

Abstract
Abstract. Objective: Benzoylaminoalkanohydroxamic acids, including 5‐(4‐dimethylaminobenzoyl)aminovaleric acid hydroxamide (4‐Me2N‐BAVAH), are structural analogues of Trichostatin A, a naturally occurring histone deacetylase inhibitor (HDACi). 4‐Me2N‐BAVAH has been shown to induce histone hyperacetylation and to inhibit proliferation in Friend erythroleukaemia cells in vitro. However, the molecular mechanisms have remained unidentified. Materials and Methods: In this study, we evaluated the effects of 4‐Me2N‐BAVAH on proliferation in non‐malignant cells, namely epidermal growth factor‐stimulated primary rat hepatocytes. Results and Conclusion: We have found that 4‐Me2N‐BAVAH inhibits HDAC activity at non‐cytotoxic concentrations and prevents cells from responding to the mitogenic stimuli of epidermal growth factor. This results in an early G1 cell cycle arrest that is independent of p21 activity, but instead can be attributed to inhibition of cyclin D1 transcription through a mechanism involving inhibition of nuclear factor‐kappaB activation. In addition, 4‐Me2N‐BAVAH delays the onset of spontaneous apoptosis in primary rat hepatocyte cultures as evidenced by down‐regulation of the pro‐apoptotic proteins Bid and Bax, and inhibition of caspase‐3 activation.
INTRODUCTION
Temporal changes in chromatin structure play an important role in eukaryotic gene transcription by affecting the affinity of histone proteins for DNA and other chromatin‐associated proteins (Workman & Kingston 1998). In this context, acetylation of N‐terminal lysine residues in nucleosome‐associated histones has been well studied. It is a reversible process catalysed by histone acetyltransferases and histone deacetylases (HDACs). A balanced histone acetylation status is essential for correct progress of cell proliferation, apoptosis and differentiation (Workman & Kingston 1998; Kouzarides 1999). Indeed, improper HDAC recruitment or activity often results in uncontrolled cell proliferation and tumour development (Marks et al. 2001). This explains the current popularity for development of HDAC inhibitors (HDACi) as targeted anticancer drugs. In vitro experiments have revealed that these compounds effectively induce cell cycle arrest and differentiation in a wide variety of neoplastic cell lines (Marks et al. 2001; Yoshida et al. 2001; Vanhaecke et al. 2004; Marks & Jiang 2005; Papeleu et al. 2005) some of which are presently being evaluated in clinical trials (Piekarz & Bates 2004; Marks & Jiang 2005). However, in vitro and in vivo efficacy of HDACi, in particular hydroxamic acid‐based compounds like Trichostatin A (TSA), are limited due to rapid metabolic inactivation of the functional group (Elaut et al. 2002; Elaut et al. 2004; Sanderson et al. 2004). 5‐(4‐Dimethylaminobenzoyl) aminovaleric acid hydroxamide (4‐Me2N‐BAVAH), a structural analogue of TSA, appears to be more stable in rat hepatocyte suspensions (Table 1; Elaut et al. 2004) and has previously been shown to inhibit HDAC activity and proliferation in Friend erythroleukaemia cells (Jung et al. 1999). The molecular mechanisms, however, remain unidentified.
Table 1.
In vitro HDAC inhibition potency, metabolic stability and biological effects of TSA and 4‐Me2N‐BAVAH
| TSA | 4‐Me2N‐BAVAH | Reference | |
|---|---|---|---|
| HDAC inhibition potency (IC50) | 11 ± 2 nm | 1920 ± 405 nm | Elaut et al. 2007 |
| Metabolic stability in hepatocyte suspensions | 30 min | 3 h | Elaut et al. 2004 |
| Effects on differentiation in cultured hepatocytes | ↑ albumin secretion | ↑↑ albumin secretion | Papeleu et al. 2003 and this paper |
| ↑ CYP1A, 2B1, 3A2 | ↑↑ CYP1A, 2B1, 3A2 | Henkens et al. 2007 | |
| ↑ Cx32, Cx43 | ↑↑ Cx32, Cx43 | Vinken et al. 2006 | |
| ↓ Cx26 | ↓↓ Cx26 | Vinken et al. 2006 |
4‐Me2N‐BAVAH, 5‐(4‐dimethylaminobenzoyl)aminovaleric acid hydroxamide; HDAC, histone deacetylase; TSA, Trichostatin A.
The majority of studies dealing with the elucidation of molecular mechanisms associated with HDACi‐induced inhibition of cell proliferation have been performed in tumour‐derived cell lines. In general, cancer cell proliferation is suppressed due to the stimulating effect of HDACi on expression of growth‐inhibitory genes, including p21 (Marks et al. 2001; Yoshida et al. 2001; Vanhaecke et al. 2004). Cancer cells, however, exhibit severely disturbed regulation of the cell cycle due to mutations in tumour‐suppressor genes or overexpression of oncogenes. Primary cultured hepatocytes, stimulated with appropriate mitogens, including epidermal growth factor (EGF), have proven to be a valuable alternative for studying cell cycle‐related signal transduction (Papeleu et al. 2004). Although cell division rarely occurs in the adult healthy liver, hepatocytes quickly respond to injury by initiating an adaptive proliferative process (Koniaris et al. 2003). One of the best‐characterized in vivo models in this respect is 70% partial hepatectomy (Koniaris et al. 2003). An analogous proliferative response occurs during the two‐step collagenase perfusion used to isolate hepatocytes from the liver (Loyer et al. 1996). Responsiveness to growth factors is ensured in a pre‐replicative priming phase and is essential for cells to pass the G1 restriction point and subsequent progression through the cell cycle (Koniaris et al. 2003; Mitchell & Gilgenkrantz 2003). EGF is a known in vivo and in vitro hepatocyte mitogen and its role in the control of hepatocyte proliferation has been studied extensively. Upon binding to its receptor, EGF activates a number of signalling pathways that ultimately converge on expression of cyclin D1, a pivotal player in G1 phase progression (Talarmin et al. 1999; Rescan et al. 2001; Coutant et al. 2002). Induction of this d‐type cyclin appears to be a rate‐limiting step in hepatocyte proliferation (Loyer et al. 1996; Albrecht & Hansen 1999; Talarmin et al. 1999; Rescan et al. 2001; Coutant et al. 2002; Nelsen et al. 2003). In the present study, we investigated the effects of the TSA‐like hydroxamic acid‐containing HDACi 4‐Me2N‐BAVAH on EGF‐induced proliferation in primary rat hepatocytes.
MATERIALS AND METHODS
Reagents
Minimal essential medium, medium 199 and crude collagenase type I came from Sigma‐Aldrich (Bornem, Belgium). [methyl‐3H]‐thymidine (25 Ci/mmol) and [α‐32P]‐dCTP (3000 Ci/mmol) came from Amersham Pharmacia Biotech (Buckinghamshire, UK). Recombinant human EGF was from Promega (Leiden, The Netherlands). 4‐Me2N‐BAVAH (purity ≥ 96%) was synthesized as previously described (Jung et al. 1999) and stock solutions of 100 mm were prepared in ethanol. Final ethanol concentrations in the media did not exceed 0.05% (v/v) (used as solvent control). All other reagents were readily commercially available and of molecular biology grade.
Cell isolation and culture
Procedures for housing rats and isolation and culture of primary rat hepatocytes were approved by the local ethical committee for animal experiments of the Vrije Universiteit Brussel, Brussels, Belgium. Hepatocytes were isolated from male outbred Sprague‐Dawley rats (200–300 g, Charles River Laboratories, Brussels, Belgium) (Papeleu et al. 2006). They were kept under controlled environmental conditions (a 12‐h light : dark cycle) and were fed a standard diet (Animalabo A04, water ad libitum). Cells (viability > 90%; assessed by trypan blue exclusion) were plated at a density of 0.4 × 105 cells/cm2 in a mixture of 75% minimal essential medium and 25% medium 199 supplemented with bovine serum albumin (1 mg/mL), bovine insulin (5 µg/mL), 10% foetal bovine serum and per millilitre: 7.3 IU benzyl penicillin, 50 µg streptomycin sulfate, 50 µg kanamycin monosulfate and 10 µg sodium ampicillin (Papeleu et al. 2004). Four hours after plating, fresh serum‐free medium containing 0.5 µg/mL hydrocortisone sodium hemisuccinate and 50 ng/mL human recombinant EGF was added to the cells. Exposure to the HDACi 4‐Me2N‐BAVAH began at time of plating unless indicated otherwise. Media were renewed daily.
Fluorescence‐activated cell sorting
Ploidy of hepatocyte nuclei was measured by fluorescence‐activated cell sorting (FACS) analysis. Cells were washed twice with ice‐cold phosphate‐buffered saline and subsequently were incubated in a hypotonic fluorochrome solution (50 µg/mL propidium iodide in 0.1% sodium citrate and 0.1% Triton X‐100) for 1 h at room temperature in the dark. Propidium iodide fluorescence of individual cells was analysed on a FACStarPlus (Becton Dickinson Biosciences, San José, CA, USA).
Western blotting
At the indicated time points, cultured hepatocytes were harvested in ice‐cold phosphate‐buffered saline. For total cell protein extraction, cell pellets were prepared as described (Loyer et al. 1996) and histones were prepared according to Cousens et al. (1979). Total protein (25 or 50 µg/lane) or histone (20 µg/lane) were resolved on sodium dodecyl sulfate‐polyacrylamide gel electrophoresis, blotted on to nitrocellulose membranes (Amersham Pharmacia Biotech) and visualized with the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech) as recommended by the manufacturer.
Antibodies used were rabbit polyclonal antiphospho‐p44/42 MAP kinase (Thr202/Tyr204) (Cell Signalling Technologies, Beverly, MA, USA), anticdk1 and anti‐IκBα (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anticaspase‐3 (Calbiochem‐Novabiochem, San Diego, CA, USA) and anti‐acetylated histone H4 (CamproScientific, Amsterdam, The Netherlands); goat polyclonal antip21 and anti‐Bax were from Santa Cruz Biotechnology and anti‐Bid was from R & D Systems, Abingdon, Oxfordshire, UK. Mouse monoclonal antibodies used were anticyclin D1 (Neomarkers, Fremont, CA, USA), anti‐GAPDH (Abcam, Cambridge, UK) and antipan‐ERK (Transduction Laboratories, Lexington, KY, USA).
DNA synthesis
Cells were cultured in triplicate and were incubated with [methyl‐3H]‐thymidine (25 Ci/mmol; 2 µCi/mL) for 24 h prior harvesting. Incorporation of the radiolabel was measured by liquid scintillation counting (Wallac 1410; counting efficiency: 67.5%) after overnight precipitation of the DNA in 15% trichloroacetic acid (Loyer et al. 1996).
Northern blotting
Total RNA was extracted with the simian virus total RNA isolation system (Promega, Leiden, The Netherlands). Northern blotting and visualization of RNA by autoradiography was performed as described previously (Loyer et al. 1996). cDNA probes used were the purified 1.3‐kb EcoRI mouse cyclin D1 insert from the pcBZ plasmid (Dr Sherr, Memphis, TN, USA) and the purified 0.75‐kb EcoRI–HindIII rat waf1 insert from the pcRW0.8–pBluescript SK plasmid (Dr Ilyin, Rennes, France).
Luciferase reporter gene assay for nuclear factor‐kappaB activity
Nuclear factor‐kappaB (NF‐κB) activation was analysed by measuring the expression of an NF‐κB‐dependent luciferase reporter gene that was transfected by adenoviral gene transfer as described previously (El Bakkouri et al. 2005; Wullaert et al. 2005). Primary hepatocytes were seeded in triplicate in 24‐well plates at a density of 0.4 × 105 cells/cm2 and were infected the day after with a replication‐deficient adenovirus at a total multiplicity of infection of 75. The adenovirus mixture comprised 50% adenovirus expressing an NF‐κB‐dependent luciferase reporter gene (AdNF‐κBluc) (gift from Dr B. McGray, University of Iowa College of Medicine, Iowa City, IA, USA; Sanlioglu et al. 2001) and 50% adenovirus expressing a β‐galactosidase gene under a constitutive active cytomegalovirus promoter (AdLacZ). To test the effect of the inhibitor of κBα (IκBα) super‐repressor (IκBαsr), the adenovirus mixture was comprised of 25% AdNF‐κBLuc, 25% AdLacZ and either 50% of control adenovirus without transgene (AdRR5) or 50% virus expressing the IκBαsr transgene (AdIκBαsr) (gift from Dr R. Hay, University of St. Andrews, St. Andrews, Scotland, UK; Grempler et al. 2004). One day after infection, cells were either untreated or were stimulated with EGF for 6 h. Recombinant adenoviruses were generated in HEK293 cells as previously described (Wullaert et al. 2005). NF‐κB promoter activity was assessed by measuring luciferase (Luc) activity in cell extracts (De Valck et al. 1996). β‐galactosidase (Gal) activity was measured using the Galactostar reporter gene assay system (Applied Biosystems, Foster City, CA, USA). Luc values were normalized for Gal values to correct for differences in infection efficiency and were plotted as fold NF‐κB activation (relative to the basal level of Luc in the absence of EGF, after normalization with Gal).
Cell viability
Lactate dehydrogenase (LDH) leakage was measured using the Merckotest (VWR International, Leuven, Belgium) and was calculated (in percentage) by the ratio: [100 × LDH activity in supernatant]/[LDH activity in (supernatant + cells)].
Albumin secretion
Medium samples were collected at indicated time points and were analysed for their albumin content by ELISA (Dunn et al. 1991).
Identification and separation of 4‐Me2N‐BAVAH metabolites
Hepatocyte monolayer cultures were incubated with 50 µm 4‐Me2N‐BAVAH for 20 h. Medium samples were collected every 20 min during the first hour and every hour during the next 6 h of culture; final samples were taken 8 and 20 h after administration of the HDACi. Separation and identification of 4‐Me2N‐BAVAH and its acid and amide metabolites were performed as previously described (Elaut et al. 2004).
Statistical analysis
Results were expressed as the mean ± SD. Statistical analyses were performed using a Student's t‐test with the significance level being set at 0.05.
RESULTS
4‐Me2N‐BAVAH inhibits EGF‐induced cell proliferation in primary hepatocytes
To determine whether the selected doses of HDACi 4‐Me2N‐BAVAH effectively block HDAC activity, primary hepatocytes were cultured for 48 h either in the absence (0 µm) or presence of 10, 25 and 50 µm 4‐Me2N‐BAVAH. Figure 1 shows that 4‐Me2N‐BAVAH enhanced histone H4 acetylation in a concentration‐dependent way. Ten and twenty‐five micromolars of the HDACi induced a slight to moderate increase in histone H4 acetylation (2.35 ± 0.05‐ and 3.56 ± 0.23‐fold induction, respectively), whereas a 7‐fold (6.91 ± 0.63) increase in acetylation levels was found upon exposure to 50 µm 4‐Me2N‐BAVAH.
Figure 1.
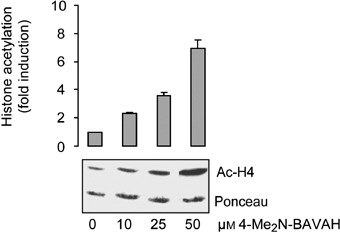
Effect of 4‐Me2N‐BAVAH on histone H4 acetylation. Primary rat hepatocytes were cultured in EGF‐containing medium (50 ng/mL, added 4 h after plating) either in the absence (0 µm) or presence of 10, 25 or 50 µm 4‐Me2N‐BAVAH (added from time plating during 48 h). After 48 h of culture, nuclear histones were extracted and histone H4 acetylation levels were evaluated by Western blotting, using a polyclonal antibody, specifically directed against hyperacetylated forms of histone H4. A representative blot of three independent experiments is shown. Ponceau S staining is shown as a control for sample loading and transfer efficiency. Quantification of Western blots by densitometry is from three independent experiments (means ± SD are shown), control values were arbitrarily set at 1.
Next, we evaluated whether 4‐Me2N‐BAVAH could effectively inhibit EGF‐induced hepatocyte proliferation. Thus, the same concentrations of HDACi were added to the culture media from time of plating, for 3 consecutive days. DNA synthesis was measured by means of 3H‐thymidine incorporation every 24 h (Fig. 2a). In addition, induction of cyclin D1, a rate‐limiting step in EGF‐induced hepatocyte proliferation, was evaluated by Western blot analysis (Fig. 2b). Exposure of EGF‐stimulated hepatocytes to increasing concentrations of 4‐Me2N‐BAVAH revealed a clear dose‐response relationship. Inhibition of DNA synthesis and cyclin D1 protein accumulation in the presence of 50 µm 4‐Me2N‐BAVAH correlated well with the observation that a significantly higher proportion of cells had a G0/G1 DNA content (P < 0.001, Student's t‐test) and a significantly lower number remained in G2/M (P < 0.05, Student's t‐test) (Fig. 2c). LDH leakage into the culture medium was not increased in hepatocytes that were exposed to 50 µm 4‐Me2N‐BAVAH, indicating that cytotoxicity did not contribute to inhibition of cell proliferation (Fig. 2d). Albumin secretion, reflecting overall cell functionality, was even significantly increased upon continuous exposure of the cells to 4‐Me2N‐BAVAH (Fig. 2e).
Figure 2.
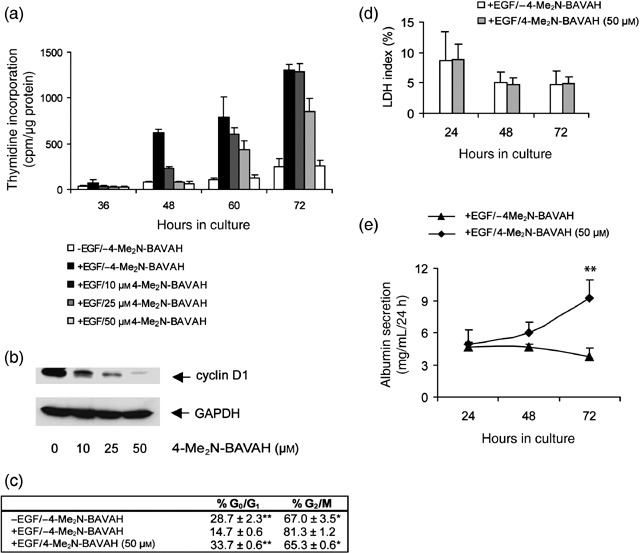
(a) 4‐Me2N‐BAVAH dose‐dependently inhibits EGF‐induced DNA synthesis in primary hepatocytes. Hepatocytes were cultured in triplicate as indicated. 3H‐thymidine incorporation was measured at the indicated time points. Data shown are the means ± SD of triplicate cultures from one experiment and are representative for four other experiments. Cells cultured in the absence of EGF serve as a negative control for hepatocyte proliferation whereas cells stimulated with EGF (50 ng/mL) 4 h after plating serve as a positive control for hepatocyte proliferation. Treatment with increasing concentrations of the HDACi started from time of plating. (b) 4‐Me2N‐BAVAH dose‐dependently inhibits cyclin D1 protein accumulation. Hepatocytes were cultured in the presence of EGF alone (added 4 h after plating) and treated or not with 10, 25 or 50 µm 4‐Me2N‐BAVAH from time of plating until 72 h of culture. Cells were then harvested and analysed for cyclin D1 protein expression (50 µg of total cellular proteins were loaded on SDS‐PAGE) (n = 3). The membranes were re‐probed with anti‐GAPDH antibody to verify sample loading. (c) Effect of 4‐Me2N‐BAVAH on cell cycle distribution of primary hepatocytes. Primary hepatocytes were cultured as indicated and their cell cycle distribution was evaluated at 72 h of culture by FACS analysis as described under Materials and Methods (n = 3). The relative high percentage of G2/M hepatocytes in the non‐proliferating conditions (– EGF/–4‐Me2N‐BAVAH and +EGF/50 µm 4‐Me2N‐BAVAH) can be explained by the fact that mononuclear tetraploid hepatocytes (1 × 4n G2/M) could not be discriminated from binuclear diploid hepatocytes (2 × 2n G0/G1). *P < 0.05 or **P < 0.001 for significant differences from EGF‐stimulated untreated control cultures according to a Student's t‐test. (d) Effect of 4‐Me2N‐BAVAH on hepatocyte viability. LDH leakage data are expressed as LDH index (%) and means ± SD are given (n = 5). (e) Effect of 4‐Me2N‐BAVAH on albumin secretion. Medium samples were taken at the indicated time points and analysed for their albumin content by ELISA. Data shown are the means ± SD of five experiments, **P < 0.01.
4‐Me2N‐BAVAH induces an early G1 cell cycle arrest by inhibiting cyclin D1 transcription
Figure 3a illustrates that 4‐Me2N‐BAVAH had less or no inhibitory effect on DNA synthesis when added either in mid‐G1 (corresponding to 24 h of culture) or after the restriction point (corresponding to 48 h of culture). In these conditions, cells were able to pass the restriction point, as evidenced by the presence of cyclin D1, and to subsequently proceed through S phase as evidenced by the presence of cdk1, an S/G2/M marker (Fig. 3b). These data suggest that 4‐Me2N‐BAVAH interferes with signals that trigger early cell cycle events. Cells that have progressed already into the mid‐ and mid‐late‐G1 phase were no longer sensitive towards the antiproliferating activity of 4‐Me2N‐BAVAH.
Figure 3.
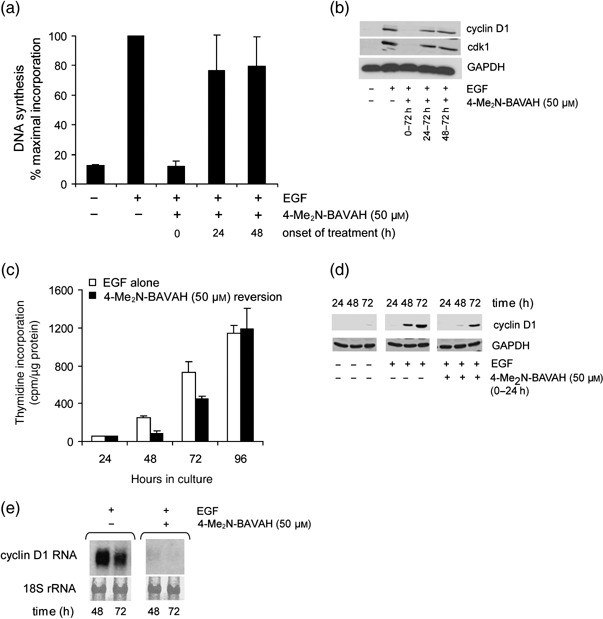
4‐Me2N‐BAVAH induces an early G1 cell cycle arrest by inhibiting cyclin D1 transcription. (a) Inhibition of DNA synthesis depends on time of onset of treatment. Freshly isolated hepatocytes were cultured either in the absence of EGF or stimulated with EGF 4 h after plating. Exposure to 50 µm 4‐Me2N‐BAVAH started at 0, 24 or 48 h after plating and was maintained until 72 h after plating. DNA synthesis was measured at 72 h of culture by means of 3H‐thymidine incorporation. Results are expressed as the percentage of maximal incorporation observed in hepatocytes cultured in the presence of EGF alone. The values obtained in this condition were arbitrarily set at 100% (n = 3). (b) Freshly isolated hepatocytes were cultured as described under (a). At 72 h of culture, cells were harvested and total cell protein extracts were prepared. 25 µg of proteins were loaded on gel and analysed for their cyclin D1 and cdk1 protein expression (n = 3). GAPDH was used as a loading control. (c) Reversion experiments. Freshly isolated hepatocytes were cultured in triplicate either in the presence of EGF alone or in the presence of both EGF and 4‐Me2N‐BAVAH (50 µm) for 24 h. Thereafter, cells were cultured for an additional 48 h in the presence of EGF alone. 3H‐thymidine incorporation was measured at the indicated time points. Data shown are the means ± SD of triplicate measurements of three independent experiments (except for values measured at 96 h where n = 2). (d) Hepatocytes were cultured in the presence of 50 µm 4‐Me2N‐BAVAH for 24 h. Thereafter, cells were cultured for an additional 48 h in the presence of EGF alone. Cyclin D1 expression was analysed by Western blotting at 24, 48 and 72 h of culture (25 µg of proteins/lane, n = 3). GAPDH was used as loading control. (e) Cyclin D1 RNA expression was analysed by Northern blotting. Hepatocytes were cultured in the presence of EGF (50 ng/mL; added 4 h after plating) and either exposed or not to 4‐Me2N‐BAVAH (50 µm). At 48 and 72 h of culture, cells were harvested and RNA was extracted. 10 ng of total RNA was loaded on a 1.1% (w/v) agarose gel. 18S rRNAs dyed by methylene blue staining was used as a loading control (n = 3).
As shown in Fig. 2b, 4‐Me2N‐BAVAH inhibits cyclin D1 protein expression in EGF‐stimulated hepatocytes in a dose‐dependent manner. Cyclin D1 is a key mediator of mitogenic signalling in primary hepatocytes. In these cells, cyclin D1 is only expressed following mitogenic stimulation and is indicative for passage of the restriction point, located in mid‐late‐G1 (Loyer et al. 1996). Its absence in the presence of 50 µm 4‐Me2N‐BAVAH thus indicates that cells were blocked somewhere in the G1 phase prior to the restriction point. Reversion experiments were performed to more precisely define where in G1 cells were arrested. For this purpose, primary proliferating hepatocytes were exposed to 50 µm 4‐Me2N‐BAVAH for 24 h from time of plating. Thereafter, cells were cultured in medium deprived of 4‐Me2N‐BAVAH for 3 consecutive days. DNA replication was analysed at the indicated time points. Hepatocytes in these reversion experiments started to incorporate 3H‐thymidine with a delay of 24–48 h (Fig. 3c), which corresponds with the lag period for initiation of DNA synthesis in EGF‐stimulated control cultures. A concomitant delay in cyclin D1 protein expression was observed (Fig. 3d). Next, Northern blots were performed to investigate whether down‐regulation of cyclin D1 occurred at the pre‐translational level. These experiments revealed that 4‐Me2N‐BAVAH suppressed cyclin D1 expression at the RNA level in EGF‐stimulated rat primary hepatocytes (Fig. 3e).
4‐Me2N‐BAVAH down‐regulates cyclin D1 RNA expression through inhibition of NF‐κB activation
Different EGF‐activated pathways, including the extracellular signal‐regulated kinase (ERK) (Rescan et al. 2001; Coutant et al. 2002) and the p70S6 kinase (Nelsen et al. 2003) pathways, mediate the expression of cyclin D1 and can as such trigger early cell cycle events. These kinases are often only transiently induced shortly after growth factor stimulation (Nelsen et al. 2003). Therefore, in order to investigate the potential role of both pathways in the HDACi‐induced cyclin D1 down‐regulation, hepatocytes were first synchronized in the G1 phase by culturing them in the absence of EGF for 48 h. They were subsequently stimulated with EGF and either exposed or not exposed to 4‐Me2N‐BAVAH (50 µm). Activation of both pathways was analysed 0.5, 1 and 3 h after growth factor stimulation. No substantial effects on the phosphorylation of the ribosomal subunit S6 were observed, at least not at residues Ser235/236 (data not shown). Because S6 phosphorylation by the p70S6 kinase plays a role in the translation of cyclin D1 (Chou & Blenis 1995), these findings confirm our observation that 4‐Me2N‐BAVAH inhibits cyclin D1 expression at the RNA level (Fig. 3e). Shortly after stimulation of synchronized primary hepatocytes with EGF, rapid and transient phosphorylation of ERK1/2 occurred (Fig. 4). 4‐Me2N‐BAVAH did not inhibit EGF‐mediated ERK activation. Instead, the HDACi slightly enhanced ERK1/2 phosphorylation levels during the initial 30 min, which decreased rapidly thereafter to control levels (Fig. 4). These data indicate that inhibition of the ERK pathway most likely is not involved in the inhibition of cyclin D1 transcription by 4‐Me2N‐BAVAH.
Figure 4.

Effect of 4‐Me2N‐BAVAH on ERK1/2 phosphorylation. As signal transduction kinases are often only transiently induced after growth factor stimulation, hepatocytes were first synchronized in the G1 phase by culturing them in the absence of EGF for 48 h. Hepatocytes were subsequently either stimulated or not with EGF (50 ng/mL) and exposed to 50 µm 4‐Me2N‐BAVAH where indicated. The level of ERK1/2 phosphorylation was analysed by Western blotting (50 µg proteins/lane) 0.5, 1 and 3 h later. Total ERK staining serves as a loading control (n = 3).
Previous reports have shown that cyclin D1 gene expression can be activated by NF‐κB in transformed cells and in synchronized mouse embryo fibroblasts (reviewed in Joyce et al. 2001). To verify whether induction of cyclin D1 by EGF in primary rat hepatocytes was NF‐κB‐dependent, cells were infected with adenovirus encoding the dominant negative, IκBα super‐repressor gene (AdIκBαsr) (Fig. 5a). IκBαsr is a mutated IκBα protein in which Ser32 and 36 are replaced with Ala. Consequently, IκBαsr can no longer be phosphorylated and therefore is not degraded by the proteasome. As such, IκBαsr behaves as a constitutively active inhibitor of NF‐κB activation in different cell types, both in vitro and in vivo (Roff et al. 1996; El Bakkouri et al. 2005; Wullaert et al. 2005). IκBαsr has also been shown to delay and reduce cyclin D1 expression in mouse embryo fibroblasts, HeLa cells and C2C12 myoblasts (Joyce et al. 2001). Concomitantly, we show here that AdIκBαsr‐infected primary hepatocytes have significantly reduced EGF‐induced cyclin D1 protein levels (Fig. 5a; P < 0.05). As this observation is similar to the effect of 4‐Me2N‐BAVAH on cyclin D1 expression, it was investigated whether this HDACi could inhibit EGF‐induced NF‐κB activation. Using a luciferase reporter gene assay, we could show that, like adenoviral infection with IκBαsr, exposure to the HDACi 4‐Me2N‐BAVAH (50 µm) substantially inhibited EGF‐induced NF‐κB activation in primary hepatocytes (Fig. 5b).
Figure 5.
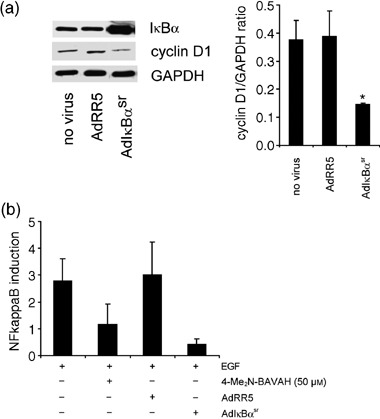
4‐Me2N‐BAVAH inhibits EGF‐mediated NF‐κB activation in primary hepatocytes. (a) Effect of AdIκBαsr on cyclin D1 expression in primary rat hepatocytes. Primary rat hepatocytes were either not infected or infected with AdRR5 or AdIκBαsr. The day after, hepatocytes were stimulated with EGF for 6 h. Cell extracts were prepared and analysed for IκBα and cyclin D1 expression by Western blot (15 µg proteins/lane). Cyclin D1 protein expression was quantified by densitometry. Data are expressed as the ratio cyclin D1/GAPDH. GAPDH was used as a control for transfer efficiency and equal sample loading, *P < 0.05. (b) Primary rat hepatocytes were infected with AdNF‐κBluc and AdLacZ alone, or in combination with either AdRR5 or AdIκBαsr. 24 h after infection, hepatocytes were stimulated with EGF (50 ng/mL), with or without the HDACi 4‐Me2N‐BAVAH (50 µm), for 6 h. Relative (Luc/Gal) NF‐κB activity was assessed by measuring Luc (luciferase) and Gal (β‐galactosidase) activities, and plotted as fold NF‐κB activation (relative to the basal level of Luc in the absence of EGF, after normalization with Gal). Values shown are the mean ± SD of triplicate measurements.
4‐Me2N‐BAVAH‐induced G1 cell cycle arrest is not mediated by p21
Induction of proliferation arrest by up‐regulating the p21 cdk inhibitor protein is a main trait of HDACi in tumour cells (Sambucetti et al. 1999; Chiba et al. 2004; Vanhaecke et al. 2004). As previously described (Albrecht & Hansen 1999; Nelsen et al. 2001), p21 expression is induced upon EGF stimulation and tightly correlates with the expression of cyclin D1 in these cells during proliferation (Fig. 6a). In primary hepatocytes, however, EGF‐induced p21 expression, was not further enhanced upon exposure to 4‐Me2N‐BAVAH; instead, p21 protein expression was prevented (Fig. 6a).
Figure 6.
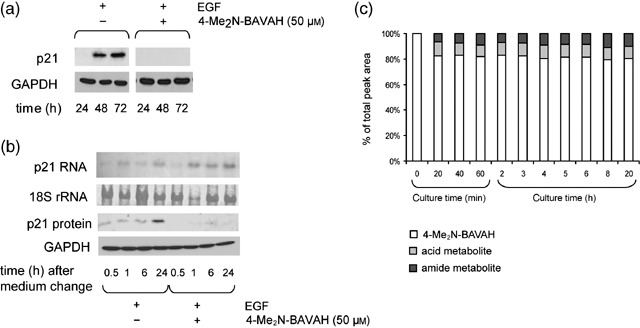
4‐Me2N‐BAVAH‐induced cell cycle arrest occurs in the absence of p21 protein induction. (a) Hepatocytes were cultured in the presence of EGF (50 ng/mL; added 4 h after plating) and either treated or not with 50 µm 4‐Me2N‐BAVAH from time of plating. Cells were harvested at the indicated time points. p21 protein expression was analysed by Western blotting after SDS‐PAGE of 50 µg of total cellular proteins (n = 3). Blots were reprobed with anti‐GAPDH antibody to verify equal sample loading and transfer efficiency. (b) Hepatocytes were cultured in the presence of EGF (50 ng/mL; added 4 h after plating) and either treated or not with 50 µm 4‐Me2N‐BAVAH from time of plating. At 24 h of culture, cells were provided with fresh medium of the same composition. Cells were then harvested at 0.5, 1, 6 and 24 h post medium renewal, corresponding to 24.5, 25, 30 and 48 h of culture. For analysis of p21 RNA expression, 10 ng of total RNA was loaded on a 1.1% (w/v) agarose gel. 18S rRNAs dyed by methylene blue staining was used as a loading control. p21 protein expression (25 µg proteins/lane) was evaluated by Western blotting. Membranes were re‐probed with anti‐GAPDH antibody to verify transfer efficiency and equal sample loading (n = 3). (c) Metabolic stability of 4‐Me2N‐BAVAH in rat hepatocyte cultures. The initial concentration of 4‐Me2N‐BAVAH was 50 µm. Aliquots were taken from the culture medium immediately after addition of the HDACi (0 min), and every 20 min during the first hour of culture. Thereafter, aliquots were taken every hour during a period of 6 h; final aliquots were taken at 8 and 20 h of culture. 4‐Me2N‐BAVAH and its major phase I metabolites were quantified by high‐pressure liquid chromatography combined with electrospray ionization mass spectrometry/ultraviolet detection. Results are expressed as percentage of the total peak area in the samples taken at the indicated time points.
We investigated whether p21 might be only transiently induced due to either instability of the HDACi in the culture medium or to rapid biotransformation of the HDACi by the hepatocytes; both leading to low levels of HDACi in the culture medium. As induction of p21 by EGF occurs between 24 and 48 h of culture, cells were provided with fresh EGF‐containing medium supplemented or not with 4‐Me2N‐BAVAH at 24 h and p21 RNA and protein levels were monitored at short intervals (0.5, 1, 6 and 24 h) within this time frame. In untreated, control, cultures, p21 RNA and protein became detectable 0.5 h after medium renewal. Peak levels were reached 24 h later, corresponding to 48 h of culture. In the presence of the HDACi, a strong decrease in p21 protein, but not RNA expression was seen (Fig. 6b). In addition, it could be shown that no extensive or rapid biotransformation of 4‐Me2N‐BAVAH into inactive metabolites occurred (Fig. 6c).
4‐Me2N‐BAVAH delays the onset of spontaneous apoptosis in primary hepatocytes
In Fig. 5b, we show that the HDACi 4‐Me2N‐BAVAH inhibits EGF‐mediated NF‐κB activation. Inhibition of NF‐κB activity has previously been reported to enhance the pro‐apoptotic activity of HDACi (Mayo et al. 2003). As spontaneous apoptosis is a well‐known pernicious event in primary hepatocyte cultures (Baily‐Maitre et al. 2002), we wished to determine whether 4‐Me2N‐BAVAH, through its action on NF‐κB activity, facilitated this spontaneous cell death or not. Data reported in Fig. 7 show that, in proliferating primary hepatocytes, 4‐Me2N‐BAVAH decreased the expression of the pro‐apoptotic proteins Bid and Bax. In addition, cleavage of pro‐caspase‐3 into the smaller active p18 fragment was reduced from 24 h of culture onwards (Fig. 7b).
Figure 7.
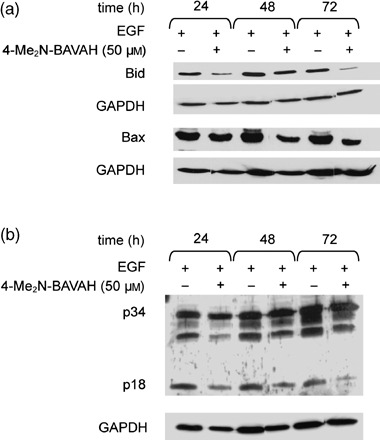
4‐Me2N‐BAVAH delays the onset of spontaneous apoptosis in primary rat hepatocytes. Hepatocytes were cultured in the presence of EGF alone (50 ng/mL; added 4 h after plating) and exposed or not to 50 µm 4‐Me2N‐BAVAH (from time of plating). Cells were harvested at the indicated time points and analysed for (a) Bid and Bax protein expression (50 µg proteins/lane) and (b) caspase‐3 activation (50 µg proteins/lane) (n = 3).
DISCUSSION
4‐Me2N‐BAVAH is an amide‐based hydroxamic acid, containing HDACi with structural similarity to TSA. Despite its higher metabolic stability (Elaut et al. 2004, Table 1), 4‐Me2N‐BAVAH appeared less potent than TSA in HDAC inhibition assays (Table 1). However, given the complex role and regulation of HDACs in gene transcription, it is clear that the selection of a suitable compound should not be based solely on IC50 values (reflecting enzyme inhibition activity) but also on its biological activity and efficacy. Some clear advantages of 4‐Me2N‐BAVAH over TSA with respect to metabolic stability and biological activity are summarized in Table 1. Jung et al. (1999) have previously reported the antiproliferative activity of 4‐Me2N‐BAVAH in a malignant cell line, yet no attention was paid to the underlying molecular mechanisms. In this study, we have investigated the effects of 4‐Me2N‐BAVAH on EGF‐induced proliferation in rat primary hepatocytes. The latter is widely recognized as a valuable alternative to cell lines for studying cell cycle‐related signal transduction (Papeleu et al. 2004).
Our data reveal that primary hepatocytes exposed to non‐cytotoxic concentrations of 4‐Me2N‐BAVAH become irresponsive to the mitogenic activity of EGF. This is evidenced by the inhibition of cyclin D1 gene transcription, and corresponds with cell cycle arrest in early G1 phase of the cell cycle. Cyclin D1 plays a pivotal role in the regulation of hepatocyte proliferation by growth factors, including EGF (Loyer et al. 1996; Nelsen et al. 2001). Transfection of hepatocytes with cyclin D1 was found to promote their proliferation in vivo as well as in vitro (Albrecht & Hansen 1999; Nelsen et al. 2001). One of the key players in transcriptional regulation of cyclin D1 is the MEK/ERK pathway. Talarmin et al. (1999), for instance, described that ERK activation preceded the induction of cyclin D1 mRNA expression after partial hepatectomy. Yet, 4‐Me2N‐BAVAH had no inhibitory effect on ERK1/2 phosphorylation, excluding a role for this pathway in the transcriptional down‐regulation of cyclin D1 by the HDACi.
Another link between EGF stimulation and G1‐to‐S‐phase progression is provided by NF‐κB activation. In unstimulated cells, NF‐κB is predominantly located in the cytoplasm through interaction with IκB inhibitory proteins, such as IκBα (Moynagh 2005). Upon activation by pro‐inflammatory cytokines, including tumour necrosis factor and IL‐1, IκBα is phosphorylated and subsequently degraded by the proteasome (Joyce et al. 2001; Moynagh 2005). This allows rapid and transient translocation of NF‐κB to the nucleus, where it can activate transcription of a wide variety of genes that are involved in cell proliferation, including cyclin D1 (Joyce et al. 2001). We provide evidence that activation of cyclin D1 gene transcription by EGF in rat primary hepatocytes is NF‐κB‐dependent. Indeed, hepatocytes infected with adenovirus expressing IκBαsr showed reduced expression of NF‐κB‐dependent luciferase reporter gene upon EGF stimulation, and concomitant down‐regulation of EGF‐induced cyclin D1 expression. Exposure of EGF‐stimulated hepatocytes to 4‐Me2N‐BAVAH inhibited NF‐κB activation in a manner similar to AdIκBαsr, arguing that HDACi inhibits cyclin D1 gene transcription through a mechanism involving NF‐κB inhibition. Although in most cases, HDACi exposure has been associated with activation of NF‐κB (Ashburner et al. 2001; Mayo et al. 2003), we show that, as in colonic epithelial cells (Yin et al. 2001), the opposite occurs in proliferating rat primary hepatocytes.
In contrast to cancer cell lines, in which HDACi induces reversible cell cycle arrest by up‐regulating expression of cyclin‐dependent kinase inhibitor p21 (Sambucetti et al. 1999; Lagger et al. 2003), 4‐Me2N‐BAVAH‐induced G1 cell cycle arrest was not associated with p21 protein accumulation. Although this could be due to impaired translation and/or enhanced proteolysis, the fact that none of the functions of p21 (such as assembly of cyclin D1/cdk4 complexes and inhibition of their activity) are required might also in part explain this finding. In addition, we show that lack of p21 induction is not due to metabolic instability of 4‐Me2N‐BAVAH in primary hepatocyte cultures. HDACi‐mediated p21‐independent cell cycle arrests have also been described in other non‐tumourous cell systems, including p21 null mouse fibroblasts (Wharton et al. 2000) and normal human dermal fibroblasts (Glaser et al. 2003). This could thus represent a general mechanism of action of HDACi in non‐malignant cells, which might contribute to their tumour‐selective apoptotic features (Papeleu et al. 2005). Indeed, primary rat hepatocytes exposed to 4‐Me2N‐BAVAH show delayed onset of spontaneous apoptosis, which could be a result of decreased p21 protein levels. In primary hepatocytes and models of liver regeneration, for example, forced expression of p21 has been shown to favour apoptosis (Kobayashi & Tsukamoto 2001; Papakyriakou et al. 2002; Qiao et al. 2002), whereas significant reduction in apoptosis has been observed in p21–/– primary mouse hepatocytes (Sheahan et al. 2004).
In conclusion, 4‐Me2N‐BAVAH induces early G1 cell cycle arrest in EGF‐stimulated primary rat hepatocytes by inhibiting NF‐κB activation and subsequent cyclin D1 transcription. In contrast to frequently observed tumour cells, HDACi‐induced cell cycle arrest in primary rat hepatocytes occurs independently of p21 induction, which could represent a general mechanism of action of HDACi in non‐malignant cells.
ACKNOWLEDGEMENTS
We thank Mr G. Stangé from the Diabetes Research Center of the Vrije Universiteit Brussel for his assistance with FACS analysis. We greatly appreciated the dedicated technical assistance of Mrs S. Coppens, Mr C. Bornauw, Mrs S. Ramaeckers, Ms. H. Mertens, Mrs G. De Pauw and Mr E. Quartier. Dr G. Baffet and Dr A. Coutant from the INSERM institute U522, Rennes, France, are deeply acknowledged for helpful comments on the manuscript. We thank Dr R. Hay and Dr B. McGray for providing recombinant adenoviruses.
This work was supported by grants from the Research Council (OZR) of the Vrije Universiteit Brussel, Brussels, Belgium and the Fund for Scientific Research (FWO), Vlaanderen, Belgium and the European Union (6th Framework project PREDICTOMICS number 504761), the Geoconcerteerde Onderzoek Acties (GOA) of the University of Gent (grant 01G06B6) and the IAP6/18 network.
REFERENCES
- Albrecht JH, Hansen LK (1999) Cyclin D1 promotes mitogen‐independent cell cycle progression in hepatocytes. Cell Growth Differ. 10, 397–404. [PubMed] [Google Scholar]
- Ashburner BP, Westerheide SD, Baldwin AS Jr (2001) The p65 (RelA) subunit of NF‐kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 21, 7065–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly‐Maitre B, De Sousa G, Zucchini N, Gugenheim J, Boulukos KE, Rahmani R (2002) Spontaneous apoptosis in primary cultures of human and rat hepatocytes: molecular mechanisms and regulation by dexamethasone. Cell Death Differ. 9, 945–955. [DOI] [PubMed] [Google Scholar]
- Chiba T, Yokosuka O, Arai M, Tada M, Fukai K, Imazeki F, Kato M, Seki N, Saisho H (2004) Identification of genes up‐regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J. Hepatol. 41, 436–445. [DOI] [PubMed] [Google Scholar]
- Chou MM, Blenis J (1995) The 70 kDa S6 kinase: regulation of a kinase with multiple roles in mitogenic signalling. Curr. Opin. Cell Biol. 7, 806–814. [DOI] [PubMed] [Google Scholar]
- Cousens LS, Gallwitz D, Alberts BM (1979) Different accessibilities in chromatin to histone acetylase. J. Biol. Chem. 254, 1716–1723. [PubMed] [Google Scholar]
- Coutant A, Rescan C, Gilot D, Loyer P, Guguen‐Guillouzo C, Baffet G (2002) PI3K‐FRAP/mTOR pathway is critical for hepatocyte proliferation whereas MEK/ERK supports both proliferation and survival. Hepatology 36, 1079–1088. [DOI] [PubMed] [Google Scholar]
- De Valck D, Heyninck K, Van Criekinge W, Contreras R, Beyaert R, Fiers W (1996) A20, an inhibitor of cell death, self‐associates by its zinc finger domain. FEBS Lett. 384, 61–64. [DOI] [PubMed] [Google Scholar]
- Dunn JC, Tompkins RG, Yarmush ML (1991) Long‐term in vitro function of adult hepatocytes in a collagen sandwich configuration. Biotechnol. Prog. 7, 237–245. [DOI] [PubMed] [Google Scholar]
- El Bakkouri K, Wullaert A, Haegman M, Heyninck K, Beyaert R (2005) Adenoviral gene transfer of the NF‐kappa B inhibitory protein ABIN‐1 decreases allergic airway inflammation in a murine asthma model. J. Biol. Chem. 280, 17938–17944. [DOI] [PubMed] [Google Scholar]
- Elaut G, Laus G, Alexandre E, Richert L, Bachellier P, Tourwé D, Rogiers V, Vanhaecke T (2007) A metabolic screening study of Trichostatin A (TSA) and TSA‐like histone deacetylase inhibitors in rat and human primary hepatocyte cultures. J. Pharmacol. Exp. Ther. 321, 400–408. [DOI] [PubMed] [Google Scholar]
- Elaut G, Török G, Papeleu P, Vanhaecke T, Laus G, Tourwe D, Rogiers V (2004) Rat hepatocyte suspensions as a suitable in vitro model for studying the biotransformation of histone deacetylase inhibitors. Altern. Lab. Anim. 32, 105–112. [DOI] [PubMed] [Google Scholar]
- Elaut G, Török G, Vinken M, Laus G, Papeleu P, Tourwe D, Rogiers V (2002) Major phase I biotransformation pathways of Trichostatin A in rat hepatocytes and in rat and human liver microsomes. Drug Metab. Dispos. 30, 1320–1328. [DOI] [PubMed] [Google Scholar]
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK (2003) Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2, 151–163. [PubMed] [Google Scholar]
- Grempler R, Kienitz A, Werner T, Meyer M, Barthel A, Ailett F, Sutherland C, Walther R, Schmoll D (2004) Tumour necrosis factor alpha decreases glucose‐6‐phosphatase gene expression by activation of nuclear factor kappaB. Biochem. J. 382, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkens T, Papeleu P, Elaut G, Vinken M, Rogiers V, Vanhaecke T (2007) Trichostatin A, a critical factor in maintaining the functional differentiation of primary cultured rat hepatocytes. Toxicol. Appl. Pharmacol. 218, 64–71. [DOI] [PubMed] [Google Scholar]
- Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG (2001) NF‐kappaB and cell‐cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 12, 73–90. [DOI] [PubMed] [Google Scholar]
- Jung M, Brosch G, Kolle D, Scherf H, Gerhauser C, Loidl P (1999) Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J. Med. Chem. 42, 4669–4679. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Tsukamoto I (2001) Prolonged Jun N‐terminal kinase (JNK) activation and the upregulation of p53 and p21 (WAF1/CIP1) preceded apoptosis in hepatocytes after partial hepatectomy and cisplatin. Biochim. Biophys. Acta 1537, 79–88. [DOI] [PubMed] [Google Scholar]
- Koniaris LG, McKillop IH, Schwartz SI, Zimmers TA (2003) Liver regeneration. J. Am. Coll. Surg. 197, 634–659. [DOI] [PubMed] [Google Scholar]
- Kouzarides T (1999) Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 9, 40–48. [DOI] [PubMed] [Google Scholar]
- Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, Tischler J, Chiocca S, Suske G, Rotheneder H, Wintersberger E, Seiser C (2003) The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin‐dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol. Cell. Biol. 23, 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyer P, Cariou S, Glaise D, Bilodeau M, Baffet G, Guguen‐Guillouzo C (1996) Growth factor dependence of progression through G1 and S phases of adult rat hepatocytes in vitro. Evidence of a mitogen restriction point in mid‐late G1 . J. Biol. Chem. 271, 11484–11492. [DOI] [PubMed] [Google Scholar]
- Marks PA, Jiang X (2005) Histone deacetylase inhibitors in programmed cell death and cancer therapy. Cell Cycle 4, 549–551. [DOI] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK (2001) Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 1, 194–202. [DOI] [PubMed] [Google Scholar]
- Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, Jones DR (2003) Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF‐kappa B through the Akt pathway. J. Biol. Chem. 278, 18980–18989. [DOI] [PubMed] [Google Scholar]
- Mitchell C, Gilgenkrantz H (2003) Transcriptional profiling of liver regeneration: new approaches to an old trick! J. Hepatol. 38, 847–849. [DOI] [PubMed] [Google Scholar]
- Moynagh PN (2005) The NF‐kappaB pathway. J. Cell Sci. 118, 4589–4592. [DOI] [PubMed] [Google Scholar]
- Nelsen CJ, Rickheim DG, Timchenko NA, Stanley MW, Albrecht JH (2001) Transient expression of cyclin D1 is sufficient to promote hepatocyte replication and liver growth in vivo . Cancer Res. 61, 8564–8568. [PubMed] [Google Scholar]
- Nelsen CJ, Rickheim DG, Tucker MM, Hansen LK, Albrecht JH (2003) Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J. Biol. Chem. 278, 3656–3663. [DOI] [PubMed] [Google Scholar]
- Papakyriakou P, Tzardi M, Valatas V, Kanavaros P, Karydi E, Notas G, Xidakis C, Kouroumalis E (2002) Apoptosis and apoptosis related proteins in chronic viral liver disease. Apoptosis 7, 133–141. [DOI] [PubMed] [Google Scholar]
- Papeleu P, Loyer P, Vanhaecke T, Henkens T, Elaut G, Guguen‐Guillouzo C, Rogiers V (2004) Proliferation of epidermal growth factor‐stimulated hepatocytes in a hormonally defined serum‐free medium. Altern. Lab. Anim. 32, 57–64. [DOI] [PubMed] [Google Scholar]
- Papeleu P, Loyer P, Vanhaecke T, Elaut G, Geerts A, Guguen‐Guillouzo C, Rogiers V (2003) Trichostatin A induces differential cell cycle arrests but does not induce differential cell cycle arrests but does not induce apoptosis in primary cultures of mitogen‐stimulated rat hepatocytes. J. Hepatol. 39, 374–382. [DOI] [PubMed] [Google Scholar]
- Papeleu P, Vanhaecke T, Elaut G, Vinken M, Henkens T, Snykers S, Rogiers V (2005) Differential effects of histone deacetylase inhibitors in tumor and normal cells‐what is the toxicological relevance? Crit. Rev. Toxicol. 35, 363–378. [DOI] [PubMed] [Google Scholar]
- Papeleu P, Vanhaecke T, Henkens T, Elaut G, Vinken M, Snykers S, Rogiers V (2006) Isolation of rat hepatocytes In: Philips IR, Shephard EA, eds. Methods in Molecular Biology, pp. 229–237. Totowa, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Piekarz R, Bates S (2004) A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr. Pharm. Des. 10, 2289–2298. [DOI] [PubMed] [Google Scholar]
- Qiao L, McKinstry R, Gupta S, Gilfor D, Windle JJ, Hylemon PB, Grant S, Fisher PB, Dent P (2002) Cyclin kinase inhibitor p21 potentiates bile acid‐induced apoptosis in hepatocytes that is dependent on p53. Hepatology 36, 39–48. [DOI] [PubMed] [Google Scholar]
- Rescan C, Coutant A, Talarmin H, Theret N, Glaise D, Guguen‐Guillouzo C, Baffet G (2001) Mechanism in the sequential control of cell morphology and S phase entry by epidermal growth factor involves distinct MEK/ERK activations. Mol. Biol. Cell 12, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roff M, Thompson J, Rodriguez MS, Jacque JM, Baleux F, Arenzana‐Seisdedos F, Hay RT (1996) Role of IkappaBalpha ubiquitination in signal‐induced activation of NFkappaB in vivo . J. Biol. Chem. 271, 7844–7850. [DOI] [PubMed] [Google Scholar]
- Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D (1999) Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J. Biol. Chem. 274, 34940–34947. [DOI] [PubMed] [Google Scholar]
- Sanderson L, Taylor GW, Aboagye EO, Alao JP, Latigo JR, Coombes RC, Vigushin DM (2004) Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin A after intraperitoneal administration to mice. Drug Metab. Dispos. 32, 1132–1138. [DOI] [PubMed] [Google Scholar]
- Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PB Jr, Ritchie TC, Hunninghake GW, Zandi E, Engelhardt JF (2001) Lipopolysaccharide induces Rac1‐dependent reactive oxygen species formation and coordinates tumor necrosis factor‐alpha secretion through IKK regulation of NF‐kappa B. J. Biol. Chem. 276, 30188–30198. [DOI] [PubMed] [Google Scholar]
- Sheahan S, Bellamy CO, Treanor L, Harrison DJ, Prost S (2004) Additive effect of p53, 21 and Rb deletion in triple knockout primary hepatocytes. Oncogene 23, 1489–1497. [DOI] [PubMed] [Google Scholar]
- Talarmin H, Rescan C, Cariou S, Glaise D, Zanninelli G, Bilodeau M, Loyer P, Guguen‐Guillouzo C, Baffet G (1999) The mitogen‐activated protein kinase kinase/extracellular signal‐regulated kinase cascade activation is a key signalling pathway involved in the regulation of G (1) phase progression in proliferating hepatocytes. Mol. Cell. Biol. 19, 6003–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaecke T, Papeleu P, Elaut G, Rogiers V (2004) Trichostatin A‐like hydroxamate histone deacetylase inhibitors as therapeutic agents: toxicological point of view. Curr. Med. Chem. 11, 1629–1643. [DOI] [PubMed] [Google Scholar]
- Vinken M, Henkens T, Vanhaecke T, Papeleu P, Geerts A, Van Rossen E, Chipman JK, Meda P, Rogiers V (2006) Trichostatin A enhances gap junctional intercellular communication in primary cultures of adult rat hepatocytes. Toxicol. Sci. 91, 484–492. [DOI] [PubMed] [Google Scholar]
- Wharton W, Savell J, Cress WD, Seto E, Pledger WJ (2000) Inhibition of mitogenesis in Balb/c‐3T3 cells by Trichostatin A. Multiple alterations in the induction and activation of cyclin‐cyclin‐dependent kinase complexes. J. Biol. Chem. 275, 33981–33987. [DOI] [PubMed] [Google Scholar]
- Workman JL, Kingston RE (1998) Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 67, 545–579. [DOI] [PubMed] [Google Scholar]
- Wullaert A, Wielockx B, Van Huffel S, Bogaert V, De Geest B, Papeleu P, Schotte P, El Bakkouri K, Heyninck K, Libert C, Beyaert R (2005) Adenoviral gene transfer of ABIN‐1 protects mice from TNF/galactosamine‐induced acute liver failure and lethality. Hepatology 42, 381–389. [DOI] [PubMed] [Google Scholar]
- Yin L, Laevsky G, Giardina C (2001) Butyrate suppression of colonocyte NF‐kappa B activation and cellular proteasome activity. J. Biol. Chem. 276, 44641–44646. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Furumai R, Nishiyama M, Komatsu Y, Nishino N, Horinouchi S (2001) Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother. Pharmacol. 48 (Suppl. 1), S20–S26. [DOI] [PubMed] [Google Scholar]