

The riminophenazine agent clofazimine (CFZ) is repurposed as an important component of the new short-course multidrug-resistant tuberculosis regimen and significantly shortens first-line regimen for drug-susceptible tuberculosis in mice. However, CFZ use is hampered by its unwelcome skin discoloration in patients.
Keywords: riminophenazine, TBI-166, clofazimine, in vivo, Mycobacterium tuberculosis
ABSTRACT
The riminophenazine agent clofazimine (CFZ) is repurposed as an important component of the new short-course multidrug-resistant tuberculosis regimen and significantly shortens first-line regimen for drug-susceptible tuberculosis in mice. However, CFZ use is hampered by its unwelcome skin discoloration in patients. A new riminophenazine analog, TBI-166, was selected as a potential next-generation antituberculosis riminophenazine following an extensive medicinal chemistry effort. Here, we evaluated the activity of TBI-166 against Mycobacterium tuberculosis and its potential to accumulate and discolor skin. The in vitro activity of TBI-166 against both drug-sensitive and drug-resistant M. tuberculosis is more potent than that of CFZ. Spontaneous mutants resistant to TBI-166 were found at a frequency of 2.3 × 10−7 in wild strains of M. tuberculosis. TBI-166 demonstrates activity at least equivalent to that of CFZ against intracellular M. tuberculosis and in low-dose aerosol infection models of acute and chronic murine tuberculosis. Most importantly, TBI-166 causes less skin discoloration than does CFZ despite its higher tissue accumulation. The efficacy of TBI-166, along with its decreased skin pigmentation, warrants further study and potential clinical use.
INTRODUCTION
Tuberculosis (TB) is the leading cause of death worldwide from a single infectious agent, ranking above HIV/AIDS. An estimated 10.4 million people developed active tuberculosis in 2016, and 1.67 million people died from it (1). The increased number of multidrug-resistant TB (MDR-TB) and extensively drug resistant TB (XDR-TB) strains has created a great challenge for the treatment and control of tuberculosis. Repurposed antituberculosis drugs as well as new drugs are urgently needed to treat drug-resistant tuberculosis more safely and effectively (2, 3). Among the repurposed drugs, clofazimine (CFZ) (Fig. 1), a riminophenazine antibiotic, has been used in the treatment of leprosy since 1962. In 2010, a trial in Bangladesh reported that a CFZ-containing regimen can cure MDR-TB in 9 to 12 months (4). The efficacy of this regimen was confirmed by other clinical trials (5). As a result, CFZ is now recommended by the WHO as an important component of the new short-course MDR-TB regimen (6). CFZ was also demonstrated to shorten the duration of treatment for drug-susceptible TB in a mouse model (7). The shortening of treatment may be related to targeting of slowly replicating/nonreplicating bacterial populations by CFZ (8–10). The bactericidal activity occurs via reactive oxygen species (ROS) production following the reduction of CFZ by mycobacterial electron transport chain enzyme type 2 NADH dehydrogenase (NDH-2) (11). CFZ kills M. tuberculosis synergistically with bedaquiline and Q203 (imidazopyridine amide compound, blocking M. tuberculosis growth by targeting the respiratory cytochrome bc1 complex [12]) through increased production of reactive oxygen species (13), suggesting potential applications of inhibiting energy metabolism for combating tuberculosis (14).
FIG 1.
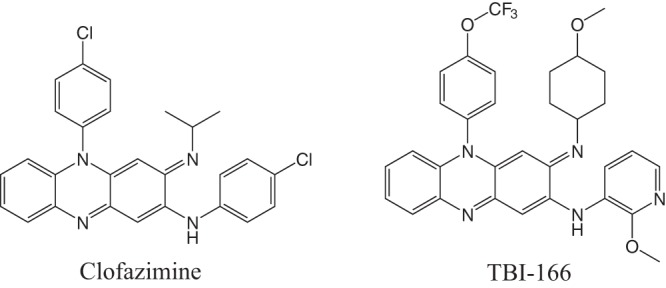
Structures of clofazimine and TBI-166.
Unfortunately, the physicochemical properties of extremely high lipophilicity and consequent side effects limit clofazimine’s use in the treatment of TB (15). An excessively long half-life (t1/2) (over 70 days in humans) and accumulation in fat and skin tissues lead to unwelcome skin discoloration (16–18). A systematic structure-activity study of more than 500 newly synthesized CFZ analogs was conducted to identify new generation compounds with equivalent or better efficacy than CFZ and at the same time incur reduced skin pigmentation potential (19–21).
Among these compounds, 12 prioritized compounds, including TBI-166 (Fig. 1), demonstrated enhanced in vitro activity compared to CFZ against replicating M. tuberculosis H37Rv (17). Dosed at 20 mg/kg of body weight/day for 3 weeks, TBI-166 showed efficacy (i.e., CFU reduction) superior to that of 20 mg/kg CFZ in an acute murine TB model (17). Improved physicochemical properties with reduced lipophilicity, maybe due to the replacement of the C-2 phenyl ring in CFZ with 2-methoxypyridyl group, suggest that TBI-166 may decrease the potential for tissue discoloration.
Here, we further profile the antimicrobial activity of TBI-166, including in vitro activity against M. tuberculosis clinical isolates and other mycobacterial species, as well as efficacy against TB in murine models. We also describe the accumulation in different tissues and lack of discoloration of TBI-166 after repeated dosing.
RESULTS
In vitro antimicrobial profile and frequency of spontaneous resistance.
The MIC of TBI-166 against replicating M. tuberculosis H37Rv is 0.063 μg/ml, which is 4-fold lower than that of CFZ. The MICs for TBI-166 against 16 drug-sensitive clinical isolates range from <0.005 to 0.15 μg/ml, lower than those for CFZ (0.026 to 0.633 μg/ml) (Table 1). Likewise, for a panel of 28 drug-resistant clinical M. tuberculosis isolates, the TBI-166 MIC was lower than that of CFZ against each individual strain. The MIC range for TBI-166 among these 28 isolates was 0.01 to 0.2 μg/ml compared to 0.075 to 0.844 μg/ml for CFZ (Table 1). The lack of cross-resistance with currently used anti-TB agents indicates the potential utility of TBI-166 against MDR strains, including strains resistant to isoniazid (INH), rifampin (RIF), streptomycin (STR), ethambutol (EMB), and levofloxacin (LVX).
TABLE 1.
MICs of TBI-166 against different mycobacterial species
| Mycobacterial species and/or straina | No. of strains | MIC data (μg/ml) by drugb |
|||
|---|---|---|---|---|---|
| TBI-166 |
CFZ |
||||
| Range | Median | Range | Median | ||
| M. tuberculosis H37Rv | 1 | NA | 0.063 | NA | 0.25 |
| M. tuberculosis fully susceptible clinical isolates | 16 | <0.005–0.15 | 0.038 | 0.026–0.633 | 0.227 |
| M. tuberculosis resistant to RIF | 1 | NA | 0.099 | NA | 0.224 |
| M. tuberculosis resistant to INH and RIF | 2 | 0.017–0.025 | 0.021 | 0.15–0.264 | 0.207 |
| M. tuberculosis resistant to INH and STR | 1 | 0.018 | 0.018 | 0.386 | 0.386 |
| M. tuberculosis resistant to RIF and EMB | 1 | NA | 0.2 | NA | 0.3 |
| M. tuberculosis resistant to INH, RIF, and LVX | 1 | NA | 0.026 | NA | 0.064 |
| M. tuberculosis resistant to INH, RIF, and STR | 1 | NA | 0.1 | NA | 0.3 |
| M. tuberculosis resistant to RIF, STR, and EMB | 1 | NA | 0.1 | NA | 0.3 |
| M. tuberculosis resistant to INH, RIF, and EMB | 3 | 0.013–0.098 | 0.025 | 0.075–0.3 | 0.143 |
| M. tuberculosis resistant to RIF, STR, LVX, and EMB | 2 | 0.1–0.164 | 0.132 | 0.15–0.544 | 0.347 |
| M. tuberculosis resistant to INH, RIF, LVX, and EMB | 4 | 0.019–0.05 | 0.0385 | 0.095–0.486 | 0.2425 |
| M. tuberculosis resistant to INH, RIF, STR, and EMB | 5 | 0.01–0.2 | 0.1 | 0.15–0.3 | 0.219 |
| M. tuberculosis resistant to INH, RIF, STR, LVX, and EMB | 6 | 0.039–0.2 | 0.1125 | 0.118–0.844 | 0.4095 |
| M. kansasii | 1 | NA | 0.183 | NA | 1.776 |
| M. scrofulaceum | 1 | NA | 1.49 | NA | 0.292 |
| M. fortuitum | 1 | NA | 0.688 | NA | 0.276 |
RIF, rifampin; INH, isoniazid; STR, streptomycin; EMB, ethambutol; LVX, levofloxacin.
NA, not applicable.
TBI-166 was also assessed for activity against selected nontuberculous mycobacteria (NTM) and was found to be approximately 10 times more potent against Mycobacterium kansasii than was CFZ (Table 1). Interestingly, TBI-166 maintained activity against Mycobacterium fortuitum and Mycobacterium scrofulaceum despite having higher MICs against these species than those of CFZ. Mutations in rv0678 are a major mechanism of clofazimine resistance (22) and cause cross-resistance between CFZ and bedaquiline (23). To identify cross-resistance between TBI-166 and CFZ/bedaquiline, we tested the MICs of TBI-166, CFZ, and bedaquiline against three rv0678 mutants using the microplate alamarBlue assay (MABA) method. On average, the fold change in MIC of TBI-166 against rv0678 mutants compared to M. tuberculosis H37Rv was lower than that of CFZ and bedaquiline (TBI-166 versus CFZ versus bedaquiline, 3:10:8) (Table 2), suggesting there is part cross-resistance between TBI-166 and CFZ/bedaquiline. Spontaneous mutants resistant to TBI-166 at 0.25 μg/ml (4 times the MIC) were found at a frequency of 2.3 × 10−7 in wild strains of M. tuberculosis.
TABLE 2.
MICs for CFZ and TBI-166 against rv0678 mutant and wild-type M. tuberculosis strains
| Strain | Sequence or mutation by gene |
MIC (μg/ml) by drug |
||||
|---|---|---|---|---|---|---|
| rv0678 | pepQ | atpE | Bedaquiline | CFZ | TBI-166 | |
| BDQ-1 | C305T | WT | WT | 0.49 | 1.34 | 0.18 |
| BDQ-2 | 292–316 nt deletion | WT | WT | 0.47 | 1.29 | 0.19 |
| BDQ-3 | C403T | WT | WT | 0.47 | 1.62 | 0.19 |
| H37Rv | WT | WT | WT | 0.06 | 0.14 | 0.060 |
TBI-166 inhibits intracellular M. tuberculosis.
We previously evaluated the cytotoxicity of TBI-166 on Vero cells using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTT) assay (17). The concentration of TBI-166 causing 50% loss of viability (CC50) was more than 64 μg/ml. We tested the activity of TBI-166 against M. tuberculosis H37Rv infecting J774A.1 cells. After 3 days of incubation, TBI-166 exhibited activity in a dose-dependent manner compared to the untreated control cells, i.e., CFU reductions of 0.54, 0.61, and 1.13 log10 were observed at TBI-166 concentrations of 0.0625, 0.25, and 1 μg/ml, respectively (Fig. 2). In macrophages, TBI-166 had activity comparable to or greater than that of CFZ.
FIG 2.

Activity of TBI-166 against intracellular M. tuberculosis H37Rv.
In vivo efficacy, discoloration, and toxicity in mice.
Prior to conducting efficacy testing, a preliminary in vivo toxicity study was carried out. All mice tolerated TBI-166 well at the doses administered. No adverse events were identified. The dose causing death in at least 50% of mice (LD50) was more than 3,000 mg/kg.
In the acute infection model, the mean (± standard deviation [SD]) lung bacterial burden of the untreated control group increased from 2.97 ± 0.17 log10 CFU to 7.00 ± 0.17 log10 CFU over the 20-day course of chemotherapy. At the end of the treatment period, the mean lung CFU counts for all doses of TBI-166-treated groups (including the TBI-166 tablet) were 2.2- to 4.2-log units lower than those for the untreated control group and the RIF-treated group (P < 0.001) (Fig. 3). TBI-166 exhibited dose-dependent antituberculosis activity at doses ranging from 10 to 80 mg/kg. All TBI-166-treated groups except the 80-mg/kg tablet TBI-166 group had lung CFU counts above the pretreatment levels in this experiment, suggesting primarily bacteriostatic rather than bactericidal activity for TBI-166 and for CFZ. Capsule CFZ had better activity than did powder CFZ (P < 0.01). This could be because capsule CFZ administered as a microcrystalline suspension in an oil-wax base improved absorption due to its lipophilicity (24, 25). The activity of 20 mg/kg TBI-166 was no different from that of an equivalent dose of CFZ and the TBI-166 tablet and 10 mg/kg TBI-166. TBI-166 at 40 mg/kg effected a larger reduction in CFU than did TBI-166 at 20 mg/kg (P = 0.02) and CFZ at 20 mg/kg (P < 0.001).
FIG 3.

Efficacy of TBI-166 in the acute mouse infection model. Dosages are in milligrams per kilogram of body weight. At the beginning of treatment at day 10 postinfection, there were 2.97 ± 0.17 log10 CFU in the lungs of untreated mice. Values were determined after 20 days of treatment in BALB/c mice infected with M. tuberculosis H37Rv.
Next, we evaluated the efficacy of TBI-166 against chronic murine TB infected by low-dose aerosol with M. tuberculosis H37Rv, with lung CFU determined after 2, 4, and 8 weeks of treatment. In this study, TBI-166 demonstrated time-dependent killing of M. tuberculosis. During the first 2 weeks of administration, there was no statistically significant difference in CFU counts between mice receiving TBI-166 at 10, 20, and 80 mg/kg and the untreated controls (Fig. 4). Thus, despite the high plasma and tissue drug concentrations, 20 mg/kg TBI-166 did not exhibit early bactericidal activity. Mice treated with 40 mg/kg TBI-166 exhibited only a 0.4-log10 reduction in lung CFU relative to the untreated controls. From weeks 4 to 8 of treatment, bactericidal activity in all mice receiving TBI-166 at 10, 20, 40, and 80 mg/kg was significant compared to that in untreated mice (P < 0.001) (Fig. 4). After 4 weeks of treatment, a 1.7- to 2.2-log10 reduction in CFU was observed in 10- to 80-mg/kg dose of TBI-166-treated mice compared to untreated mice, and there was no significant difference compared to CFZ at a dose of 20 mg/kg. In mice treated for 8 weeks with any dose of TBI-166, the lung CFU continued to decline. Although TBI-166 at 10 and 20 mg/kg had somewhat lower activity than did the dose of 20 mg/kg CFZ, there was no significant difference in activity between 40 and 80 mg/kg TBI-166 and CFZ at 20 mg/kg. No toxicity as assessed by clinical observations and monitoring of body weight was observed during any of these efficacy studies (data not shown).
FIG 4.

Dose-response and kill kinetics of TBI-166 in the chronic mouse infection model. Eight weeks of treatment with vehicle (CMC), 10 mg/kg RIF, 20 mg/kg CFZ, or 10 to 80 mg/kg TBI-166; treatment was initiated on day 28 postinfection. Dosages are in milligrams per kilogram of body weight. All mice were sacrificed prior to treatment or after 2, 4, and 8 weeks of treatment.
Discoloration.
TBI-166 and CFZ-induced discoloration was observed in ears (Fig. 5; see also Fig. S1 in the supplemental material) and fat (Fig. 6 and S2) of mice during 2, 4, and 8 weeks of treatment. The discoloration of TBI-166 occurred in a dose-dependent manner at 10, 20, 40, and 80 mg/kg. For example, after 4 weeks of daily TBI-166, the coloration of adipose tissue ranged from normal to orange as the dose increased from 10 to 80 mg/kg (Fig. 6). Importantly, even 80 mg/kg of TBI-166 treatment produced less or equal discoloration of adipose tissue than did CFZ at 20 mg/kg at each time point. Similarly, TBI-166 reduced pigmentation in ears compared to CFZ.
FIG 5.

Riminophenazine-induced discoloration of ears in the chronic mouse infection model after 4 weeks of treatment. Ear discoloration of mice after 2 and 8 weeks of administration are presented in Fig. S2 in the supplemental material.
FIG 6.

Riminophenazine-induced discoloration of subcutaneous adipose tissue in the chronic infection model after 4 weeks of treatment. Fat discoloration of the mice after 2 and 8 weeks of administration is presented in Fig. S1.
Distribution and accumulation of TBI-166 in tissues.
TBI-166 possessed a shorter plasma half-life (about 45 h [17]) than did CFZ (about 65 h; our unpublished data) in a single-dose pharmacokinetics study. As tissue accumulation and slow elimination of riminophenazines can cause discoloration, we performed a parallel experiment in uninfected mice after 2, 4, and 8 weeks of TBI-166 or CFZ administration to assess the accumulation. The mouse strain, formulation, and dosing route were chosen to match those employed in the mouse efficacy studies. Compared with the plasma concentration before dose, the increased concentration of TBI-166 3 h after dosing was larger than that of CFZ (Fig. 7), suggesting that TBI-166 is eliminated faster than is CFZ. Over 2 to 8 weeks of administration, the plasma TBI-166 concentration in mice receiving 20 mg/kg reached steady state, which is well above the MIC for M. tuberculosis. Although the TBI-166 concentrations in the fat, skin, lungs, liver, and spleen were higher than concentrations after the same dose of CFZ after 2, 4, and 8 weeks of administration (Table 3), TBI-166-treated mice had reduced discoloration of adipose tissue and skin. Among the tested tissues, concentrations were highest in fat tissue and skin compared to other tissues in mice receiving TBI-166 as well as CFZ.
FIG 7.

Plasma concentrations before dose and 3 h postdose after repeated dosing.
TABLE 3.
Distribution and accumulation of 20 mg/kg TBI-166 and CFZ in uninfected mice after 2, 4, and 8 weeks of exposure
| Drug | Infection site | Concn (μg/g) by exposure time |
||
|---|---|---|---|---|
| 2 wk | 4 wk | 8 wk | ||
| TBI-166 | Lung | 27.12 ± 6.15 | 28.91 ± 12.33 | 44.28 ± 12.62 |
| CFZ | 7.92 ± 7.10 | 31.87 ± 2.82 | 32.88 ± 10.28 | |
| TBI-166 | Liver | 32.29 ± 9.68 | 53.60 ± 42.66 | 108.17 ± 46.93 |
| CFZ | 2.01 ± 0.92 | 4.94 ± 2.02 | 12.75 ± 0.87 | |
| TBI-166 | Spleen | 27.98 ± 11.17 | 16.83 ± 15.39 | 36.54 ± 19.52 |
| CFZ | 13.18 ± 3.35 | 33.30 ± 0.97 | 36.47 ± 4.71 | |
| TBI-166 | Fat | 576.45 ± 307.52 | 1,336.34 ± 503.18 | |
| CFZ | 180.07 ± 88.66 | 253.27 ± 28.50 | ||
| TBI-166 | Skin | 103.51 ± 71.50 | 169.23 ± 15.91 | |
| CFZ | 47.78 ± 19.67 | 41.57 ± 13.88 | ||
DISCUSSION
The riminophenazine CFZ has shown promise for the shortening of TB treatment in both humans (4) and in mice (7) and is now included in the WHO-recommended shorter regimen for the treatment of MDR-TB (6). However, animal and clinical studies indicate that discoloration occurs in many tissues, especially skin, fat, and organs of the mononuclear phagocyte system by intracellular accumulation and crystallization of this bright red compound due to its extremely high lipophilicity and long half-life (16, 26, 27). Several groups have aimed to discover riminophenazine analogues with activity equal to or better than that of CFZ and with reduced side effects, but development has not progressed beyond preliminary in vivo efficacy (28–31). Previously, we reported 12 riminophenazine analogues with lower MIC and superior activity at 20 mg/kg in an acute mouse infection model compared to CFZ. Herein, we reported the efficacy and pigmentation profiles of the most promising riminophenazine, TBI-166.
The potency advantage of TBI-166 compared to that of CFZ has been seen against M. tuberculosis H37Rv and clinical isolates with different drug susceptibility profiles. CFZ/BDQ-resistant mutants harboring mutations in rv0678 were partly resistant to TBI-166, which needs further investigation. The in vitro antimicrobial profile of TBI-166 indicates a low frequency of resistance development and activity against M. kansasii that is superior to that of CFZ. M. kansasii is the third most common cause of pulmonary mycobacterial disease with mostly indistinguishable clinical syndromes from that of M. tuberculosis (32). The 10-fold lower MIC of TBI-166 than that of CFZ suggests the potential clinical use of TBI-166 on M. kansasii. CFZ accumulates within cells of the mononuclear phagocyte system (MPS) and kills intracellular bacteria (17, 33). Here, TBI-166 demonstrated activity against M. tuberculosis replicating inside macrophages similar to that of CFZ.
A key finding of this study is that the in vivo efficacy of TBI-166 is equivalent to that of CFZ at the same dose with significantly reduced skin discoloration. TBI-166 reduced pigmentation in the skin compared to CFZ at all time points despite achieving higher concentrations in tested tissues. Unexpectedly, TBI-166, with lower lipophilicity (logP = 4.52) and shorter half-life (t1/2, 41.25 h) compared to CFZ (logP = 5.34; t1/2, 65.38 h), accumulated more in tissues. This may be attributed to the unique physicochemical properties. The distribution of TBI-166 in tissues showed that fat tissue and skin accumulated most among fat, skin, lungs, liver, and spleen, which is similar to that of CFZ. CFZ accumulates in high levels in tissues, has a long half-life, and retains antimicrobial activity after treatment is stopped (34). The duration of sustained antimicrobial activity is associated with the time that serum CFZ levels remained at or above the MIC for M. tuberculosis (34). Likewise, the tissue accumulation of TBI-166 could potentially be exploited to provide antimicrobial activity after treatment cessation. Like CFZ, TBI-166 shows primarily bacteriostatic rather than bactericidal activity in the acute infection model, with dose-dependent activity at doses ranging from 10 to 80 mg/kg. The activity of the 20-mg/kg TBI-166 tablet is similar to those of the TBI-166 powder and CFZ powder at equivalent doses, implying that TBI-166 could be used in the future in tablet formulation to treat TB patients. Because tablets have a lower cost than capsules, the application of TBI-166 in tablet form may help decrease the financial burden of TB treatment.
TBI-166 exhibits delayed antimicrobial activity in the chronic infection model, similar to that of CFZ. Although the plasma TBI-166 concentration in uninfected mice receiving 20 mg/kg approached steady-state levels well above the 0.063 μg/ml MIC for M. tuberculosis, TBI-166 lacked early bactericidal activity (EBA) during the first 2 weeks. Such an observation is similar to observations with CFZ and may be related to its mechanism of action, which needs further investigation. It could due to involvement of the respiratory chain of M. tuberculosis similar to CFZ and is determined by factors such as the time needed to disrupt the proton motive force and/or deplete energy stores before antimicrobial activity is observed (11, 35). In addition to reducing skin discoloration compared to CFZ, TBI-166 had higher maximum concentration of drug in serum (Cmax) and area under the concentration-time curve (AUC) (17), and it possessed a shorter plasma half-life than CFZ, as well as good safety.
In future studies, a comparison of the efficacy of TBI-166 and CFZ against TB in other mouse models, such as C3HeB/FeJ mice, and with other infecting M. tuberculosis strains will be important to corroborate our findings. Irwin et al. reported that CFZ alone exhibited decreased activity against M. tuberculosis in the lungs of C3HeB/FeJ mice compared with that in the lungs of BALB/c mice (9). The limited activity was associated with the presence of hypoxic pulmonary granulomas with caseous necrosis in the C3HeB/FeJ mice, whereas BALB/c mice form cellular inflammatory lesions harboring largely intracellular bacilli (9). It may also be related to near-neutral pH of caseum, as CFZ has greater activity at lower pH (36).
CFZ not only contributes substantially in second-line regimens (37) but also shortens the duration of the first-line regimen for TB (7). Thus, it is important to investigate the activity of TBI-166 substituted for CFZ in these treatment-shortening regimens to reduce side effects, such as discoloration. The evaluation of TBI-166-containing regimens is ongoing and will be reported upon its completion. TBI-166 has been approved by the Chinese FDA for clinical trial application in November 2016 and advanced to a phase 1 trial in January 2018. Overall, the data presently suggest that TBI-166 has the potential to be a next-generation riminophenazine for TB and warrant further studies.
MATERIALS AND METHODS
Compounds.
Isoniazid (INH), rifampin (RIF), streptomycin (STR), levofloxacin (LVX), and ethambutol (EMB) were purchased from Sigma. CFZ and CFZ capsules (50 mg, micronized CFZ suspended in an oil-wax base) were provided by Nanjing Liye (China). TBI-166 powder (purity, 99.56%) and TBI-166 tablets (100 mg) were synthesized by the Institute of Materia Medica.
Strains.
Forty-eight clinical M. tuberculosis isolates and nontuberculous mycobacteria (NTM) were obtained from the National Clinical Laboratory on Tuberculosis, Beijing Chest Hospital. Among clinical M. tuberculosis isolates, 16 strains were fully drug susceptible and 28 were drug resistant, as determined using the microplate alamarBlue assay (MABA) (17, 38). Drug resistance was defined as resistance to any of the drugs INH, RIF, STR, LVX, or EMB, using 1, 1, 2, 1, and 5 μg/ml, respectively, as the critical concentrations. The M. tuberculosis and nontuberculous mycobacterial strains were grown in Middlebrook 7H9 broth (Difco, USA) supplemented with 0.2% (vol/vol) glycerol, 0.05% Tween 80, and 10% (vol/vol) oleic acid-albumin-dextrose-catalase (OADC; Becton, Dickinson, USA). Rv0678 mutants (BDQ-1, BDQ-2, and BDQ-3) were isolated from M. tuberculosis H37Rv on selective 7H11 medium containing bedaquiline at 0.125 μg/ml. They are resistant to CFZ and bedaquiline.
MIC determinations.
TBI-166 and CFZ MICs were determined by MABA, using 2-fold dilutions ranging from 0.4 to 0.00625 μg/ml and 0.96 to 0.015 μg/ml, respectively (17, 39). Briefly, bacteria (100 μl containing 2 × 105 CFU) were added to wells, yielding a final testing volume of 200 μl. The plates were incubated at 37°C; on day 7 of incubation, 12.5 μl of 20% Tween 80 and 20 μl of alamarBlue were added to all wells. After incubation at 37°C for another 24 h, the fluorescence was measured at an excitation wavelength of 530 nm and an emission wavelength of 590 nm. The MIC was defined as the lowest concentration eliciting a reduction in fluorescence of ≥90% relative to the mean fluorescence of replicate drug-free controls. M. tuberculosis H37Rv was used as a drug-susceptible control.
Measurement of the frequency of spontaneous resistance.
A 20-ml starting culture was prepared by inoculating wild-type M. tuberculosis H37Rv from a single colony to minimize the presence of spontaneous mutants. The cultures were incubated in a shaking incubator at 37°C for 2 weeks, and bacterial growth reached approximately 108 CFU/ml. A total of 108 bacteria were plated onto selective Middlebrook 7H11 agar (with OADC and glycerol) containing 0.25 μg/ml (4× the MIC) TBI-166. The number of total CFU was estimated by making serial dilutions and plating onto nonselective Middlebrook 7H11 agar (with OADC and glycerol). After 4 weeks of incubation, the colonies were counted, and the frequency of spontaneous resistance was calculated.
Intracellular activity assay.
J774A.1 cells were grown to confluence in 75-cm2 cell culture flasks in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), as described previously (17). The cells were detached with trypsin digestion and centrifuged at 800 × g for 5 min at room temperature, and the pellet was resuspended to a final concentration of 4 × 105 cells/ml. Aliquots (1 ml) of cell suspension were distributed into 24-well plates, and the plates were incubated at 37°C in a 5% CO2 incubator for 16 h. Mid-log-growth-phase M. tuberculosis H37Rv cultures were passed through an 8 μm-pore-size filter to remove clumps and diluted to infect macrophages at a multiplicity of infection (MOI) of 5 bacteria per cell. Infection was carried out for 4 h, followed by three washes with DMEM without FBS to remove the extracellular mycobacteria. The contents of the wells were replaced with DMEM containing 10% FBS and different concentrations of the compounds (1.0, 0.25, and 0.0625 μg/ml). The control wells received drug-free medium. After 3 days of incubation, monolayers were visually inspected under the microscope to ensure that they remained intact, and then the medium was removed and the macrophages were lysed with 200 μl of 0.1% sodium dodecyl sulfate. Then, the lysates were diluted with fresh medium and plated onto 7H11 plates supplemented with 10% OADC to enumerate the CFU (40).
Acute toxicity study.
TBI-166 was administered as a single dose to BALB/c mice (female) weighing 18 to 21 g, with 10 mice in each dose group. The number of mice which survived after an oral administration of a single dose of compounds at 500, 1,000, 1,500, 2,000, and 3,000 mg/kg, followed by a 7-day observation, was recorded.
Aerosol infection with M. tuberculosis.
Specific-pathogen-free (SPF) BALB/c male mice (18 to 20 g) were infected via aerosol with M. tuberculosis H37Rv using a Glas-Col inhalation system. The acute infection model used aerosol infection with a suspension of 5 × 106 CFU/ml to deposit 50 to 100 bacilli into the lungs of each mouse, as previously described (17). The chronic infection model used aerosol infection with a suspension of 2 × 106 CFU/ml to deposit 20 to 50 bacilli into the lungs. Five mice (acute and chronic model) were humanely killed 3 days after infection and on the day of treatment initiation (D0) to determine the number of bacteria implanted in the lungs and at the start of treatment, respectively.
Chemotherapy.
Treatment was initiated 10 days after infection in the acute model and 28 days after infection in chronic model. The drugs and compounds were prepared weekly by suspension in 0.5% (wt/vol) carboxymethylcellulose (CMC), except for capsule CFZ, and administered once daily by gavage in a 200-μl suspension. Mice were dosed for 5 consecutive days each week. The scheme of the acute model experiment is shown in Table S1, and the scheme of the chronic model experiment is shown in Table S2.
In the acute model, the vehicle control mice received an equal volume of 0.5% CMC, and the positive-control mice received INH (25 mg/kg), RIF (10 mg/kg), or CFZ (20 mg/kg). Capsule CFZ (20 mg/kg) was dissolved in soybean oil and used as a positive control as the formulation of CFZ in clinical treatment. The test mice received TBI-166 alone (10, 20, 40, or 80 mg/kg) or the TBI-166 tablet (20 or 80 mg/kg) as the intended clinical formulation. The TBI-166 tablet was crushed and suspended in 0.5% CMC. Treatment was administered for up to 3 weeks.
In the chronic model, the vehicle control mice received CMC, and the positive-control mice received RIF (10 mg/kg) or CFZ (20 mg/kg). The test mice received TBI-166 at 10, 20, 40, or 80 mg/kg. The treatment durations were 2, 4, and 8 weeks.
Assessment of treatment efficacy.
Efficacy was assessed on the basis of the lung CFU counts. Ten mice per treatment group in the acute model and 8 mice per treatment group in the chronic model were sacrificed at each time point. Lungs were removed, homogenized, and serially diluted in 10-fold steps in phosphate-buffered saline (PBS). Portions (100 μl) were spread onto 0.4% charcoal-containing selective 7H11 plates in duplicate to reduce carryover effects (41). The plates were incubated for up to 6 weeks at 37°C before final CFU counts were determined.
Tissue distribution after repeated exposure.
In the multiple-dose experiment, uninfected BALB/c male mice received TBI-166 or CFZ at 20 mg/kg in 0.5% CMC daily (5 days/week) for up to 8 weeks. Groups of 3 mice were sacrificed immediately before the Friday dose (expected trough) and 3 h after Friday’s dose (expected peak) after 2, 4, and 8 weeks of administration. Plasma, lung, spleen, liver, subcutaneous fat, and skin samples were selected to measure drug concentrations. Skin was taken from the back of the mice after shaving off their hair. Drug concentrations in samples were determined by high-performance liquid chromatography (HPLC), as previously described (17).
Statistical analysis.
Lung CFU counts were log transformed before analysis, and the mean CFU counts were compared by one-way analysis of variance with Dunnett’s posttest to control for multiple comparisons. The Mann-Whitney test was used to test for significance on nonnormally distributed CFU data. All analyses were performed with GraphPad Prism version 5 (GraphPad, San Diego, CA). A P value of 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Chinese Academy of Medical Sciences & Peking Union Medical College for providing the drugs used in this study. We thank Eric L. Nuermberger for reviewing the manuscript and giving us suggestions.
This study was supported by National Science and Technology Project of China (grant 2015ZX09102007-015) and the Global Alliance for TB Drug Development and National Natural Science Foundation of China (grant 81803588).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02155-18.
REFERENCES
- 1.World Health Organization. 2017. Global tuberculosis report 2017. World Health Organization, Geneva, Switzerland: https://www.who.int/tb/publications/global_report/gtbr2017_main_text.pdf. [Google Scholar]
- 2.Tiberi S, Du Plessis N, Walzl G, Vjecha MJ, Rao M, Ntoumi F, Mfinanga S, Kapata N, Mwaba P, McHugh TD, Ippolito G, Migliori GB, Maeurer MJ, Zumla A. 2018. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect Dis 18:E183–E198. doi: 10.1016/s1473-3099(18)30110-5. [DOI] [PubMed] [Google Scholar]
- 3.Sharma D, Dhuriya YK, Deo N, Bisht D. 2017. Repurposing and revival of the drugs: a new approach to combat the drug resistant tuberculosis. Front Microbiol 8:2452. doi: 10.3389/fmicb.2017.02452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Deun A, Maug AK, Salim MA, Das PK, Sarker MR, Daru P, Rieder HL. 2010. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med 182:684–692. doi: 10.1164/rccm.201001-0077OC. [DOI] [PubMed] [Google Scholar]
- 5.Cholo MC, Mothiba MT, Fourie B, Anderson R. 2017. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J Antimicrob Chemother 72:338–353. doi: 10.1093/jac/dkw426. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. 2016. WHO treatment guidelines for drug-resistant tuberculosis (2016 update). World Health Organization, Geneva, Switzerland: http://www.who.int/tb/areas-of-work/drug-resistant-tb/treatment/resources/en/. [Google Scholar]
- 7.Tyagi S, Ammerman NC, Li SY, Adamson J, Converse PJ, Swanson RV, Almeida DV, Grosset JH. 2015. Clofazimine shortens the duration of the first-line treatment regimen for experimental chemotherapy of tuberculosis. Proc Natl Acad Sci U S A 112:869–874. doi: 10.1073/pnas.1416951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu J, Lu Y, Fu L, Zhu H, Wang B, Mdluli K, Upton AM, Jin H, Zheng M, Zhao W, Li P. 2012. In vitro and in vivo activity of clofazimine against Mycobacterium tuberculosis persisters. Int J Tuber Lung Dis 16:1119–1125. doi: 10.5588/ijtld.11.0752. [DOI] [PubMed] [Google Scholar]
- 9.Irwin SM, Gruppo V, Brooks E, Gilliland J, Scherman M, Reichlen MJ, Leistikow R, Kramnik I, Nuermberger EL, Voskuil MI, Lenaerts AJ. 2014. Limited activity of clofazimine as a single drug in a mouse model of tuberculosis exhibiting caseous necrotic granulomas. Antimicrob Agents Chemother 58:4026–4034. doi: 10.1128/AAC.02565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. 2007. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yano T, Kassovska-Bratinova S, Teh JS, Winkler J, Sullivan K, Isaacs A, Schechter NM, Rubin H. 2011. Reduction of clofazimine by mycobacterial type 2 NADH: quinone oxidoreductase a pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem 286:10276–10287. doi: 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim SA, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SP, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, et al. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 13.Lamprecht DA, Finin PM, Rahman MA, Cumming BM, Russell SL, Jonnala SR, Adamson JH, Steyn AJ. 2016. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat Commun 7:12393. doi: 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bald D, Villellas C, Lu P, Koul A. 2017. Targeting energy metabolism in mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 8:e00272-17. doi: 10.1128/mBio.00272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Job CK, Yoder L, Jacobson RR, Hastings RC. 1990. Skin pigmentation from clofazimine therapy in leprosy patients: a reappraisal. J Am Acad Dermatol 23:236–241. doi: 10.1016/0190-9622(90)70204-U. [DOI] [PubMed] [Google Scholar]
- 16.Swanson RV, Adamson J, Moodley C, Ngcobo B, Ammerman NC, Dorasamy A, Moodley S, Mgaga Z, Tapley A, Bester LA, Singh S, Grosset JH, Almeida DV. 2015. Pharmacokinetics and pharmacodynamics of clofazimine in the mouse model of tuberculosis. Antimicrob Agents Chemother doi: 10.1128/aac.00260-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y, Zheng M, Wang B, Fu L, Zhao W, Li P, Xu J, Zhu H, Jin H, Yin D, Huang H, Upton AM, Ma Z. 2011. Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob Agents Chemother 55:5185–5193. doi: 10.1128/AAC.00699-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang S, Yao L, Hao X, Liu Y, Zeng L, Liu G, Li M, Li F, Wu M, Zhu Y, Sun H, Gu J, Wang X, Zhang Z. 2015. Clofazimine for the treatment of multidrug-resistant tuberculosis: prospective, multicenter, randomized controlled study in China. Clin Infect Dis 60:1361–1367. doi: 10.1093/cid/civ027. [DOI] [PubMed] [Google Scholar]
- 19.Liu B, Liu K, Lu Y, Zhang D, Yang T, Li X, Ma C, Zheng M, Wang B, Zhang G, Wang F, Ma Z, Li C, Huang H, Yin D. 2012. Systematic evaluation of structure-activity relationships of the riminophenazine class and discovery of a C2 pyridylamino series for the treatment of multidrug-resistant tuberculosis. Molecules 17:4545–4559. doi: 10.3390/molecules17044545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D, Lu Y, Liu K, Liu B, Wang J, Zhang G, Zhang H, Liu Y, Wang B, Zheng M, Fu L, Hou Y, Gong N, Lv Y, Li C, Cooper CB, Upton AM, Yin D, Ma Z, Huang H. 2012. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant tuberculosis. J Med Chem 55:8409–8417. doi: 10.1021/jm300828h. [DOI] [PubMed] [Google Scholar]
- 21.Zhang D, Liu Y, Zhang C, Zhang H, Wang B, Xu J, Fu L, Yin D, Cooper CB, Ma Z, Lu Y, Huang H. 2014. Synthesis and biological evaluation of novel 2-methoxypyridylamino-substituted riminophenazine derivatives as antituberculosis agents. Molecules 19:4380–4394. doi: 10.3390/molecules19044380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang S, Chen J, Cui P, Shi W, Zhang W, Zhang Y. 2015. Identification of novel mutations associated with clofazimine resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 70:2507–2510. doi: 10.1093/jac/dkv150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartkoorn RC, Uplekar S, Cole ST. 2014. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2979–2981. doi: 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yawalkar SJ, Vischer W. 1979. Lamprene (clofazimine) in leprosy. Basic information. Lepr Rev 50:135–144. [DOI] [PubMed] [Google Scholar]
- 25.Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. 2011. Clofazimine: current status and future prospects. J Antimicrobial Chemother 67:290–298. doi: 10.1093/jac/dkr444. [DOI] [PubMed] [Google Scholar]
- 26.Mansfield RE. 1974. Tissue concentrations of clofazimine (B663) in man. Am J Trop Med Hyg 23:1116–1119. doi: 10.4269/ajtmh.1974.23.1116. [DOI] [PubMed] [Google Scholar]
- 27.Venkatesan K, Deo N, Gupta UD. 2007. Tissue distribution and deposition of clofazimine in mice following oral administration with or without isoniazid. Arzneimittelforschung 57:472–474. doi: 10.1055/s-0031-1296634. [DOI] [PubMed] [Google Scholar]
- 28.O’Sullivan JF, Conalty ML, Morrison NE. 1988. Clofazimine analogues active against a clofazimine-resistant organism. J Med Chem 31:567–572. doi: 10.1021/jm00398a013. [DOI] [PubMed] [Google Scholar]
- 29.Jagannath C, Reddy MV, Kailasam S, O’Sullivan JF, Gangadharam PR. 1995. Chemotherapeutic activity of clofazimine and its analogues against Mycobacterium tuberculosis. In vitro, intracellular, and in vivo studies. Am J Respir Crit Care Med 151:1083–1086. doi: 10.1164/ajrccm.151.4.7697235. [DOI] [PubMed] [Google Scholar]
- 30.Reddy VM, Nadadhur G, Daneluzzi D, O’Sullivan JF, Gangadharam PR. 1996. Antituberculosis activities of clofazimine and its new analogs B4154 and B4157. Antimicrob Agents Chemother 40:633–636. doi: 10.1128/AAC.40.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Rensburg CEJ, Jooné GK, Sirgel FA, Matlola NM, O’Sullivan JF. 2000. In vitro investigation of the antimicrobial activities of novel tetramethylpiperidine-substituted phenazines against Mycobacterium tuberculosis. Chemotherapy 46:43–48. doi: 10.1159/000007255. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava S, Gumbo T. 2018. Clofazimine for the treatment of Mycobacterium kansasii. Antimicrob Agents Chemother 62:e00248-18. doi: 10.1128/AAC.00248-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barry VC, Belton JG, Conalty ML, Denneny JM, Edward DW, O’Sullivan JF, Twomey D, Winder F. 1957. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 179:1013–1015. doi: 10.1038/1791013a0. [DOI] [PubMed] [Google Scholar]
- 34.Swanson RV, Ammerman NC, Ngcobo B, Adamson J, Moodley C, Dorasamy A, Moodley S, Mgaga Z, Bester LA, Singh SD, Almeida DV, Grosset JH. 2016. Clofazimine contributes sustained antimicrobial activity after treatment cessation in a mouse model of tuberculosis chemotherapy. Antimicrob Agents Chemother 60:2864–2869. doi: 10.1128/AAC.00177-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ammerman NC, Swanson RV, Tapley A, Moodley C, Ngcobo B, Adamson J, Dorasamy A, Moodley S, Mgaga Z, Bester LA, Singh SD, Almeida DV, Grosset JH. 2017. Clofazimine has delayed antimicrobial activity against Mycobacterium tuberculosis both in vitro and in vivo. J Antimicrob Chemother 72:455–461. doi: 10.1093/jac/dkw417. [DOI] [PubMed] [Google Scholar]
- 36.Lanoix JP, Ioerger T, Ormond A, Kaya F, Sacchettini J, Dartois V, Nuermberger E. 2016. Selective inactivity of pyrazinamide against tuberculosis in C3HeB/FeJ mice is best explained by neutral pH of caseum. Antimicrob Agents Chemother 60:735–743. doi: 10.1128/AAC.01370-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grosset JH, Tyagi S, Almeida DV, Converse PJ, Li SY, Ammerman NC, Bishai WR, Enarson D, Trebucq A. 2013. Assessment of clofazimine activity in a second-line regimen for tuberculosis in mice. Am J Respir Crit Care Med 188:608–612. doi: 10.1164/rccm.201304-0753OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins L, Franzblau SG. 1997. Microplate alamarBlue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother 41:1004–1009. doi: 10.1128/AAC.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Wang B, Hu M, Huo F, Guo S, Jing W, Nuermberger E, Lu Y. 2017. Primary clofazimine and bedaquiline resistance among isolates from patients with multidrug-resistant tuberculosis. Antimicrob Agents Chemother 61:e00239-17. doi: 10.1128/AAC.00239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, Wang J-x, Zhou J-m, Xu C-l, Huang B, Xing Y, Wang B, Luo R, Wang Y-c, You X-f, Lu Y, Yu L-y. 2017. A novel protein kinase inhibitor IMB-YH-8 with anti-tuberculosis activity. Sci Rep 7:5093. doi: 10.1038/s41598-017-04108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tasneen R, Li SY, Peloquin CA, Taylor D, Williams KN, Andries K, Mdluli KE, Nuermberger EL. 2011. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 55:5485–5492. doi: 10.1128/AAC.05293-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.