

Fungal infections pose a substantial threat to the human population. They can cause either mild and relatively harmless infections or invasive and often lethal diseases in patients with a weakened immune system.
KEYWORDS: Candida albicans, Candida glabrata, Saccharomyces cerevisiae, antifungal drug resistance, antifungal drug tolerance, fluconazole, vesicle transport
ABSTRACT
Fungal infections pose a substantial threat to the human population. They can cause either mild and relatively harmless infections or invasive and often lethal diseases in patients with a weakened immune system. The majority of these human fungal infections are caused by Candida species. The limited amount of available therapies, together with the development of resistance against these drugs, strongly emphasizes the need for novel therapeutic strategies. As it is quite time-consuming to introduce completely new drugs to the market, potentiating the efficacy of existing drugs would be a better strategy. Therefore, it is important to identify cellular pathways involved in the development of drug resistance. We found that vesicular transport is involved in fungal susceptibility to the most widely used antifungal drug, fluconazole. We identified specific complexes in the vesicular transport pathway which contribute to fluconazole resistance or tolerance in the model organism Saccharomyces cerevisiae. Furthermore, we confirmed our findings in the clinically relevant fungi Candida albicans and Candida glabrata. Finally, we show that the combination of fluconazole with a specific inhibitor of the vesicular transport pathway increases the susceptibility of Candida species, indicating the potential of using vesicular transport as a target in combination therapy.
INTRODUCTION
There are over 600 different fungal pathogens able to cause either minor or invasive infections in humans (1, 2). The majority of human fungal infections are caused by species of the Candida genus, including Candida albicans and Candida glabrata (3). In most healthy individuals, they are both harmless commensals of the oral cavity, gastrointestinal tract, and/or urogenital tract. However, under conditions involving a reduced immune competence or an imbalance of the microflora, they are able to cause infections (2, 4). Superficial infections are relatively harmless and affect approximately 25% of the worldwide population (5). However, the incidence of severe invasive fungal infections is still rising due to the increase in the number of immunocompromised patients (1). The lack of effective antifungal drugs and the emergence of fungal resistance against these drugs pose a significant threat to human health (6).
The most commonly used antifungal drugs are azoles (7). These fungistatic drugs, with the most studied member being fluconazole, target the ergosterol biosynthesis pathway. Fluconazole binds to the active site of the lanosterol 14α-demethylase enzyme (Erg11), thereby inhibiting the conversion of lanosterol into ergosterol, which is an essential component of the fungal cell membrane (8, 9). However, long-term exposure of fungal cells to antifungal drugs has different consequences. The cells can develop resistance mechanisms to overcome the inhibitory actions of the drug. Known mechanisms of resistance against azoles include alteration of the target enzyme, overexpression of the gene encoding this target, and increased drug efflux (10, 11). Resistance mechanisms allow the cells to grow at higher drug concentrations than susceptible cells. Alternatively, cells can develop tolerance to the drug to allow trailing growth at inhibitory concentrations (12). This phenomenon is alternatively referred to as FoG, for fraction of growth (13). The development of new antifungal drugs is important to overcome antifungal resistance. Alternatively, therapeutic strategies can be improved by enhancing the efficacy of existing drugs, such as fluconazole. Therefore, it is important to identify the genes and cellular pathways involved in the development of drug resistance or tolerance. Combination therapy has shown great potential to exert synergism and reduce resistance (14). However, there is still a lack of knowledge concerning the pharmacodynamics, pharmacokinetics, and clinical success of drug combinations (15, 16).
In this study, we used the baker’s yeast Saccharomyces cerevisiae as a model organism to examine the role of the vesicular transport pathway in fluconazole susceptibility. We screened a library of S. cerevisiae deletion strains for decreased growth on fluconazole compared to the wild-type strain and found that impairment of vesicular transport increases susceptibility to this drug. The highly regulated vesicular transport pathway is involved in transporting cargo, such as lipids and proteins, between membrane-bound compartments (17). Proteins are transported from the endoplasmic reticulum (ER) to the Golgi apparatus via vesicles during biosynthesis. Afterwards, proteins are targeted to the plasma membrane, to the vacuole via endosomes, or to the external medium. In the reverse process, proteins can be internalized by endocytosis and transported to the endosomes. Here, they are targeted to the vacuole for degradation or to the Golgi apparatus for recycling (18). Vesicular transport and the correct targeting of vesicles are highly controlled by different proteins at the fusion sites. Soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) complexes mediate the fusion of cargo-loaded vesicles to acceptor compartments. The vesicles from donor compartments are coated with tethering complexes, which regulate the specific targeting to the acceptor compartment (19).
Although a link between fluconazole sensitivity and vesicular transport was already suggested previously (20–23), in this project, we identified the involvement of particular complexes in this process. We selected six susceptible S. cerevisiae deletion strains which were affected in vesicular trafficking: the S. cerevisiae pep3Δ (Scpep3Δ), Scpep5Δ, Scpep7Δ, Scvps45Δ, Scvps51Δ, and Scvps54Δ strains. The ScPep3 and ScPep5 proteins are members of the endosomal tethering complex called the class C core vacuole/endosome tethering (CORVET) complex. The CORVET complex is a multimeric protein complex which is required for endocytosis and for the attachment of vesicles to and from the endosome. In addition, they are also members of the homotypic fusion and vacuole protein sorting (HOPS) tethering complex, which is involved in the fusion of vesicles to the vacuole (24). The ScVps51 and ScVps54 proteins are both members of the Golgi-associated retrograde protein (GARP) complex, which is involved in retrograde trafficking of vesicles from the endosomes to the Golgi apparatus (25). Finally, the ScPep7 and ScVps45 proteins physically interact with each other and are important for vesicle fusion at the endosome (26). For all six deletion strains, we confirmed increased susceptibility by analysis of the fluconazole (FLC) MIC (MICFLC) and showed a concomitant decrease in retained ergosterol after treatment. In addition, we could confirm the involvement of vesicular transport in fluconazole efficacy against C. albicans and C. glabrata. Finally, we tested the effect of sorting inhibitors (sortins), which inhibit vesicular transport (27–30), on fluconazole efficacy against several C. glabrata and C. albicans isolates and showed a synergistic interaction with sortin2.
RESULTS
Interference in vesicular transport increases susceptibility to fluconazole in S. cerevisiae.
In order to identify the regulatory processes involved in susceptibility to fluconazole, we screened the S. cerevisiae haploid MATα deletion collection for reduced growth in the presence of fluconazole (10 or 20 μg/ml). As described in earlier publications, we added the iron chelator doxycycline to the minimal testing medium (50 or 100 μg/ml) to decrease the background growth or tolerance at supra-MICs of the drug and increase the sensitivity of the screening system (31, 32). Strains with enhanced fluconazole susceptibility, being the deletion strains that showed increased susceptibility to the drug, were selected by visually monitoring reduced growth after 3 and 4 days of incubation at 30°C compared to the growth of the BY4742 wild-type laboratory strain (see Table S1 in the supplemental material). The genes deleted in all enhancer strains were clustered, using the online tool FunSpec (33). A list of these genes, classified by gene ontology (GO) annotation, is displayed in Table S2. Using a P value cutoff of 0.001, processes mainly related to intracellular vesicle transport were identified as biological processes regulating susceptibility to fluconazole, including Golgi apparatus-to-endosome transport (ScPEP7, ScVPS45, ScPEP3, and ScPEP5), vesicle docking involved in exocytosis (ScPEP7, ScVPS45, ScPEP3, and ScPEP5), Golgi apparatus-to-vacuole transport (ScVPS54, ScPEP7, and ScVPS45), and regulation of SNARE complex assembly (ScPEP3 and ScPEP5). Many of the involved proteins were localized to the GARP complex (ScVPS54 and ScVPS51), the HOPS complex (ScPEP3 and ScPEP5), or the CORVET complex (ScPEP3 and ScPEP5). Increasing this P value (P < 0.01) also showed the involvement of other known processes, such as sterol biosynthesis and the general drug response. These processes have been shown to be involved in fluconazole susceptibility by earlier screening setups (22, 34). Parsons et al. performed a similar screening in S. cerevisiae, though they used a different experimental setup (22). They found that the ScPDR16, ScPDR5, and ScERG genes, when deleted, confer susceptibility to this drug, as did we. They also identified a number of genes related to vesicular transport to be of importance in drug susceptibility. This partial overlap endorses the accuracy of our screening method, yet it also shows that differences in results occur due to differential experimental setups.
To verify the involvement of vesicular transport in susceptibility to fluconazole, we selected six deletion strains from the mutant collection, the Scpep3Δ, Scpep5Δ, Scpep7Δ, Scvps45Δ, Scvps51Δ, and Scvps54Δ strains, and created at least three independent transformants of each genotype. These strains were used as biological replicates in the following experiments. Initially, spot assays on fluconazole- and doxycycline-containing media were repeated (Fig. 1), confirming increased susceptibility to drugs.
FIG 1.

Deletion of genes encoding vesicular transport-regulating proteins increases susceptibility to fluconazole in S. cerevisiae. Serial dilutions of six deletion strains and the BY4742 wild-type strain were spotted on SDglu medium containing fluconazole (10 μg/ml) and/or doxycycline (50 μg/ml). Pictures were taken after 72 h of incubation at 30°C. At least three biological repeats were tested, and representative results are shown.
To determine whether the increase in susceptibility of the strains impaired in vesicular transport could be linked to a decrease in the MIC of fluconazole (MICFLC) for these strains, we performed Etest (Fig. S1a) and broth microdilution assays (BDAs) (Fig. S1b). All experiments were performed with at least three independent biological replicates. As can be seen from Table 1, the MICFLC values for all six deletion strains were lower than those for the BY4742 control strain, indicating lower resistance to the drug. Viewing from the Etest assays, the Scvps51Δ, Scvps54Δ, Scpep3Δ, and Scpep5Δ strains showed a decrease in trailing growth or tolerance, defined as the supra-MIC growth inside the halo of growth retardation/inhibition. The Scvps45Δ and Scpep7Δ strains did not show a similar decrease in tolerance. In contrast to the BY4742 and pep7Δ strains, the vps45Δ strain also showed high trailing growth in the broth microdilution assay; however, this method is less suitable to determine trailing growth due to the insensitivity of the optical density (OD) reading.
TABLE 1.
MICFLC values for the analyzed strains determined by Etest and broth microdilution assay analysisa
| Strain | MICFLC (μg/ml) |
||
|---|---|---|---|
| Etest | Broth microdilution assay |
||
| 50% | 90% | ||
| S. cerevisiae pep3Δ | 2–3 | 4 | 4–8 |
| S. cerevisiae pep5Δ | 1–2 | 4 | 4–8 |
| S. cerevisiae pep7Δ | 0.75–1.5 | 2 | 2 |
| S. cerevisiae pep45Δ | 1.5–3 | 0.5–1 | >128 |
| S. cerevisiae pep51Δ | 0.5–0.75 | 4 | 4–8 |
| S. cerevisiae pep54Δ | 0.75–1 | 4 | 4–8 |
| S. cerevisiae BY4742 | 6–8 | 16–32 | 32 |
| C. albicans vps11Δ/Δ | 0.5 | 0.125 | 0.5 |
| C. albicans YJB6284 | 0.25 | 0.125 | 0.25 |
| C. glabrata vps45Δ/Δ | 8 | 4 | 8 |
| C. glabrata ATCC 2001 | 16 | 8 | >128 |
The MICFLC was determined by Etest (after 48 h or 72 h) or broth microdilution assay (after 48 h) of 6 deletion strains of S. cerevisiae (the pep3Δ, pep5Δ, pep7Δ, vps45Δ, vps51Δ, and vps54Δ strains) and the wild-type strain (BY4742), the C. albicans vps11Δ/Δ and C. glabrata vps45Δ/Δ deletion strains, and the respective wild-type strains (YJB6284 and ATCC 2001).
To eliminate the possible influence of the auxotrophies in the BY4742 background on our phenotype, we created the deletion genotypes in the prototrophic S288c strain and repeated the MICFLC analyses. As seen in Fig. S2, the effect of these deletions on growth in the presence of fluconazole remained persistent, indicating that the auxotrophies of the BY4742 reference strain did not interfere in the observed phenotypes.
Interference in vesicular transport lowers the cellular ergosterol content in S. cerevisiae.
Fluconazole targets the lanosterol 14α-demethylase enzyme, encoded by ERG11, which is involved in the biosynthesis of ergosterol. To determine whether vesicular transport affects the level of this sterol in the cell and could thereby link this process to fluconazole susceptibility, we measured the level of ergosterol using an assay described in the literature (35). In a previous report, we showed that this method yields results very comparable to those obtained by gas chromatography-mass spectrometry-based sterol analysis (32), indicating that it is justifiable to use the technically simpler spectrophotometric technique for the analysis of the cellular ergosterol content. Figure 2 shows the ergosterol level in the BY4742 wild-type strain and mutants in the absence and presence of fluconazole. In the absence of fluconazole, the vesicular transport mutants seemed to accumulate slightly, yet significantly, more ergosterol than the wild-type strain. However, when fluconazole was added, the ergosterol levels of these mutants dropped far below the level for the wild-type strain, linking ergosterol levels to the increased susceptibility to fluconazole. Since the level of ergosterol in the mutants was not lowered in the absence of fluconazole, ergosterol synthesis as such is not a target of vesicular transport. We speculate that upon impairment of vesicular transport, fluconazole somehow accumulates in the cell, thereby lowering the residual amount of ergosterol.
FIG 2.
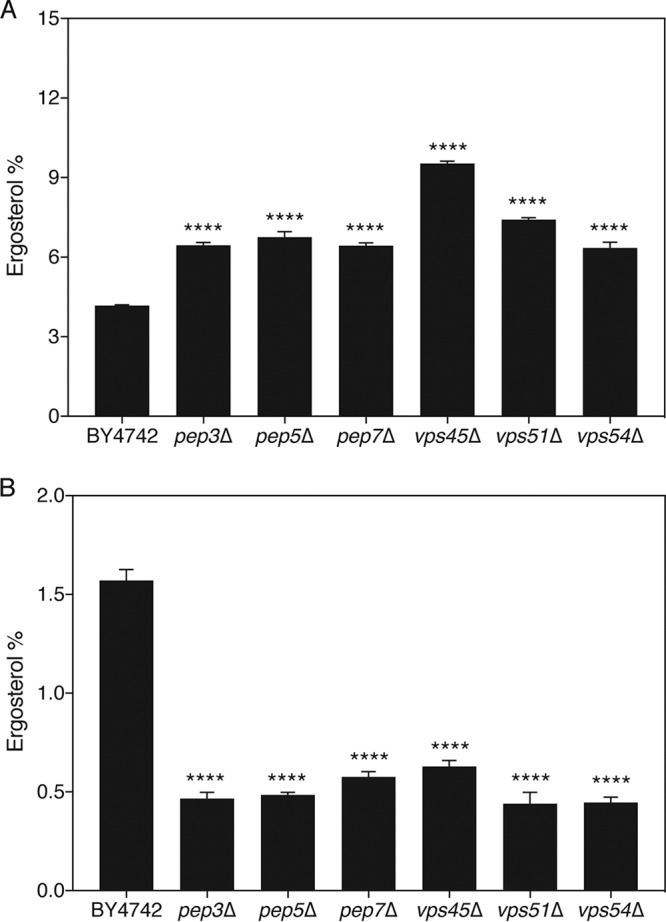
Proper vesicular transport is essential to retain large amounts of ergosterol upon fluconazole treatment in S. cerevisiae. The deletion strains and wild-type strain were grown in SDglu medium for 48 h at 30°C in the absence (A) or the presence (B) of 20 μg/ml fluconazole. Sterols were extracted, and the level of ergosterol was monitored spectrophotometrically. Average levels together with the standard error of the mean (SEM) are displayed. Statistical analysis was conducted by one-way analysis of variance with the Bonferroni correction. ****, P < 0.0001.
Vesicular transport mutants with increased fluconazole susceptibility show lowered expression of genes encoding drug efflux pumps.
We showed that, specifically in the presence of fluconazole, the ergosterol content of the six vesicular transport mutants was lower than that in the wild-type strain. We hypothesized that a relative increase in the intracellular drug concentration compared to that in the wild type would be caused by a lower level of drug efflux. It was reported earlier that deletion of ScVPS3, which encodes a protein involved in transporting proteins to the vacuole as a member of the CORVET complex, lowers drug efflux in S. cerevisiae (36). We postulated that the same would be true for other mutants with mutations in the vesicular transport pathway. We aimed to determine whether vesicular transport affects the expression of genes encoding known drug efflux pumps in S. cerevisiae. We tested expression of the main genes encoding drug efflux pumps, ScPDR5, ScPDR10, ScPDR11, ScPDR15, ScYCF1, ScYOR1, and ScSNQ2, or regulators thereof, ScPDR3 and ScPDR1, in the absence and presence of fluconazole (37). Remarkably, expression of most of these genes was significantly lower in the mutant strains than in the wild-type strain under both conditions (Fig. 3 and S3). This indicates that impairment of vesicular transport affects drug efflux gene expression, drug efflux, and, possibly, fluconazole susceptibility in this manner. We cannot exclude the possibility, however, that other processes also contribute to the process.
FIG 3.

Expression of drug efflux-related genes is downregulated in S. cerevisiae vesicular transport mutant strains. The deletion strains, as well as the wild-type strain, were incubated in SDglu medium for 24 h at 30°C in the absence of fluconazole. Gene expression was analyzed using quantitative reverse transcriptase PCR. Average results relative to those for the wild-type control together with the SEM are displayed. Statistical analysis was conducted on log2(y)-transformed values using one-way analysis of variance with the Bonferroni correction. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Candida albicans PEP5 is involved in tolerance to fluconazole.
C. albicans is a commonly isolated human-pathogenic fungus. To verify whether vesicular transport could similarly affect susceptibility to fluconazole in this fungus, as we described for S. cerevisiae, we tested the C. albicans vps11Δ/Δ (Cavps11Δ/Δ) strain (kindly provided by Glen Palmer) on medium containing fluconazole (38). CaVPS11 is the orthologue of the ScPEP5 gene of S. cerevisiae. Neither the Etest assay nor the broth microdilution assay showed a decrease in the MICFLC for the deletion mutant compared to that for the wild type, indicating that different processes might be involved in both organisms (Table 1 and Fig. S4). Despite the absence of an altered MICFLC, we could, however, show that the tolerance to fluconazole or growth in the presence of supra-MICFLCs of the drug was lower for the Cavps11Δ/Δ mutant strain than for the background strain (Fig. 4). The starting inoculum of approximately 1,830 cells per ml dropped to 0 at higher fluconazole concentrations in the mutant, while it doubled in the wild-type strain. The complete lack of survival at higher fluconazole concentrations indicates that the mutation renders the drug fungicidal in this organism, as cells initially inoculated do not survive treatment. To determine whether vesicular transport also affects expression of drug efflux-related genes in C. albicans, we tested the expression of four genes encoding drug efflux pumps. Figure S5A shows the effect of CaVPS11 deletion on drug efflux gene expression. CaCDR1, CaMDR1, and CaSNQ2 gene expression was decreased in the mutant, while CaCDR2 expression was increased.
FIG 4.
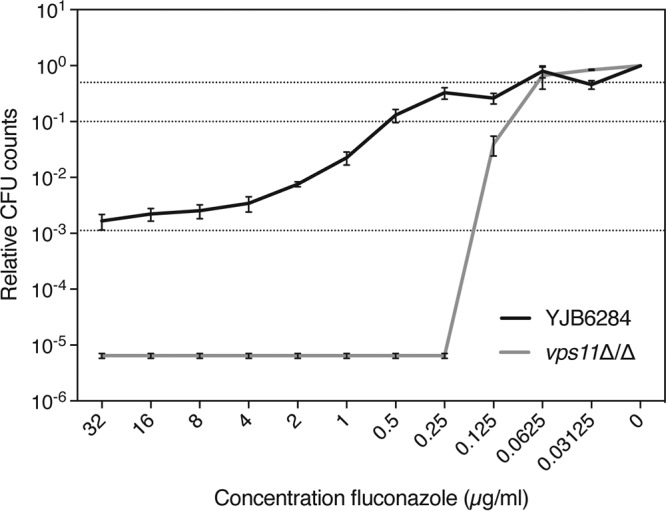
Tolerance to fluconazole is reduced in a C. albicans vps11Δ/Δ mutant compared to the wild-type control. Tolerance assay results for the vps11Δ/Δ mutant and the YJB6284 wild-type control strain are displayed as dose-response curves after 48 h of incubation at 37°C. Relative CFU counts compared to those achieved under the 0-μg/ml fluconazole condition are shown. Dotted lines indicate 50% (upper line) and 90% (middle line) growth inhibition and the initial inoculum (lower line).
Candida glabrata VPS45 is involved in resistance and tolerance to fluconazole.
As described above for C. albicans, we tested a C. glabrata mutant with a mutation in vesicular transport for altered growth in the presence of fluconazole. The C. glabrata VPS45 (CgVPS45) gene is the orthologue of the ScVPS45 gene in S. cerevisiae. Both the Etest analysis and the broth microdilution assay showed a reduced MICFLC for the deletion mutant compared to that for the wild-type strain (Table 1 and Fig. S6). Moreover, we found that the tolerance to fluconazole was also decreased in the vps45Δ/Δ strain (Fig. 5). This indicates that susceptibility to fluconazole, encompassing both resistance and tolerance, is affected by vesicular transport in C. glabrata. To verify if vesicular transport also affects expression of drug efflux-related genes in C. glabrata, we tested the expression of genes encoding drug efflux pumps by the background strains and the Cgvps45Δ/Δ strain (Fig. S5B). Only CgCDR2 expression was slightly decreased in the mutant strain compared to the background strain.
FIG 5.
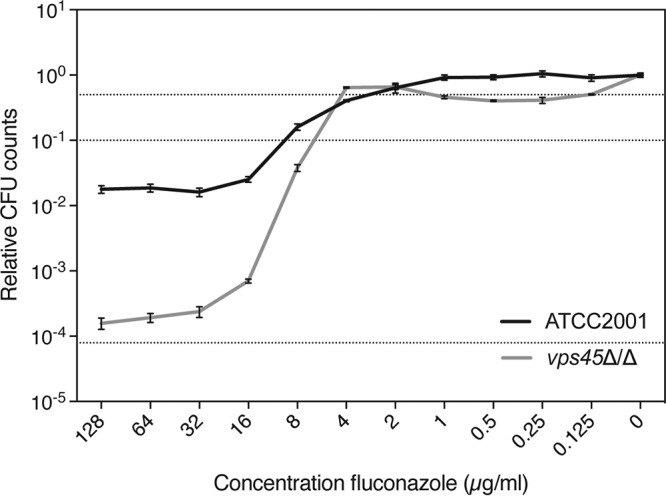
Tolerance to fluconazole is reduced in a C. glabrata vps45Δ/Δ mutant compared to the wild-type control. Tolerance assay results for the vps45Δ/Δ mutant and the ATCC 2001 wild-type control strain are displayed as dose-response curves after 48 h of incubation at 37°C. Relative CFU counts compared to those achieved under the 0-μg/ml fluconazole condition are shown. Dotted lines indicate 50% (upper line) and 90% (middle line) growth inhibition and the initial inoculum (lower line).
An inhibitor of vesicular transport exhibits synergistic interactions with fluconazole against C. albicans and C. glabrata.
Identifying chemical compounds that selectively inhibit vesicular transport similarly to the mutants and, as such, that mimic their phenotypic interaction with fluconazole would hold great potential for combination therapy. We selected two compounds that were earlier identified to be modulators of vacuolar trafficking, termed sortins, for sorting inhibitors (30). Although the exact mechanism is not yet known, it has been shown in S. cerevisiae, as well as in Arabidopsis thaliana, that sortin2 and sortin3 affect vacuolar trafficking, potentially by inhibiting members of the endomembrane complex (27, 28). We tested the effect of the two small compounds both on the MICFLC and on tolerance to fluconazole in C. albicans and C. glabrata and calculated the fractional inhibitory concentration (FIC) values of the drugs as well as the FIC index (∑FIC) of the interaction (Table 2). Figures 6A and 7A show the results of the checkerboard assay carried out for both pathogenic fungi. Sortin3 did not seem to have any effect on the MIC for fluconazole in C. glabrata (Fig. 6A), yet in C. albicans increasing concentrations gave rise to increased MICFLC levels, indicating an antagonistic relationship (Fig. 7A). According to the CLSI guidelines, the ∑FIC for C. glabrata of 1.5 or 2 for the MIC50 and MIC90, respectively, indicates an indifferent interaction (39). The ∑FIC for C. albicans of 5 and 17 for the MIC50 and MIC90, respectively, indicates a clear antagonistic relationship. This apparent antagonism was also visible in the tolerance assay, where higher levels of sortin3 clearly stimulated survival in the presence of higher concentrations of fluconazole specifically in C. albicans (Fig. 7B) and not in C. glabrata (Fig. 6B). Sortin2, in general, acted synergistically with fluconazole against both fungi. This was especially visible in the tolerance assay (Fig. 6B and 7B). The MICFLC of C. glabrata was also mildly lower with higher concentrations of the compound, while this was not clearly visible in C. albicans. The ∑FIC values confirmed this relation. For C. glabrata, the ∑FIC for the MIC50 was 0.275, indicating synergism, and the ∑FIC for the MIC90 was 0.625, indicating an indifferent yet nearly synergistic relationship. For C. albicans, the ∑FIC for the MIC50 was 2.05, indicating indifference, and the ∑FIC for the MIC90 was 0.5, indicating synergism. These data confirm, at least for C. albicans, that the synergistic effect of sortin2 with fluconazole becomes clearer at higher fluconazole concentrations. We can conclude that sortin2 and sortin3 clearly distinguishably act with fluconazole against different species. Sortin2 acted synergistically with fluconazole at higher concentrations of the latter drug, as such inhibiting tolerance. Specifically for C. glabrata, an effect on the MICFLC could also be observed. Sortin3 did not elicit any clear effect in C. glabrata, yet for C. albicans, a clear antagonistic relation both on tolerance and on resistance could be observed. We also tested the effect of sortin2 and sortin3 against our model system, S. cerevisiae, yet found effects less significant than those against the pathogenic fungi (Table 2). Figure S7 shows that sortin2 increased the susceptibility to fluconazole. Remarkably, we saw that although sortin2 did lower the tolerance to fluconazole, this effect was restored at higher concentrations of the sorting inhibitor. While sortin3 did not alter the MICFLC, it did lower the tolerance to fluconazole mildly. Taken together, these data indicate that fine-tuning the exact role of vesicular transport in fluconazole susceptibility is of pivotal importance to further explore its potential as a target in combinatorial therapy with fluconazole.
TABLE 2.
Analysis of interaction between fluconazole and sortins for the three speciesa
| Strain | Sortin | MIC (μg/ml) |
FIC |
∑FIC | ||||
|---|---|---|---|---|---|---|---|---|
| FLC | FLC + sortin | Sortin | Sortin + FLC | FLC | Sortin | |||
| MIC50 | ||||||||
| S. cerevisiae | 2 | 16 | 16 | 200 | 10 | 1 | 0.05 | 1.05 |
| 3 | 16 | 16 | 200 | 25 | 1 | 0.125 | 1.125 | |
| C. albicans | 2 | 0.0625 | 0.125 | 200 | 10 | 2 | 0.05 | 2.05 |
| 3 | 0.125 | 0.5 | 200 | 200 | 4 | 1 | 5 | |
| C. glabrata | 2 | 32 | 8 | 200 | 5 | 0.25 | 0.025 | 0.275 |
| 3 | 32 | 16 | 200 | 200 | 0.5 | 1 | 1.5 | |
| MIC90 | ||||||||
| S. cerevisiae | 2 | 16 | 16 | 200 | 200 | 1 | 1 | 2 |
| 3 | 16 | 16 | 200 | 25 | 1 | 0.125 | 1.125 | |
| C. albicans | 2 | 4 | 1 | 200 | 50 | 0.25 | 0.25 | 0.5 |
| 3 | 4 | 64 | 200 | 200 | 16 | 1 | 17 | |
| C. glabrata | 2 | 64 | 32 | 200 | 25 | 0.5 | 0.125 | 0.625 |
| 3 | 128 | 128 | 200 | 200 | 1 | 1 | 2 | |
The MIC was determined at 50 or 90% inhibition. The fractional inhibitory concentration (FIC) was determined by dividing the MIC of the drug in the combination by the MIC of the drug alone. The FIC index (∑FIC) was determined by adding the FIC for fluconazole to the FIC for sortin.
FIG 6.

Sortin2 acts synergistically with fluconazole against C. glabrata. (A) Broth microdilution assay results for the BG2 wild-type strain in the presence of several concentrations of sortin2 or sortin3 after 48 h of growth at 37°C. Relative OD600 values compared to those achieved under the 0-μg/ml fluconazole condition are shown. Dotted lines indicate 50% (upper line) and 90% (lower line) growth inhibition. The MIC50 and MIC90 values can be deduced from the cross-section between the respective dotted lines and the data curves. (B) Tolerance assay results for the BG2 wild-type strain in the presence of several concentrations of sortin2 or sortin3. CFU counts were compared in the absence and the presence of fluconazole (64 μg/ml) relative to those achieved under the 0-μg/ml sortin condition. Statistical analysis was conducted using one-way analysis of variance with the Bonferroni correction. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
FIG 7.

Sortin2 acts synergistically and sortin3 acts antagonistically with fluconazole against C. albicans. (A) Broth microdilution assay results for the SC5314 wild-type strain in the presence of several concentrations of sortin2 or sortin3 after 48 h of growth at 37°C. Relative OD600 values compared to those achieved under the 0-μg/ml fluconazole condition are shown. Dotted lines indicate 50% (upper line) and 90% (lower line) growth inhibition. The MIC50 and MIC90 values can be deduced from the cross-section between the respective dotted lines and the data curves. (B) Tolerance assay results for the SC5314 wild-type strain in the presence of several concentrations of sortin2 or sortin3. CFU counts were compared in the absence and the presence of fluconazole (32 μg/ml) relative to those achieved under the 0-μg/ml sortin condition. Statistical analysis was conducted using one-way analysis of variance with the Bonferroni correction. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Sortin2 lowers the tolerance of C. glabrata and C. albicans in clinically relevant isolates.
To explore the clinical relevance of synergy between sortin2 and fluconazole, we tested the effect of this drug combination on a number of clinically retrieved isolates of C. albicans and C. glabrata. The effect of sortin2 both on the MIC of fluconazole and on tolerance was monitored. The MICFLC values of the various isolates in the presence (100 μg/ml) and absence of sortin2 are depicted in Table 3. We show that, overall, the MIC50 of fluconazole in the presence of sortin2 remained similar to that when this compound was not added, while the MIC90 mostly decreased. Figure S8 depicts the effect of sortin2 on overall growth in the presence of various concentrations of fluconazole. Figure 8 confirms that, especially in C. glabrata isolates, the tolerance to fluconazole dropped dramatically when vesicular transport was inhibited by sortin2. In C. albicans isolates, a similar trend could be observed, yet the difference was not always significant due to larger variation between biological repeats.
TABLE 3.
Effect of sortin2 on MICFLC of clinical isolatesa
| Isolate | MIC (μg/ml) |
|||
|---|---|---|---|---|
| 50% |
90% |
|||
| FLC | FLC + sortin2 | FLC | FLC + sortin2 | |
| C. glabrata | ||||
| 1 | 16 | 8 | 32 | 16 |
| 2 | 8 | 8 | 64 | 32 |
| 3 | 16 | 16 | 64 | 32 |
| 4 | 8 | 8 | 32 | 16 |
| C. albicans | ||||
| 1 | 0.25 | 0.125 | 1 | 0.5 |
| 2 | 16 | 8 | 128 | 16 |
| 3 | 0.125 | 0.0624 | 128 | 1 |
| 4 | 16 | 8 | 128 | 16 |
The MIC values were determined at 50 or 90% inhibition in the absence and presence of 100 μg/ml sortin2.
FIG 8.
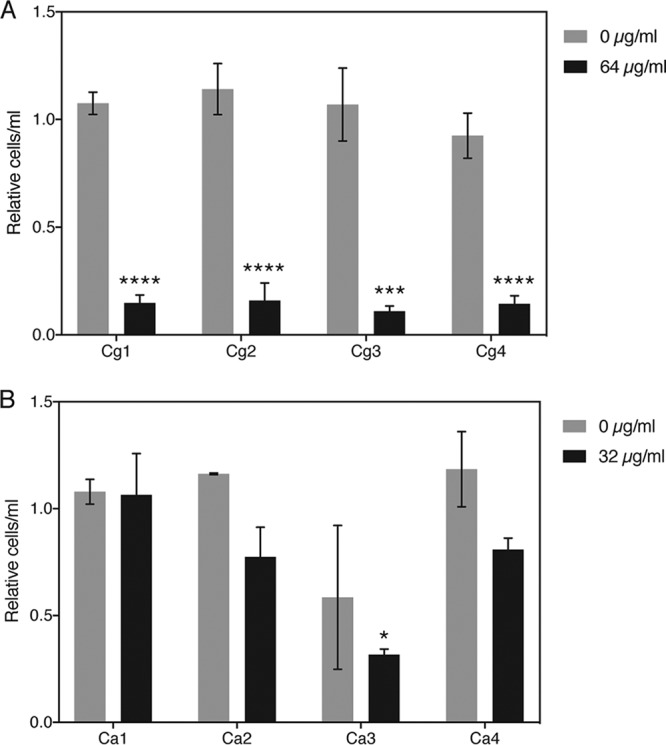
Sortin2 lowers tolerance to fluconazole in clinical C. glabrata (A) and C. albicans (B) isolates. Tolerance to fluconazole, indicated as CFU counts at 64 and 32 μg/ml fluconazole for C. glabrata and C. albicans, respectively, was determined in the absence and the presence of 100 μg/ml sortin2. All values were compared to those under the condition without sortin. Statistical analysis was conducted using two-way analysis of variance with the Bonferroni correction. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
In this study, we found that impairment of vesicular transport in fungal cells potentiates the antifungal effect of fluconazole.
In the model organism S. cerevisiae, six deletion mutants affected in vesicular transport were identified to have increased susceptibility to this widely used drug. We showed that upon treatment, these strains retained significantly less ergosterol than the wild-type control strain, leading ultimately to a lower MICFLC and/or tolerance to fluconazole. Remarkably, in the absence of the drug, a slight yet significant increase in ergosterol levels could be observed in the mutants compared to the wild-type strain. This indicates that the mutant genotypes as such do not negatively affect ergosterol metabolism but that the phenotype strictly depends on the presence of the drug. We expect that the inhibition of vesicular transport positively influences the concentration of the drug inside the cell, as such leading to stronger inhibition of the Erg11 enzyme and increased susceptibility. It was reported earlier that an Scvps3Δ mutant of S. cerevisiae displays lower drug efflux rates than the corresponding wild-type strain (36). ScVps3 is a member of the CORVET complex, to which ScPep3 and ScPep5 also belong, and regulates tethering of vesicles to and from the endosome. To verify whether drug efflux would also be affected in the six deletion mutants resulting from our screening, we decided to measure the expression levels of seven genes encoding drug efflux pumps and two genes encoding regulators thereof. Remarkably, we found that expression of nearly all genes was significantly downregulated in the six deletion mutants in both the absence and the presence of fluconazole. When taking a closer look at the genes which were analyzed, we noticed that the expression of all of them is directly or indirectly regulated by the transcription factor ScPdr1 (40, 41). ScPDR1 expression itself is also reported to be a target of regulation by its own gene product in C. glabrata (42). Any effect of impaired vesicular transport on nuclear localization or expression of this protein/gene would thereby also likely lead to the observed effects on the expression of genes encoding drug efflux pumps or ScPDR3.
We confirmed a role for vesicular transport in fluconazole susceptibility in C. albicans and C. glabrata. Apart from the small and potentially insignificant effect on the MICFLC, homozygous deletion of the CaVPS11 gene, the orthologue of the ScPEP5 gene, caused the nearly complete absence of trailing growth at supra-MICFLCs of fluconazole. The Cgvps45Δ/Δ strain in a C. glabrata background similarly showed a decrease in tolerance yet also a lowered MICFLC. This indicates that the observations that we made in S. cerevisiae are, at least to some extent, relevant for other fungi as well. Expression of drug efflux genes seems altered to a lesser extent in these fungal pathogens than in S. cerevisiae. In both organisms, the expression of some efflux pumps was mildly decreased, while that of others was not. We hypothesize that the stronger effect of vesicular transport inhibition on the MICFLC in S. cerevisiae correlates specifically with altered levels of drug efflux gene expression.
Sortins are small compounds that interfere with vesicular trafficking to or from the vacuole and endosomes (30). Based on our observations on the role of vesicular transport in fluconazole susceptibility in C. albicans and C. glabrata, we verified whether these chemicals could synergize with fluconazole in these two organisms. For both C. albicans and C. glabrata, we observed a synergistic effect of sortin2 with higher concentrations of fluconazole. The level of tolerance or trailing growth at supra-MICFLCs of fluconazole was significantly reduced with higher levels of sortin2. Specifically, in C. glabrata, the MICFLC also dropped mildly when higher concentrations of sortin2 were added to the culture medium. We also showed that sortin2 lowered the tolerance to fluconazole in our model organism, S. cerevisiae, yet this effect seemed to be concentration dependent. These results indicate that inhibition of vesicular transport indeed synergizes with fluconazole against several fungi and, thus, emphasizes the potential of targeting this process in combination therapy. Contrary to the results obtained with sortin2, sortin3 did not affect fluconazole efficacy in C. glabrata and even antagonized its effect in C. albicans regarding both the MICFLC and tolerance. Contrary to what we found in Candida species, sortin3 acted mildly synergistically on tolerance with S. cerevisiae, yet only at high concentrations of sortin. No effect on the MICFLC could be distinguished. It thus seems to be of pivotal importance to elucidate how both sortins function.
Another aspect that needs to be considered when proposing sortin2 or an analogous compound for combination therapy with fluconazole is its potency against clinically relevant isolates. Therefore, we tested the effect of sortin2 against several clinical C. glabrata and C. albicans isolates with various MICFLC values. For nearly all isolates, sortin2 lowered the tolerance to fluconazole and the MIC90 of this drug. Especially in C. glabrata, the reduction was dramatic. The effect of the compound on the MIC50 of fluconazole was minimal, confirming that sortin2 specifically alters the growth of the pathogens at higher fluconazole concentrations. Although the reduction of growth upon treatment with sortin2 strictly depended on the presence of fluconazole, further in-depth analysis is necessary to confirm the absence of toxicity in mammalian cells, as it was shown that the effect of these sorting inhibitors is not limited to yeast cells (30).
In conclusion, we identified specific components of the vesicular transport pathway which are important for the development of resistance and/or tolerance to fluconazole in Candida species. In addition, we show here that inhibition of vesicular transport in a targeted way leads to synergism with fluconazole against several fungal species, as summarized in Fig. 9. Further analysis is necessary to elucidate the full potential of vesicular transport-inhibiting compounds, such as sortin2, in combination therapy with fluconazole. Apart from its potential clinical relevance, we show that sortin2 acts rather specifically against tolerance to fluconazole. We thus propose that this compound will be a valuable tool in further elucidation of the relevance and modes of action of this phenomenon in pathogenic fungi.
FIG 9.

Schematic overview of the role of vesicular transport in susceptibility to fluconazole. The six proteins identified to be essential for retaining resistance and/or tolerance to fluconazole in S. cerevisiae are depicted according to their function in vesicular transport. We hypothesize that a signal originating from this process induces expression of drug efflux pump-encoding genes in the nucleus, thereby increasing the efflux of fluconazole and lowering the intracellular concentration of the drug. This would result in the increased production of ergosterol and reduced susceptibility to fluconazole. Sortin2 acts synergistically with fluconazole in Candida fungi by inhibiting vesicular transport. HOPS, homotypic fusion and vacuole protein sorting; GARP, Golgi-associated retrograde protein; CORVET, class C core vacuole/endosome transport complex.
MATERIALS AND METHODS
Strains and growth conditions.
All S. cerevisiae strains used in this study were isogenic to the BY4742 laboratory wild-type strain and are listed in Table S3 in supplemental material. The deletion strains used in further assays originated from the Yeast Knockout (YKO) collection and were recreated in this study to obtain at least three biological replicates of each genotype. The primers used to generate these strains are listed in Table S4. The C. albicans vps11Δ/Δ strain was used in combination with its wild-type strain, YJB6284 (38). For the wild-type and mutant C. albicans strains, three colonies were considered biological repeats. The Cgvps45Δ/Δ strain in the ATCC 2001 background (43) was created using the pYC44 plasmid (44). For sortin-fluconazole interaction assays, the BG2 clinical isolate was used as the wild-type strain (45), and for C. albicans, the SC5314 strain was used as the wild-type strain (46).
S. cerevisiae strains were grown in synthetic dextrose medium, consisting of 0.17% Difco yeast nitrogen base without amino acids or ammonium sulfate, 0.5% ammonium sulfate, and 2% glucose (SDglu medium). The pH of the medium was adapted to 5.5 for liquid medium and 6.5 for solid medium. After addition of 1.6% agar, the medium was autoclaved. Appropriate amino acids and nucleotides were added before use. In certain experiments, fluconazole (catalog number F8929; Sigma) and doxycycline (catalog number D9891; Sigma) were added, obtaining concentrations of, respectively, 10 or 20 μg/ml and 50 or 100 μg/ml (31). Cultures containing fluconazole and/or doxycycline were always kept in the dark.
C. albicans and C. glabrata strains were pregrown in SDglu medium. Assays were carried out in filter-sterilized RPMI 1640 medium with l-glutamine (catalog number R6504; Sigma) and buffered with 0.165 M morpholinepropanesulfonic acid at pH 7. Depending on the assay, autoclave-sterilized and precooled agar and 1.8% glucose were added to the medium.
Screening the yeast knockout collection for enhancers of fluconazole susceptibility.
The deletion strains from the MATα Yeast Knockout (YKO) collection were screened according to a protocol adapted from one described previously (32, 34). The strains were initially spotted on solid SDglu medium containing 10 or 20 μg/ml fluconazole and 50 or 100 μg/ml doxycycline (47). Addition of doxycycline reduced the background growth, thereby easing the visual inspection of the growth reduction. Enhancer strains of fluconazole susceptibility, which were deletion strains that showed increased susceptibility to the drug, were selected by visually monitoring reduced growth after 3 days of incubation at 30°C compared to the growth of the BY4742 wild-type laboratory strain and were stored in 96-well microtiter plates. The genes deleted in all enhancer strains were clustered, using the online tool FunSpec (33). These enhancers of fluconazole susceptibility were later retested on the same medium, using at least three biological replicates.
Determination of fluconazole susceptibility: spot assays, MICFLC evaluation, and tolerance assay.
Spot assays were conducted by adapting the optical density at 600 nm (OD600) to 1 and making serial 1/5 dilutions. The cultures were spotted on solid SDglu medium containing different concentrations of fluconazole and/or doxycycline. After incubation of the plates at 30°C for 72 h, they were scanned.
To determine the MIC of fluconazole (MICFLC) of the strains, both the Etest (bioMérieux) and broth microdilution assay (BDA) were used (48, 49). For the Etest assay, overnight S. cerevisiae cultures were adjusted to an OD600 of 0.5 in water and spread on SDglu agar medium. After placing the strip onto the cells, the plates were incubated for 72 h at 30°C. The MICFLC was determined by identifying the concentration of fluconazole on the strip where the latter intersects the halo of growth inhibition/retardation (49). For C. albicans and C. glabrata, the same protocol was used, but the culture was diluted to an OD600 of 0.2 instead of 0.5 and plated on RPMI 1640 agar medium containing 2% glucose (32). The plates were scanned after 48 h of incubation at 37°C. The Etest method was used in parallel with the broth microdilution assay. This technique was performed using a protocol based on both CLSI (39) and EUCAST (50) standard methods. According to the CLSI protocol, cells were loaded in round-bottom, UV-sterilized 96-well microtiter plates to obtain a concentration of 0.5 × 103 to 2.5 × 103 cells per milliliter (39). Fluconazole was added to the wells, together with SDglu or RPMI 1640 medium, at concentrations ranging from 0 to 128 μg/ml or 32 μg/ml, in the case of C. albicans, in 1/2 dilutions. According to CLSI, one can determine breakpoints after 24 or 48 h of static growth. We determined the growth after 48 h of incubation at 30°C or 37°C for S. cerevisiae and the Candida strains, respectively, in order to achieve clearer breakpoints (32). The OD600 of the resuspended cultures was measured objectively using a spectrophotometer and plotted to obtain dose-response curves, according to EUCAST measures (50). We earlier showed the proper concordance of both CFU counts and spectrophotometric measurements to determine the MICFLC (32). The MICFLC was calculated by subtracting the background OD of the growth medium from all data points and normalization to the condition with 0 μg/ml fluconazole. When this relative OD drops below 50 or 10% of the initial OD with no drug, the MIC50 and MIC90, respectively, can be determined.
To evaluate drug tolerance, we generated dose-response curves based on CFU counts for the control and/or mutant strain as described by CLSI (39) and as established earlier (32). In short, we loaded the cells in round-bottom, UV-sterilized 96-well microtiter plates to obtain a concentration of 0.5 × 103 to 2.5 × 103 cells per milliliter (39). Fluconazole was added to the wells, together with SDglu or RPMI medium, at concentrations ranging from 0 to 128 μg/ml or 32 μg/ml, in the case of C. albicans, in 1/2 dilutions. After 48 h of growth at 30 or 37°C, the contents of the wells of the broth microdilution assay plate were resuspended, and each culture was diluted and plated. The drug tolerance was determined by checking the CFU counts under the conditions seen with the two highest fluconazole concentrations. All experiments were conducted with at least three biological replicates.
Fluconazole interaction analysis: checkerboard assay.
The interaction between fluconazole and sortins was determined by checkerboard analysis, generally as described previously (31). S. cerevisiae strains were incubated in SDglu medium, and Candida strains were incubated in RPMI medium supplemented with 2% glucose. Cells were added in round-bottom, UV-sterilized 96-well microtiter plates to obtain a concentration of 0.5 × 103 to 2.5 × 103 cells per milliliter. Fluconazole was added to the wells at concentrations ranging from 0 to 128 μg/ml in 1/2 dilutions in the appropriate medium. In addition, sortins were first dissolved in 20% dimethyl sulfoxide (DMSO) and were added at concentrations of 0, 2,5, 5, 10, 25, 50, and 100 μg/ml in the appropriate medium containing a final concentration of 1% DMSO. After 72 h of static growth at 30°C for S. cerevisiae or 48 h of growth at 37°C for the Candida strains, the OD600 of the resuspended cultures was measured using a spectrophotometer and plotted to obtain dose-response curves. The MICFLC was calculated by subtracting the background OD of the growth medium with sortins from all data points and normalization to the condition with 0 μg/ml fluconazole and 0 μg/ml sortin. Additionally, for each concentration of sortin, CFU were plated in one of the highest and lowest concentrations of fluconazole on yeast extract-peptone-dextrose plates to determine tolerance. ∑FIC values were determined using the following formula, based on the CLSI guidelines (39) and according to the Instructions to Authors of Antimicrobial Agents and Chemotherapy (51): (MICFLC + sortin/MICFLC) + (MICsortin + FLC/MICsortin). Based on these guidelines, a ∑FIC of ≤0.5 implies synergy, a ∑FIC of >4 implies antagonism, and the values in between denote indifference. Both the ∑FIC for an MIC50 and an ∑FIC for an MIC90 were determined, based on MIC50 and MIC90 data.
Gene expression analysis by quantitative reverse transcriptase PCR.
We performed gene expression analysis according to the protocol described previously (32). The S. cerevisiae deletion strains, as well as the wild-type strain, were incubated in SDglu medium for 24 h at 30°C in the absence or the presence of 20 μg/ml fluconazole. The C. albicans and C. glabrata strains were inoculated in RPMI 1640 medium with 2% glucose and 8 μg/ml fluconazole and incubated for 8 h at 37°C. The cells were washed with cold Milli-Q water, frozen in liquid nitrogen, and kept at −80°C as long as necessary. The pellets were resuspended in the TRIzol reagent (Thermo Fisher), followed by breaking of the cells with glass beads using a FastPrep machine. RNA was extracted using chloroform and isopropanol and was washed with 70% ethanol afterwards. To break down the residual DNA, DNase enzyme (New England Biolabs) was added. The RNA was converted into cDNA using an iScript cDNA synthesis kit from Bio-Rad. The actual quantitative PCR was performed with the GoTaq polymerase from Promega and a StepOnePlus device from Thermo Fisher. Data analysis was performed using the qBasePlus software from Biogazelle. Statistical analysis was performed using GraphPad Prism software. Prior to that, all data points were log2(y) transformed in order to allow the use of standardized statistical methods.
Sterol measurement.
Sterols were measured as described previously (35, 52). The strains under study were incubated in SDglu medium for 48 h at 30°C in the absence or the presence of 20 μg/ml fluconazole. Cells were washed with cold Milli-Q water, frozen in liquid nitrogen, and kept at −80°C as long as necessary. The cell pellets were resuspended in 3 ml saponification medium, consisting of KOH, H2O, and ethanol, and vortexed for 1 min. After incubation of the samples for 1 h at 80°C, 1 ml of water and 4 ml of hexane were added. After another 3 min of vortex mixing, the two layers were allowed to separate. The OD281.5 and OD230 of the upper layer were measured using a spectrophotometer and UV-transmittable 96-well microtiter plates (Costar; Corning). The percentage of ergosterol relative to the wet weight was measured using the formula introduced in reference 35.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ilse Palmans, Celia Lobo Romero, and Thomas Talpe for technical assistance. We also acknowledge Nico Vangoethem for help with the figures. We thank Glen Palmer for kindly sending us the Cavps11Δ/Δ mutant strain and Dominique Sanglard and Katrien Lagrou for providing the clinical C. albicans and C. glabrata isolates. Finally, we thank Erwin Swinnen and Alessandro Fiori for valuable input in the starting phase of this project.
L.D. and K.V.D. conceived of and performed all experiments. All authors contributed to discussion of the results.
L.D. and K.V.D. were supported by personal research grants from the Fund for Scientific Research Flanders (FWO; grant 11P9814N to L.D. and grant 1181818N to K.V.D.). This work was supported by grants from the Fund for Scientific Research Flanders (grant no. G0C1514) and by the Research Council of KU Leuven (grant no. C14/17/063)
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01998-18.
REFERENCES
- 1.Brown GD, Denning DW, Levitz SM. 2012. Tackling human fungal infections. Science 336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 2.Mayer FL, Wilson D, Hube B. 2013. Candida albicans pathogenicity mechanisms. Virulence 4:119–128. doi: 10.4161/viru.22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunke S, Hube B. 2013. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell Microbiol 15:701–708. doi: 10.1111/cmi.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman J. 2012. Candida albicans. Curr Biol 22:R620–R622. doi: 10.1016/j.cub.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 5.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 6.Roemer T, Krysan DJ. 2014. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med 4:a019703. doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheehan DJ, Hitchcock CA, Sibley CM. 1999. Current and emerging azole antifungal agents. Clin Microbiol Rev 12:40–79. doi: 10.1128/CMR.12.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly SL, Arnoldi A, Kelly DE. 1993. Molecular genetic analysis of azole antifungal mode of action. Biochem Soc Trans 21:1034–1038. doi: 10.1042/bst0211034. [DOI] [PubMed] [Google Scholar]
- 9.Vanden Bossche H. 1985. Biochemical targets for antifungal azole derivatives: hypothesis on the mode of action. Curr Top Med Mycol 1:313–351. doi: 10.1007/978-1-4613-9547-8_12. [DOI] [PubMed] [Google Scholar]
- 10.Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. 2014. Mechanisms of antifungal drug resistance. Cold Spring Harb Perspect Med 5:a019752. doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berkow EL, Lockhart SR. 2017. Fluconazole resistance in Candida species: a current perspective. Infect Drug Resist 10:237–245. doi: 10.2147/IDR.S118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delarze E, Sanglard D. 2015. Defining the frontiers between antifungal resistance, tolerance and the concept of persistence. Drug Resist Updat 23:12–19. doi: 10.1016/j.drup.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Gerstein AC, Rosenberg A, Hecht I, Berman J. 2016. diskImageR: quantification of resistance and tolerance to antimicrobial drugs using disk diffusion assays. Microbiology 162:1059–1068. doi: 10.1099/mic.0.000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson MD, Perfect JR. 2010. Use of antifungal combination therapy: agents, order, and timing. Curr Fungal Infect Rep 4:87–95. doi: 10.1007/s12281-010-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baddley JW, Pappas PG. 2005. Antifungal combination therapy: clinical potential. Drugs 65:1461–1480. doi: 10.2165/00003495-200565110-00002. [DOI] [PubMed] [Google Scholar]
- 16.Johnson MD, MacDougall C, Ostrosky-Zeichner L, Perfect JR, Rex JH. 2004. Combination antifungal therapy. Antimicrob Agents Chemother 48:693–715. doi: 10.1128/AAC.48.3.693-715.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubuke ML, Munson M. 2016. The secret life of tethers: the role of tethering factors in SNARE complex regulation. Front Cell Dev Biol 4:42. doi: 10.3389/fcell.2016.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feyder S, De Craene JO, Bar S, Bertazzi DL, Friant S. 2015. Membrane trafficking in the yeast Saccharomyces cerevisiae model. Int J Mol Sci 16:1509–1525. doi: 10.3390/ijms16011509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bröcker C, Engelbrecht-Vandré S, Ungermann C. 2010. Multisubunit tethering complexes and their role in membrane fusion. Curr Biol 20:R943–R952. doi: 10.1016/j.cub.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Cornet M, Bidard F, Schwarz P, Da Costa G, Blanchin-Roland S, Dromer F, Gaillardin C. 2005. Deletions of endocytic components VPS28 and VPS32 affect growth at alkaline pH and virulence through both RIM101-dependent and RIM101-independent pathways in Candida albicans. Infect Immun 73:7977–7987. doi: 10.1128/IAI.73.12.7977-7987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luna-Tapia A, Kerns ME, Eberle KE, Jursic BS, Palmer GE. 2015. Trafficking through the late endosome significantly impacts Candida albicans tolerance of the azole antifungals. Antimicrob Agents Chemother 59:2410–2420. doi: 10.1128/AAC.04239-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. 2004. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol 22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- 23.Caza M, Hu G, Nielson ED, Cho M, Jung WH, Kronstad JW. 2018. The Sec1/Munc18 (SM) protein Vps45 is involved in iron uptake, mitochondrial function and virulence in the pathogenic fungus Cryptococcus neoformans. PLoS Pathog 14:e1007220. doi: 10.1371/journal.ppat.1007220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peplowska K, Markgraf DF, Ostrowicz CW, Bange G, Ungermann C. 2007. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev Cell 12:739–750. doi: 10.1016/j.devcel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Conibear E, Cleck JN, Stevens TH. 2003. Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol Biol Cell 14:1610–1623. doi: 10.1091/mbc.e02-10-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webb GC, Hoedt M, Poole LJ, Jones EW. 1997. Genetic interactions between a pep7 mutation and the PEP12 and VPS45 genes: evidence for a novel SNARE component in transport between the Saccharomyces cerevisiae Golgi complex and endosome. Genetics 147:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zouhar J, Hicks GR, Raikhel NV. 2004. Sorting inhibitors (sortins): chemical compounds to study vacuolar sorting in Arabidopsis. Proc Natl Acad Sci U S A 101:9497–9501. doi: 10.1073/pnas.0402121101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasquez-Soto B, Manriquez N, Cruz-Amaya M, Zouhar J, Raikhel NV, Norambuena L. 2015. Sortin2 enhances endocytic trafficking towards the vacuole in Saccharomyces cerevisiae. Biol Res 48:39. doi: 10.1186/s40659-015-0032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson JS, Klionsky DJ, Banta LM, Emr SD. 1988. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol 8:4936–4948. doi: 10.1128/MCB.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishev K, Dejonghe W, Russinova E. 2013. Small molecules for dissecting endomembrane trafficking: a cross-systems view. Chem Biol 20:475–486. doi: 10.1016/j.chembiol.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Fiori A, Van Dijck P. 2012. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob Agents Chemother 56:3785–3796. doi: 10.1128/AAC.06017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demuyser L, Swinnen E, Fiori A, Herrera-Malaver B, Vestrepen K, Van Dijck P. 2017. Mitochondrial cochaperone Mge1 is involved in regulating susceptibility to fluconazole in Saccharomyces cerevisiae and Candida species. mBio 8:e00201-17. doi: 10.1128/mBio.00201-17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MD, Grigull J, Mohammad N, Hughes TR. 2002. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics 3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson JB, Sirjusingh C, Parsons AB, Boone C, Wickens C, Cowen LE, Kohn LM. 2003. Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 163:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arthington-Skaggs BA, Jradi H, Desai T, Morrison CJ. 1999. Quantitation of ergosterol content: novel method for determination of fluconazole susceptibility of Candida albicans. J Clin Microbiol 37:3332–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rutledge RM, Ghislain M, Mullins JM, de Thozee CP, Golin J. 2008. Pdr5-mediated multidrug resistance requires the CPY-vacuolar sorting protein Vps3: are xenobiotic compounds routed from the vacuole to plasma membrane transporters for efflux? Mol Genet Genomics 279:573–583. doi: 10.1007/s00438-008-0334-5. [DOI] [PubMed] [Google Scholar]
- 37.Rogers B, Decottignies A, Kolaczkowski M, Carvajal E, Balzi E, Goffeau A. 2001. The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J Mol Microbiol Biotechnol 3:207–214. [PubMed] [Google Scholar]
- 38.Palmer GE, Cashmore A, Sturtevant J. 2003. Candida albicans VPS11 is required for vacuole biogenesis and germ tube formation. Eukaryot Cell 2:411–421. doi: 10.1128/EC.2.3.411-421.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts, 3rd ed. Approved standard. CLSI M27-A3(28) Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 40.Jungwirth H, Kuchler K. 2006. Yeast ABC transporters—a tale of sex, stress, drugs and aging. FEBS Lett 580:1131–1138. doi: 10.1016/j.febslet.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 41.Wehrschütz-Sigl E, Jungwirth H, Bergler H, Högenauer G. 2004. The transporters Pdr5p and Snq2p mediate diazaborine resistance and are under the control of the gain-of-function allele PDR1-12. Eur J Biochem 271:1145–1152. doi: 10.1111/j.1432-1033.2004.04018.x. [DOI] [PubMed] [Google Scholar]
- 42.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 43.Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuvéglise C, Talla E, Goffard N, Frangeul L, Aigle M, Anthouard V, Babour A, Barbe V, Barnay S, Blanchin S, Beckerich J-M, Beyne E, Bleykasten C, Boisramé A, Boyer J, Cattolico L, Confanioleri F, De Daruvar A, Despons L, Fabre E, Fairhead C, Ferry-Dumazet H, Groppi A, Hantraye F, Hennequin C, Jauniaux N, Joyet P, Kachouri R, Kerrest A, Koszul R, Lemaire M, Lesur I, Ma L, Muller H, Nicaud J-M, Nikolski M, Oztas S, Ozier-Kalogeropoulos O, Pellenz S, Potier S, Richard G-F, Straub M-L, et al. 2004. Genome evolution in yeasts. Nature 430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 44.Yáñez-Carrillo P, Orta-Zavalza E, Gutiérrez-Escobedo G, Patrón-Soberano A, De Las Peñas A, Castaño I. 2015. Expression vectors for C-terminal fusions with fluorescent proteins and epitope tags in Candida glabrata. Fungal Genet Biol 80:43–52. doi: 10.1016/j.fgb.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 45.Cormack BP, Falkow S. 1999. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics 151:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fonzi WA, Irwin MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giaever G, Nislow C. 2014. The yeast deletion collection: a decade of functional genomics. Genetics 197:451–465. doi: 10.1534/genetics.114.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serefko A, Los R, Biernasiuk A, Malm A. 2008. Comparison of microdilution method and E-test procedure in susceptibility testing of caspofungin against Candida non-albicans species. New Microbiol 31:257–262. [PubMed] [Google Scholar]
- 49.Lass-Florl C, Perkhofer S, Mayr A. 2010. In vitro susceptibility testing in fungi: a global perspective on a variety of methods. Mycoses 53:1–11. doi: 10.1111/j.1439-0507.2009.01813.x. [DOI] [PubMed] [Google Scholar]
- 50.Subcommittee on Antifungal Susceptibility Testing (AFST) of the ESCMID European Committee for Antimicrobial Susceptibility Testing (EUCAST). 2008. EUCAST definitive document EDef 7.1: method for the determination of broth dilution MICs of antifungal agents for fermentative yeasts. Clin Microbiol Infect 14:398–405. doi: 10.1111/j.1469-0691.2007.01935.x. [DOI] [PubMed] [Google Scholar]
- 51.American Society for Microbiology. 2019. Instructions to authors. Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC. https://aac.asm.org/sites/default/files/additional-assets/AAC-ITA.pdf.
- 52.Arthington-Skaggs BA, Lee-Yang W, Ciblak MA, Frade JP, Brandt ME, Hajjeh RA, Harrison LH, Sofair AN, Warnock DW, Candidemia Active Surveillance Group. 2002. Comparison of visual and spectrophotometric methods of broth microdilution MIC end point determination and evaluation of a sterol quantitation method for in vitro susceptibility testing of fluconazole and itraconazole against trailing and nontrailing Candida isolates. Antimicrob Agents Chemother 46:2477–2481. doi: 10.1128/AAC.46.8.2477-2481.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.