended, such as accelerated or accelerated‐hyperfractionated radiotherapy, whereby multiple doses are delivered to the tumour daily, and treatment break over the weekend is reduced to 1 day, as accelerated radiotherapy assumes 6 days consecutive treatment per week instead of the conventional 5 days (discussed in the section below).
Tumour re‐growth factor (TRF) was defined as the ratio between percentages of cell re‐growth in two consecutive weekends, for the same tumour population, and the data are presented in Table 1:
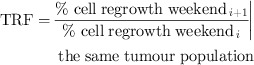 |
Table 1.
Tumour re‐growth factor (TRF) as a function of tumour stem‐cell cycle time
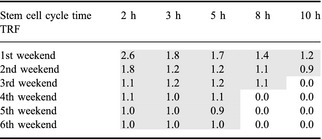
As a general trend, it can be noted that re‐growth factor is highest shortly after onset of re‐population (first weekend) and decreases slowly during subsequent weekends. Yet, for very short stem‐cell cycle times (<6 h), re‐growth is recorded even over the last weekend of treatment, indicating that the tumour population is thriving and could not be compensated for by radiation‐induced cell killing. This fact is substantiated by the Growth Rate Factor during radiotherapy (GRFRT), defined as the ratio of cell counts between two consecutive 24‐h intervals, that is, cell population growth between two consecutive dose fractions (compared to pre‐treatment GRF, which has considered ratio of cell counts between two subsequent 100 h during free growth).
As opposed to the pre‐treatment scenario (Fig. 2) when tumour growth converges towards a more stable growth rate factor independent of cell cycle time, during radiotherapy, stem‐cell cycle time has had a stronger impact on overall tumour behaviour, even when disregarding obvious growth bursts over weekends (Fig. 7). Therefore, average GRFRT varies from 1.08 for stem cycle of 10 h, to 1.64 for stem cycle of 2 h, without weekend growth, and from 1.10 to 2.23 with weekend growth, respectively. It is suggested that this difference is due to interplay between (i) accelerated stem cell division, (ii) random effect of radiation on tumour cell killing, (iii) different cell radiosensitivities through the cell cycle and (iv) uncompensated cell population growth between fractions for low cell cycle times as opposed to pre‐treatment growth, which is dictated by balanced cell proliferation, cell division and natural cell loss (that is, steadier growth rate factor).
Figure 7.
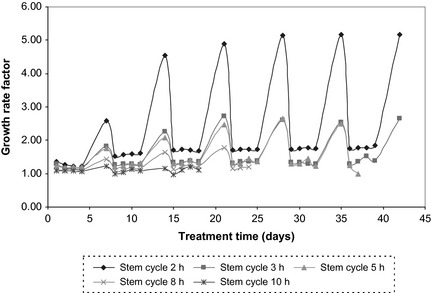
Tumour growth rate factor as a function of stem‐cell cycle time during radiotherapy.
Effect of accelerated radiotherapy on tumour re‐growth, involving accelerated stem cell division
To illustrate the potent effect of tumour re‐growth during treatment, due to short stem‐cell cycle times, the current work has simulated an accelerated treatment schedule, which delivers the same overall dose (70 Gy) as conventional radiotherapy, but over 6 weeks treatment with 6 days of 2 Gy/fraction a week. In this scenario, the advantage of accelerated radiotherapy over the conventional schedule consists of shorter re‐population times over weekends (48 h compared to 72 h), thus better control of the stem cell population (Fig. 8).
Figure 8.
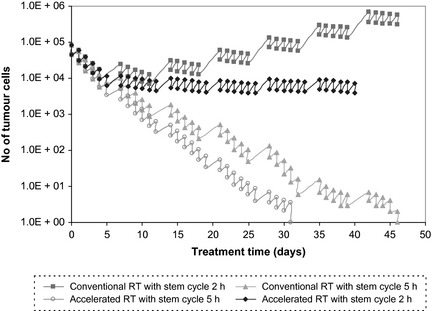
Conventional versus accelerated radiotherapy for tumours with short cell cycle times. For very short stem cycle times, radiation‐induced cell killing is overcome by the mechanism of accelerated stem cell division in both conventional and accelerated radiotherapy.
Despite the accelerated treatment regimen, for very short stem‐cell cycle times (<5 h), treatment does not qualify as more successful than conventional radiotherapy. Radiation‐induced cell killing is overcome with ease by the mechanism of accelerated stem cell division and while there is noticeable change in slope of the survival curve for accelerated treatment regimen compared to standard, tumour population cannot be controlled (Fig. 8).
However, for short but clinically more plausible stem‐cell cycle times (≥5 h), the accelerated regimen allows reduction in the number of fractions, thus decrease in overall dose for the same bio‐effect with 14 Gy (7 fractions) compared to conventional radiotherapy. This quantitative result cannot be equated with the same clinical dose, as the model has its limitations by not considering tumour microenvironment and/or interplay of other factors such tumour stage, size and adjacent normal tissue confines (to name but a few). Furthermore in clinics, accelerated radiotherapy is often planned with a treatment gap after about 30 Gy to allow for normal tissue repair and re‐population. This treatment gap can have devastating effect on tumour control, as tumour cells re‐grow freely during this resting period. Thus, treatment regimens must be developed based on radiobiological properties of individual head and neck cancers as tumour re‐population during therapy is an obvious contributor towards recurrence.
To focus merely on the aspect of accelerated tumour re‐population as a response to radiotherapy, the hypoxia factor has not been modelled in the present study. Nevertheless, hypoxia is another key parameter in management of advanced head and neck tumours, as lack of oxygen confers resistance to radiotherapy [see 15 for the effect of hypoxia on tumour control in virtual head and neck cancers]. It is known that head and neck cancers have high hypoxic content and that hypoxic cells are up to three times more radio‐resistant than cells in oxic conditions. Radioresistance, added to the above observation in which accelerated stem cell division (as a sole mechanism), could potentially lead to treatment failure, underpins the necessity to quantitatively evaluate mechanisms behind accelerated re‐population during treatment.
Furthermore, accelerated stem cell division is probably not the only mechanism responsible for accelerated re‐population during radiotherapy. Cell recruitment from the available pool of quiescent cells and also loss of asymmetrical division of stem cells leading to symmetrically dividing stem cells (that is, two daughter stems) are mechanisms which can contribute to re‐population during radiotherapy 3, 14. The graph in Fig. 9 illustrates consequences of tumour cell re‐population when interaction of all above mechanisms is modelled [see also 14]. Time ‘0’ represents the start of radiotherapy and the tumour regression curve is the result of tumour response to radiotherapy without simulating any re‐population mechanisms.
Figure 9.
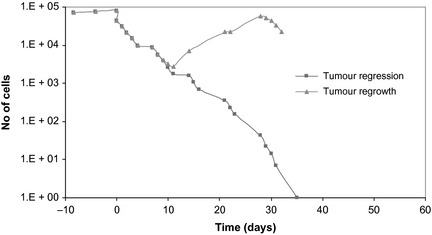
Tumour behaviour with and without radiotherapy. Theoretical modelling of tumour re‐population during radiotherapy, with accelerated stem cell division and loss of asymmetrical division of stem cells [see also 14].
While the mechanism of accelerated stem cell division is influential enough to trigger uncontrollable tumour re‐population, this effect can only be aggravated when considering additional factors, such as cell recruitment, loss of asymmetry, loss of stem cell division, tumour hypoxia and radioresistance.
Consequences of the above presented premise are translated into clinical implications of accelerated re‐population during radiotherapy.
Optimal management of head and neck cancer patients would need appropriate patient selection, based on tumour proliferation kinetics via predictive assays or PET imaging, which specifically target proliferation. While personalized medicine is becoming a widespread concept in oncology, head and neck cancer patients should particularly benefit from individualized treatment plans due to high variability of tumour‐related parameters (such as cell proliferative ability), among patients. Based on cell proliferative ability of each particular head and neck tumour, individualized treatment can be designed to optimize treatment outcome. These personalized treatments could be delivered as:
Accelerated radiotherapy, shortening overall treatment time without reducing total radiation dose. This treatment regimen is probably most commonly used to overcome accelerated re‐population. However, one drawback is the treatment gap, which is usually scheduled after delivery of around 34 Gy, to allow for normal tissue repair. This gap in treatment is a perfect opportunity for surviving tumour cells to start re‐populating the tumour. To avoid treatment interruption, radioprotectors such as amifostine should be used.
High dose rate brachytherapy, for localized tumours, either as a boost or as monotherapy.
Chemo‐radiotherapy, either concurrently or chemotherapy as a neoadjuvant treatment. However, if chemotherapy is delivered as induction, no gaps between neoadjuvant and curative treatment should be allowed, due to re‐population triggered by drug‐caused cell loss. Furthermore, for cisplatin‐based chemotherapy, cisplatin in low doses administered on a daily basis in combination with radiotherapy is more effective than given in weekly cycles, in large doses. Besides better tumour control, rate of normal tissue complications can be also reduced.
Proton therapy, where available. Proton therapy is well suited to tumour sites, such as head and neck, which need a dose higher than 70 Gy to be delivered for tumour control. Also, with the well‐focalized Bragg peak of the proton dose distribution curve, organs at risk which surround the diseased area are better spared.
Targeted molecular therapies, against epidermal growth factor receptor (EGFR) could play a key role in management of advanced head and neck tumours, as the vast majority of these overexpress EGFR, which is associated with poor prognosis.
Conclusion
Tumour cell re‐population during radiotherapy is suggested to be caused by various mechanisms, one of the most plausible being accelerated division of stem cells, in which they are able to drastically shorten the duration of their cell cycle. The presented work suggests that if accelerated stem cell division is indeed a key mechanism behind tumour re‐population, the cell cycle time can drop below 10 h. Considering this hypothesis, it is crucial to determine value range of cell cycle time to enable the design of optimal treatment, as short cycles (of a few hours) lead to re‐population, which cannot be overcome by cell killing, thus resulting in treatment failure.
References
- 1. Withers HR, Taylor JMG, Maciejewski B (1988) The hazard of accelerated tumor clonogen re‐population during radiotherapy. Acta Oncol. 27, 131–146. [DOI] [PubMed] [Google Scholar]
- 2. Withers HR (1993) Treatment‐induced accelerated human tumor growth. Semin. Radiat. Oncol. 3, 135–143. [DOI] [PubMed] [Google Scholar]
- 3. Dörr W (1997) Three A's of re‐population during fractionated irradiation of squamous epithelia: asymmetry loss, acceleration of stem‐cell divisions and abortive divisions. Int. J. Radiat. Biol. 72, 635–643. [DOI] [PubMed] [Google Scholar]
- 4. Trott K, Kummermehr J (1991) Accelerated re‐population in tumours and normal tissues. Radiother. Oncol. 22, 159–160. [DOI] [PubMed] [Google Scholar]
- 5. Denham J, Walker QJ, Lamb DS, Hamilton CS, O'Brien PC, Spry NA et al (1996) Mucosal regeneration during radiotherapy. Trans Tasman Radiation Oncology Group (TROG). Radiother. Oncol. 41, 109–118. [DOI] [PubMed] [Google Scholar]
- 6. Trott K (1999) The mechanisms of acceleration of re‐population in squamous epithelia during daily irradiation. Acta Oncol. 38, 153–157. [DOI] [PubMed] [Google Scholar]
- 7. Dörr W, Hamilton S, Boyd T, Reed B, Denham JW (2002) Radiation‐induced changes in cellularity and proliferation in human oral mucosa. Int. J. Radiat. Oncol. Biol. Phys. 52, 911–917. [DOI] [PubMed] [Google Scholar]
- 8. Bourhis J, Overgaard J, Audry H, Ang KK, Saunders M, Bernier J et al (2006) Hyperfractionated or accelerated radiotherapy in head and neck cancer: a meta‐analysis. Lancet 368, 843–854. [DOI] [PubMed] [Google Scholar]
- 9. Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300. [DOI] [PubMed] [Google Scholar]
- 10. Marcu L, van Doorn T, Olver I, Zavgorodni S (2002) Growth of a virtual tumour using probabilistic methods of cell generation. Australas. Phys. Eng. Sci. Med. 25, 155–161. [DOI] [PubMed] [Google Scholar]
- 11. Steel GG (1997) The growth rate of tumours In: Steel GG, ed. Basic Clinical Radiobiology, 2nd edn pp. 8–13. New York: Oxford University Press. [Google Scholar]
- 12. Tannock I, Hill R (1998) The Basic Science of Oncology, 3rd edn New York: McGraw‐Hill. [Google Scholar]
- 13. Hall E (2000) Radiobiology for the Radiologist, 5th edn Philadelphia: Lippincott Williams & Wilkins. [Google Scholar]
- 14. Marcu L, van Doorn T, Olver I (2004) Modelling of post irradiation accelerated re‐population in squamous cell carcinomas. Phys. Med. Biol. 49, 3676–3779. [DOI] [PubMed] [Google Scholar]
- 15. Harriss‐Phillips WM, Bezak E, Yeoh EK (2011) Monte Carlo radiotherapy simulations of accelerated re‐population and reoxygenation for hypoxic head and neck cancer. Br. J. Radiol. 84, 903–918. [DOI] [PMC free article] [PubMed] [Google Scholar]