

Infection with the free-living amoeba Naegleria fowleri leads to life-threatening primary amoebic meningoencephalitis. Efficacious treatment options for these infections are limited, and the mortality rate is very high (∼98%).
KEYWORDS: Naegleria fowleri, glucokinase
ABSTRACT
Infection with the free-living amoeba Naegleria fowleri leads to life-threatening primary amoebic meningoencephalitis. Efficacious treatment options for these infections are limited, and the mortality rate is very high (∼98%). Parasite metabolism may provide suitable targets for therapeutic design. Like most other organisms, glucose metabolism is critical for parasite viability, being required for growth in culture. The first enzyme required for glucose metabolism is typically a hexokinase (HK), which transfers a phosphate from ATP to glucose. The products of this enzyme are required for both glycolysis and the pentose phosphate pathway. However, the N. fowleri genome lacks an obvious HK homolog and instead harbors a glucokinase (Glck). The N. fowleri Glck (NfGlck) shares limited (25%) amino acid identity with the mammalian host enzyme (Homo sapiens Glck), suggesting that parasite-specific inhibitors with anti-amoeba activity can be generated. Following heterologous expression, NfGlck was found to have a limited hexose substrate range, with the greatest activity observed with glucose. The enzyme had apparent Km values of 42.5 ± 7.3 μM and 141.6 ± 9.9 μM for glucose and ATP, respectively. The NfGlck structure was determined and refined to 2.2-Å resolution, revealing that the enzyme shares greatest structural similarity with the Trypanosoma cruzi Glck. These similarities include binding modes and binding environments for substrates. To identify inhibitors of NfGlck, we screened a small collection of inhibitors of glucose-phosphorylating enzymes and identified several small molecules with 50% inhibitory concentration values of <1 μM that may prove useful as hit chemotypes for further leads and therapeutic development against N. fowleri.
INTRODUCTION
Human infection by the free-living amoeba Naegleria fowleri can lead to life-threatening illness. When trophozoites encountered in freshwater are inadvertently introduced into the nasal passages, parasites can travel to the brain and cause a deadly infection, primary amoebic meningoencephalitis (PAM). Between 1962 and 2016, 143 PAM cases were reported in the United States (Centers for Disease Control and Prevention). While the frequency of reported infection is low, the limited treatment options have historically yielded poor outcomes, with a study of 123 cases in the United States revealing that 122 infections resulted in fatalities (1). A combination therapy that included amphotericin B, miconazole, fluconazole, and ketoconazole was used to successfully treat a single case of PAM, although the contribution of this therapeutic cocktail to patient survival is unclear (2). More recently, miltefosine has shown some promise as an anti-amoebic agent (3). Nevertheless, the lack of effective therapies for this infection, which remains lethal in ∼98% of cases, makes elucidating therapeutic targets for novel drug discovery a high priority.
Mechanisms that the amoebae use to satisfy their metabolic needs are poorly resolved and are primarily based on assessment of growth under different culturing conditions and on analysis of the predicted gene content of the genome. The only N. fowleri metabolic enzyme characterized to date, phosphofructokinase (PFK), is a pyrophosphate-dependent (instead of ATP-dependent) enzyme (4). Use of the alternative phosphoryl group donor is usually associated with enzymes from anaerobic organisms, suggesting that N. fowleri inhabits niches where oxidative phosphorylation is limited.
The role of glycolysis in meeting the metabolic needs of N. fowleri during human infection remains unresolved. However, the relative abundance of glucose in both the brain and cerebrospinal fluid (CSF), as well as the correlation of parasite presence with reduced CSF glucose concentrations, suggest that glucose depletion in the brain is a consequence of pathogen consumption (5). Therefore, the carbon source may be important for parasite metabolism.
Most eukaryotic cells rely on a hexose phosphotransferase (a hexokinase, or HK) to catalyze the first enzymatic step common to both the glycolytic and the pentose phosphate pathways (PPP) to generate glucose-6-phosphate (G6P). These enzymes typically have a broad affinity for different hexoses, including glucose, mannose, fructose, and galactose. N. fowleri harbors a gene that encodes a predicted glucokinase (NfGlck) and lacks other recognizable enzymes that could catalyze the transfer of the γ-phosphoryl group of ATP to glucose to generate G6P. Glucokinases (Glcks), which are typically restricted to using glucose as a substrate, have been classified into two groups, A and B (6). Group A Glcks include enzymes found in Gram-negative bacteria, Cyanobacteria, and amitochondriate protists. Group B Glck members include enzymes from Gram-positive bacteria that can use either ATP or polyphosphate as a phosphoryl group donor. Here, we describe the biochemical and structural characterization of NfGlck. This work has revealed that the enzyme has limited hexose substrate use (like other Glcks) and shares structural similarity with the group A Glck from Trypanosoma cruzi (7). However, kinetic differences suggest the NfGlck has evolved to satisfy the metabolic needs of Naegleria in low-resource environments. Lastly, we describe the interrogation of a selected panel of potential inhibitors which has yielded the first NfGlck inhibitors disclosed to date. These compounds serve as a source of potential hits for further leads and therapeutic development.
RESULTS
The dietary requirements of the free-living amoeba Naegleria fowleri are poorly defined, with parasite growth described from cultures maintained in the presence of bacteria or human feeder cells. Axenic culture in defined media has also been established (8), but the role of individual components in the metabolic success of the parasites has not been fully explored.
Most of the described media include glucose. To assess the importance of this carbon source to the parasite, trophozoites were seeded into Nelson’s complete medium (NCM) with or without glucose or with mannose replacing the glucose (Fig. 1A and data not shown). After 2 days, culture in the absence of glucose led to reduced growth and enhanced formation of cyst-like structures compared to levels in the other two media. In either glucose- or mannose-bearing media, cyst-like structures became evident only after trophozoites reached near confluence (see Fig. S1A in the supplemental material).
FIG 1.

(A) N. fowleri trophozoites were seeded at 2 × 106 cells/ml in control medium (Nelson’s complete medium) and cultured with or without glucose. (B) Cells (1 × 106 cells/ml) were incubated with or without 3-bromopyruvate (50 mM) for 2 days prior to imaging. Images were captured on an EVOS imaging system, and data are representative of multiple experiments. Scale bar, 20 μM.
To further characterize the observed cyst-like structures, the samples were treated with detergent (0.25% SDS) at a concentration that eliminated all trophozoites (Fig. S1A). The cyst-like structures generated by growth of trophozoites to high concentrations in normal NCM were lysed by this treatment, while samples from parasites cultured in either the absence of glucose or in the presence of mannose were refractory to lysis. Resistance suggested that these samples contained authentic cysts and not pseudocysts. However, excystation following culture in detergent-free medium or on plates with Escherichia coli as a food source was only detected from the detergent-resistant cysts from the mannose-containing medium, indicating that the cysts from the glucose-free condition were in some way defective (Fig. S1B).
To further consider the importance of glucose to the parasites, trophozoites were incubated with the glycolytic poison 3-bromopyruvate (3BP) in standard glucose-bearing (9 mM glucose) NCM. Inclusion of the compound, which is known to inhibit the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (9), led to reduced cell growth (Fig. 1B), with only cyst-like structures detectable after 2 days of incubation. These structures were not viable cysts, as treatment with detergent (0.25% SDS) eliminated all of the cyst-like structures generated by the 3BP incubation, and viable trophozoites were not recovered after culture in detergent-free medium.
The absence of glucose or treatment with 3BP could be impacting N. fowleri by interfering with metabolism (through glycolysis) or sugar nucleotide biosynthesis (through the pentose phosphate pathway). Entry into either of these pathways requires glucose-6-phosphate, which is typically generated by HK or Glck enzymes, suggesting inhibitors of this activity are useful anti-amoebic compounds. The N. fowleri genome does not encode obvious HK-like enzymes but does harbor a gene for a putative Glck. Unlike HKs, which can transfer a phosphoryl group to different hexoses, Glcks have a restricted substrate range, primarily utilizing glucose as an acceptor for the γ-phosphoryl of ATP. Beyond Glcks from other Naegleria spp. and related amoebae, NfGlck shares limited identity (37%) with the Glck from Trichomonas vaginalis, a protist that lives primarily under anaerobic conditions. The enzyme was less similar to Homo sapiens Glck (HsGlck), T. cruzi Glck (TcGlck), and Leishmania donovani Glck, with sequence identities of 25%, 27%, and 26%, respectively (Data File S1 shows a comparison of the sequences of HsGlck, Kluyveromyces lactis HK, yeast hexokinase P1, TcGlcK, and NfGlcK).
Recombinant NfGlck, which was purified to ∼99% homogeneity (as determined by Coomassie staining of an SDS-PAGE gel), was determined to be a monomer by gel filtration chromatography. However, at high concentrations it eluted as a distinct shoulder of higher molecular weight. The enzyme displayed Michaelis-Menten kinetics when glucose or ATP levels were increased (Fig. 2A and B) and had apparent Km values of 42.5 ± 7.3 and 141.6 ± 9.9 μM for glucose and ATP, respectively. The enzyme had a kcat value of 36.5 s−1 and a pH optimum of 8.5 (Fig. 2C). NfGlck had a limited number of sugar substrates it could use in catalysis, with detectable activity when glucose, glucosamine, or fructose (at high concentration) was used as the sugar substrate (Fig. 3A). Glucosamine was also a competitive inhibitor of glucose, with a Ki of 186.4 ± 20.6 μM. While AMP and PPi had no impact, ADP was an inhibitor of the enzyme, with a Ki of 116.3 ± 7.0 μM (Fig. 3B). The enzyme functioned best when MgCl2 was included in the assay with MnCl2 and CaCl2, yielding 40% and 9%, respectively, of the activity observed when MgCl2 was included in the assay.
FIG 2.
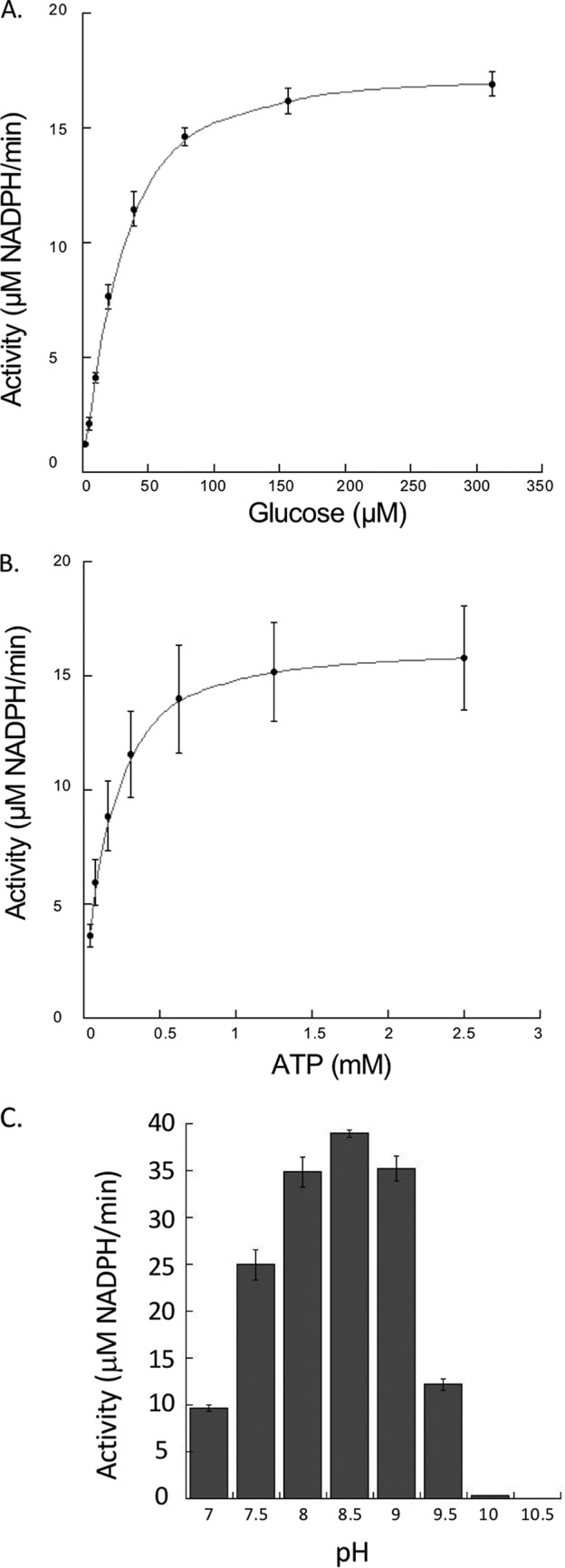
Effects of ATP, glucose, and pH on NfGlck activity. (A) NfGlck (10 nM) activity in response to increasing glucose concentrations (2.44 to 312 μM) (A), increasing ATP concentrations (0.04 to 5.0 mM) (B), and different pH levels (C). Two different buffers with buffering capacity in the range of pH tested were used for the assay, Tris-Cl (pH 7 to 8.5) and sodium carbonate (pH 9.0 to 10.5). For pH analysis, glucose and ATP concentrations were 425 μM and 1.5 mM ATP, respectively. Reactions were carried out in triplicate, and standard deviations are indicated.
FIG 3.
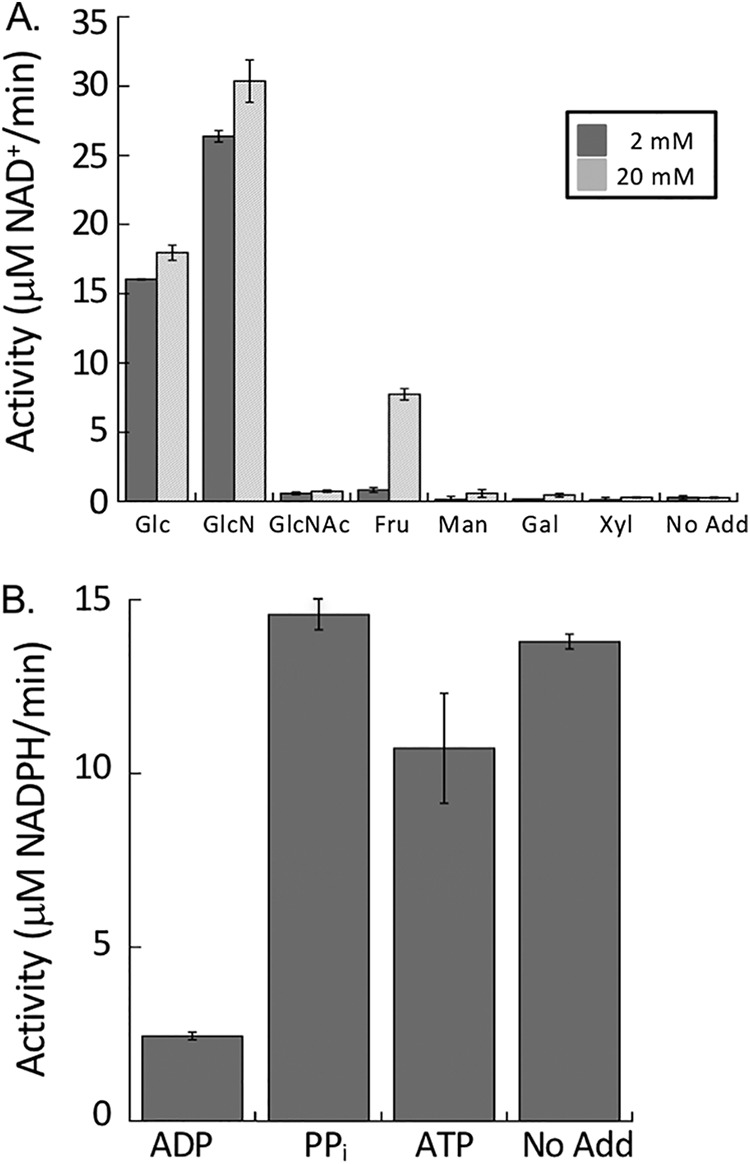
(A) NfGlck (10 nM) activity with various hexoses (5 mM). (B) Impact of phosphoryl-bearing substrates on NfGlck activity. NfGlck activity was measured in the presence of 1.5 mM ATP supplemented with ADP, PPi, and ATP (all at 5 mM). Assays were carried out in triplicate, and standard deviations are indicated.
Structural analysis of NfGlck. (i) Overall structure.
The structure of NfGlck was solved by molecular replacement despite low sequence homology with structures in the PDB. The closest homolog by sequence in the PDB is the T. cruzi Glck (TcGlck), with 27% sequence identity (Data File S1). This protein is also the source of the closest structural homologs in the PDB, which are structures of the TcGlck in complex with β-d-glucose and ADP (PDB entry 2Q2R, 1.7-Å root mean square deviations [RMSD]), in complex with carboxybenzyl glucosamine (CBZ-GLCN, 5BRE, 1.8-Å RMSD), and in complex with benzoyl glucosamine (BENZ-GLCN, 5BRD, 2.0-Å RMSD), hydroxyphenyloxopropyl glucosamine (HPOP-GLCN, 5BRF, 2.0-Å RMSD), and dioxobenzylthiophenyl glucosamine (DBT-GLCN, 5BRH, 2.0-Å RMSD) (10). The structure of HsGlck (3FGU) superimposes with an RMSD of 3.0 Å. In each of the alignments, ∼300 out of the 370 to 380 residues were aligned. Structure solution was facilitated by the domain approach of MorDa (11), which placed three domains from PDB entries 5BRH, 1SZ2, and 3AAB with an overall RMSD of 1.2 Å for 282 aligned Cα atoms. HsGlck shares little sequence homology with NfGlck. HsGlck in complex with an allosteric inhibitor (4MLH) superimposes on NfGlck with an RMSD of 2.9 Å over 244 residues. While NfGlck and HsGlck share a similar fold and glucose binding domain, the noted sequence and structural diversity between the two enzymes suggests that specific inhibitors of NfGlck could be developed.
The structure of NfGlck was refined against diffraction data up to 2.2-Å resolution (Fig. 4). The structure refines with appropriate quality indicators (Table 1), and NfGlck crystallizes with one molecule per asymmetric unit. N-terminal residues Met1-Glu22 and C-terminal residues Gly401-Lys443 do not have the sufficient electron density needed to be modeled, while a consecutive model could be built for residues Pro23-Leu400. NfGlck consists of two domains, a small α/β domain (residues Pro23-Asn171 and Asn384-Leu400) and a larger α/β domain (His174-Glu382). NfGlck was crystallized in the presence of adenylyl-imidodiphosphate (AMPPNP) and d-glucose, and both molecules could be modeled with confidence, even though AMPPNP did not appear to be fully occupied (81% refined occupancy). While d-glucose is located at the domain interface, AMPPNP mostly binds to the large domain.
FIG 4.
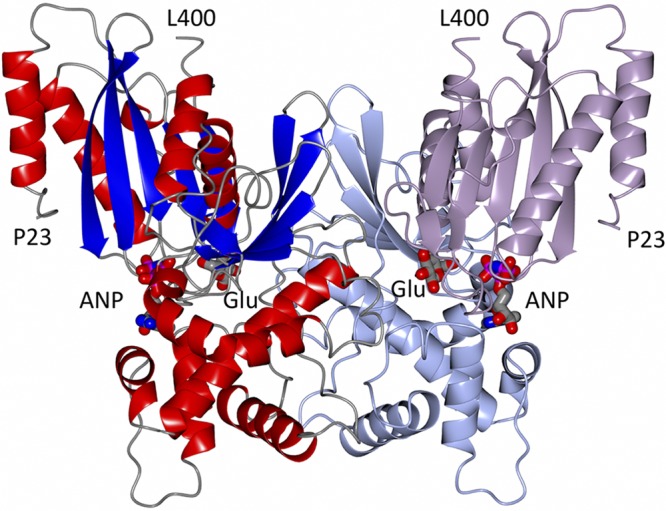
Structure of a crystallographic dimer of NfGlck bound to AMPPNP (ANP) and glucose (Glu) is shown in two color schemes. The left monomer is colored by secondary structure elements. The right monomer shows the smaller α/β domain (Pro23-Asn171 and Asn384-Leu400) in lavender and the larger α/β domain (His174-Glu382) in light blue. The glucose molecule is mostly buried, while part of AMPPNP is solvent accessible.
TABLE 1.
Crystallography parameters
| Crystal parametera | Value(s)b |
|---|---|
| Data collection statistics | |
| Space group | I422 |
| Cell dimensions | |
| a, b, c (Å) | 202.5, 202.5, 68.8 |
| α = β = γ (°) | 90.0 |
| Data set | |
| X-ray source | Rigaku F-RE+SuperBright |
| Wavelength (Å) | 1.5418 |
| Resolution (Å) | 50–2.20 (2.26–2.20) |
| Rmerge | 0.090 (0.572) |
| I/σ(I) | 17.95 (3.27) |
| CC1/2 | 99.0 (86.8) |
| Completeness (%) | 99.6 (99.7) |
| No. of reflections overall | 258,457 (13,992) |
| No. of reflections, unique | 36,362 (2,642) |
| Multiplicity | 7.1 (5.3) |
| Refinement statistics | |
| Reflections used in refinement (test set) | 36,357 (2,015) |
| Rwork | 0.1539 |
| Rfree | 0.1872 |
| RMSD | |
| Bond length (Å) | 0.007 |
| Bond angle (°) | 0.822 |
| Ramachandran plot | |
| Favored (%) | 97.61 |
| Allowed (%) | 2.39 |
| Outliers (no.) | 0 |
| No. of: | |
| Protein chains | 1 |
| Protein residues | 374 |
| Solvent molecules | 357 |
| Ligand molecules | 2 |
| Wilson B factor (Å2) | 27.1 |
| Avg B factors, all/main chain/solvent (Å2) | 35.6/31.0/44.6 |
| Molprobity clash score | 1.46 |
| Molprobity score | 1.03 |
| PDB entry | 6DA0 |
, , (where is the variance of average intensities of unique reflections of a resolution shell and is the average of all sample variances of the averaged intensities across all unique reflections), and , using a test set of 2,015 reflections, 5.5% of the data.
Values in parentheses are for the highest-resolution shell.
Despite the low sequence identity and high RMSDs, NfGlck and TcGlck have an identical topology; all secondary structure elements match up well. Notable exceptions are loops around residues Leu42-Phe47, Pro179-Gln190, and Gly238-247. None of these loops are involved in ligand binding. NfGlck appears to adopt the same nearly closed conformation as that described for inhibitor-bound TcGlck structures. The binding modes and binding environment for both d-glucose and AMPPNP or ADP, respectively, in NfGlck and TcGlck (2Q2R) are also very well conserved.
(ii) Quarternary structure.
NfGlck purifies mostly as a monomer in size exclusion chromatography; however, at higher concentrations the peak has a distinct higher-molecular-weight shoulder. NfGlck crystallizes with one molecule in the asymmetric unit. While the interface of a crystallographic dimer per PISA (12) only comprises 1,970 Å2, its shape complementarity as calculated by SC (13) is high (0.755). Furthermore, the crystallographic dimer of NfGlck is very similar to the noncrystallographic interface of TcGlck. We therefore assume that TcGlck is able to form dimers, for instance, at high concentrations in the crystal lattice.
(iii) Ligand binding.
In NfGlck (Fig. 5A), all hetero atoms of glucose form hydrogen bonds with protein residues or water molecules: O1 forms hydrogen bonds with His231 Nε2 and Glu268 Oε1/Oε2; O2 forms hydrogen bonds with Thr127 Oγ1, Glu228 Nε2, and His231 Nε2; O3 forms hydrogen bonds with Glu228 Oε2 and Asn155 Nδ2; O4 forms hydrogen bonds with Asp156 Oδ1 and water 727; O5 forms a hydrogen bond with water 838; and O6 forms hydrogen bonds with Asp156 Oε2 and water 635.
FIG 5.
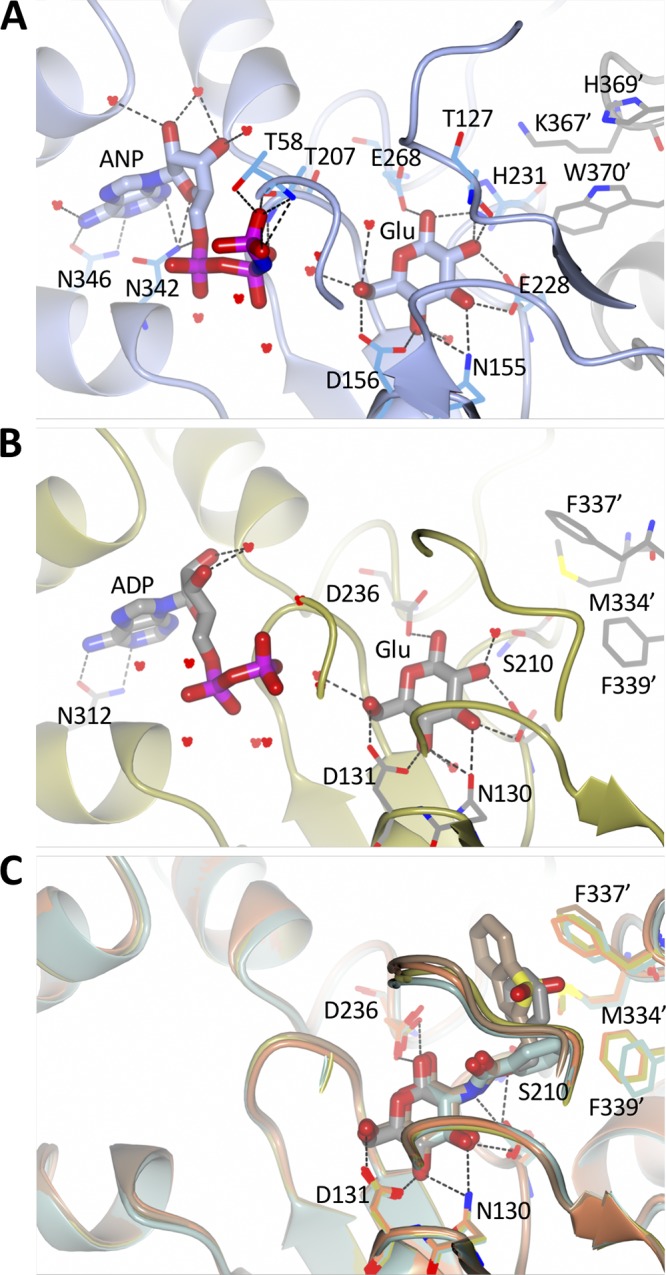
Nucleotide and glucose binding sites of various Glcks. (A) In NfGlck, AMPPNP (ANP) and glucose (Glu) are bound with a strong hydrogen bond network, including hydrogen bonds from the 1- and 2-hydroxyl groups to His231. AMPPNP and glucose are shown in sticks, while residues and water molecules interacting with the ligands are shown in thick bonds. Residues Lys367′, His369′, and Trp370 are from the other molecule in the crystallographic dimer. (B) In TcGlck, ADP and glucose are bound with a very similar hydrogen bond pattern. Ser210 replaces His231 and cannot engage in a hydrogen bond with glucose. Residues Met334′, Phe337′, and Phe339′ are located in the other chain of the dimer. (C) TcGlck bound to several glucosamine ligands (BENZ-GLCN, green, PDB entry 5BRD; CBZ-GLCN, gold, PDB entry 5BRE; HPOP-GLCN, orange, PDB entry 5BRF; BDT-GLCN, brown, PDB entry 5BRH), the amine moiety is part of the space that the His231 side chain of NfGlck occupies.
In contrast to a mostly buried glucose, the AMPPNP molecule is partially solvent accessible, especially the three phosphate groups. The adenine moiety makes a minor hydrophobic interaction with a small hydrophobic patch formed by the side chains of Leu276, Ala296, and Ala299. Most hetero atoms of AMPPNP interact with protein residues or water molecules: N1 and N6 form hydrogen bonds with Asn346 Nδ2 and Oδ1, respectively; N7 forms a hydrogen bond with water 624; O2′ forms hydrogen bonds with waters 620 and 784; O3′ forms hydrogen bonds with waters 620 and 730; O4′ forms a hydrogen bond with Asp342 Nδ2; O1A forms a hydrogen bond with water 650 and O1B with water 613; O2B forms hydrogen bonds with Thr 207 Oγ1 and N and with water 635; N3B forms hydrogen bonds with waters 660 and 788; and O1G, O2G, and O3G form hydrogen bonds with Thr 58 Oγ1 and N, Thr 207 Oγ1, and water 660.
In TcGlck (Fig. 5), there is no equivalent to the hydrogen bond of Thr127 with the 1-hydroxyl group of glucose. Similarly, in NfGlck, His231 provides additional hydrogen bonds with the 1- and 2-hydroxyl groups of glucose. This interaction is not available in TcGlck, where the equivalent Ser210 does not reach the glucose (Fig. 5A and B). The amine moiety of a glucosamine substrate would extend toward His231. This might explain the different specificities of NfGlck and TcGlck for glucosamine as a substrate. In HsGlck, the glucose binding is a blend of the NfGlck and TcGlck binding mode; there is no direct replacement for the interaction of His231. However, in HsGlck, Thr168 has a role similar to that of Thr127 in NfGlck.
Additional TcGlck structures have been described in the presence of several glucosamine analogue inhibitors (10) (Fig. 5C, CBZ-GLCN), where the glucose moiety of the ligand binds identically to glucose. The phenyl moiety of CBZ-GLCN, for instance, has hydrophobic interactions with residues Met334, Phe337, and Phe339 from the other chain in the dimer. Despite a variety of aromatic substituents, the orientation of Phe337 and Phe339 remains largely unchanged. In NfGlck, the equivalent residues are Lys334′, His369′, and Trp370′ of the other chain of the crystallographic dimer. Polar nitrogen atoms of the side chains point toward the site that the aromatic moiety of TcGlck ligands occupies. These differences in geometry and chemistry suggest a significantly different binding profile for glucosamine analogue inhibitors for NfGlck and TcGlck and options for specificity.
(iv) Identification of small-molecule inhibitors of NfGlck.
To identify inhibitors of NfGlck, a subset of known HK inhibitors and related structural analogs were tested against the N. fowleri glucokinase enzyme. For example, the screening collection assessed for this effort contained compounds selected from the Pathogen Box chemical library that were known to inhibit T. brucei hexokinase 1 (TbHK1), other structurally distinct small-molecule inhibitors identified in our TbHK1-based high-throughput screening (HTS) campaigns, and analogs subsequently developed from these hits (14). The advantage of using these compounds included the possibility that they would inhibit a different (15.2% identical at the amino acid level) parasitic metabolic enzyme through binding to conserved domains found in both proteins, including substrate binding domains. Additionally, our past experiences with validating these compounds indicated (i) that these agents did not interfere with the coupled enzyme used in the screen and (ii) that the compounds were generally nontoxic to mammalian cells (compounds were from Medicines for Malaria Venture [MMV]) (14).
NfGlck was inhibited by two structurally related bis-guanadinyl compounds from the Pathogen Box collection, MMV688179 and MMV688271, which had antiparasitic activity against African trypanosomes (15). Interestingly, MMV688179 yielded a 50% inhibitory concentration (IC50) value of 2.02 ± 0.31 μM against NfGlck and was similarly active against TbHK1 and Plasmodium falciparum hexokinase (PfHK), with IC50 values of 2.90 ± 1.13 and 0.78 ± 0.17 μM, respectively. This compound, which was a mixed inhibitor with respect to ATP, did not inhibit HsGlck at 10 μM (Table 2). Comparatively, the related compound MMV688271 was a mixed inhibitor with respect to ATP and showed modestly more potent inhibition of parasite enzymes, with IC50 values at least 2-fold better than that for MMV688179. However, the two compounds differed in that MMV688271 was less selective, inhibiting HsGlck with an IC50 value of 2.58 ± 0.53 μM, a value similar to those found for parasite enzyme inhibition. Neither Pathogen Box compound was toxic to Naegleria at 10 μM, but given their IC50 values, this was not surprising (data not shown).
TABLE 2.
Small molecule inhibitors of NfGlck
| Entry | Sample ID | Structure | IC50c (μM) |
|||
|---|---|---|---|---|---|---|
| NfGlck | TbHK1 | PfHK | HsGlck | |||
| 1 | MMV688179 | 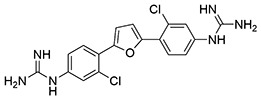 |
2.0 ± 0.31 | 2.9 ± 1.1 | 0.78 ± 0.17 | >10 |
| 2 | MMV688271 | 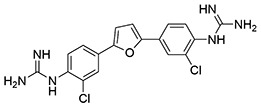 |
0.70 ± 0.09 | 1.3 ± 0.43 | 0.16 ± 0.06 | 2.6 ± 0.53 |
| 3 | SID856002 (ebselen) | 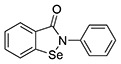 |
0.88 ± 0.06 | 0.05 ± 0.03a | 0.01 ± 0.00 b | NDd |
| 4 | SID17387000 (ebsulfur) | 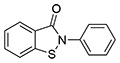 |
>10 | 2.0 ± 0.5a | 0.23 ± 0.03b | ND |
| 5 | SID144241589 (ML205) | 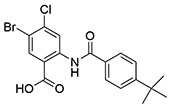 |
>10 | 0.98 ± 0.07e | >10 | >10 |
| 6 | SID162211071 | 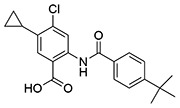 |
>10 | 6.7 ± 0.38 | ND | >10 |
| 7 | SID162211077 | 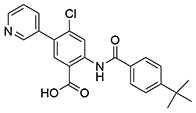 |
>10 | >10 | ND | >10 |
| 8 | SID144241590 | 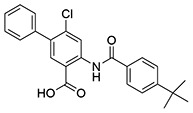 |
>10 | 0.28 ± 0.00 | >10 | >10 |
| 9 | SID162211078 | 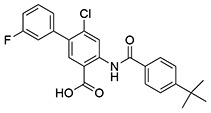 |
>10 | >10 | ND | >10 |
| 10 | SID162211083 | 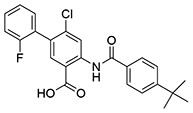 |
>10 | 6.7 ± 1.0 | ND | >10 |
| 11 | SID162211084 | 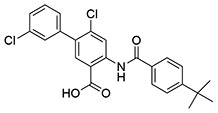 |
6.5 ± 0.61 | >10 | ND | >10 |
| 12 | SID162211073 | 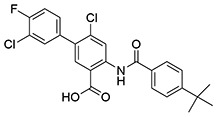 |
5.9 ± 0.26 | >10 | ND | >10 |
| 13 | SID162211080 | 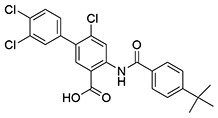 |
2.9 ± 0.32 | >10 | ND | >10 |
| 14 | SID162211075 | 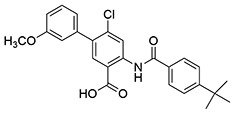 |
>10 | 3.9 ± 0.65 | ND | ND |
| 15 | SID162211076 | 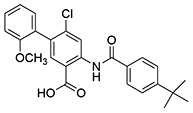 |
>10 | 9.3 ± 0.55 | ND | >10 |
| 16 | SID162211079 | 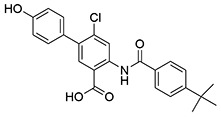 |
>10 | 3.1 ± 0.17 | ND | >10 |
| 17 | SID162211085 | 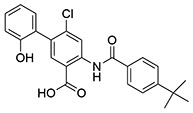 |
>10 | 1.2 ± 0.1 | ND | >10 |
| 18 | SID162211086 | 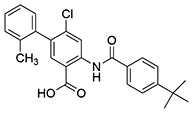 |
>10 | >10 | ND | >10 |
| 19 | SID162211072 | 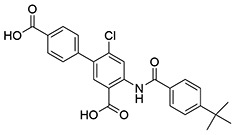 |
>10 | 3.5 ± 0.33 | ND | >10 |
| 20 | SID162211070 | 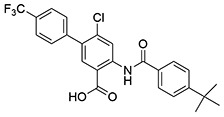 |
6.0 ± 0.20 | >10 | ND | >10 |
| 21 | SID162211074 | 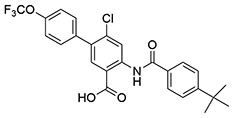 |
5.0 ± 0.26 | >10 | ND | >10 |
| 22 | SID162211081 | 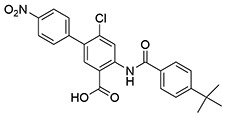 |
>10 | >10 | ND | ND |
| 23 | SID162211082 | 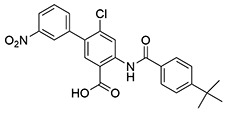 |
9.3 ± 0.74 | >10 | ND | >10 |
From the TbHK1 HTS, two distinct scaffolds were identified and pursued for analog development against TbHK1. Ebselen (SID856002) and the structurally similar isobenzothiazolinone (SID17387000) were identified as potent inhibitors of the trypanosome enzyme and are active against the Plasmodium HK (16). Of the two agents, only ebselen showed inhibition of NfGlck, with an IC50 of 0.88 μM, a notably higher concentration than that required to inhibit the other parasite HKs (Table 2). The second scaffold, a benzamidobenzoic acid-based inhibitor of TbHK1 (14), was developed through structure-activity relationship (SAR) optimization against the trypanosome to yield ML205, a potent inhibitor of TbHK1 (17). While this particular compound was not active against the Naegleria enzyme (IC50, >10 μM), evaluation of related analogs revealed some promising activity.
Analogs of ML205 tested in this work included compounds featuring substitution of the bromine atom on the benzoic acid core. One of the best modifications found for inhibition of TbHK1 involved exchange of the bromine atom of ML205 with a phenyl ring to afford SID144241590 with a TbHK1 IC50 value of 0.28 μM; however, this analog showed no significant inhibition of NfGlck. Likewise, bromine replacement with a cyclopropyl group (SID162211071), a 3-pyridyl moiety (SID162211077), or monofluorinated phenyl groups (SID162211078 and SID162211083) did not generate analogs that inhibited NfGlck, despite showing submicromolar activity in some cases against TbHK1.
While monofluorination of the phenyl appendage did not impact NfGlck, incorporation of chlorinated phenyl rings was advantageous against this enzyme. The analog bearing a 3-chlorophenyl group (SID162211084) gave moderate NfGlck inhibition (IC50 of 6.5 μM), the 3-chloro-4-fluorophenyl analog (SID162211073) trended towards marginally better potency, and the 3,4-dichlorophenyl derivative (SID162211080) showed the most improved potency against NfGlck, with an IC50 of 2.9 μM. Alternative phenyl ring substitutions, which included hydroxylation, methyl, or methoxy groups, did not afford analogs with appreciable inhibition of NfGlck, although several of these did demonstrate reasonable inhibition of TbHK1. Other analogs that inhibited NfGlck with potency better than 10 μM featured a 4-trifluoromethylphenyl or 4-trifluoromethoxyphenyl group (SID162211070 and SID162211074, respectively) or a 3-nitrophenyl group (SID162211082). Relative to that of ML205, the data suggest that elaborate phenyl rings in place of the bromine atom help to refine potency against NfGlck, and the specific substitution patterns that are favored are a result of a combination of spatial and electron-withdrawing effects. Further, this effort revealed that the structural elements that improve NfGlck inhibition appear to differ from those driving TbHK1 inhibition for this particular structural series as it relates to changes in this region of the scaffold. For instance, analogs with electron-donating substituents on the phenyl ring registered with some TbHK1 inhibition but did not do the same for NfGlck. Alternatively, analogs with more electron-deficient phenyl rings appended to the core favored NfGlck inhibition and not TbHK1.
DISCUSSION
As a thermophilic free-living amoeba, N. fowleri is found worldwide in or nearby warm freshwater. In the freshwater habitat, free glucose in the water column is likely limited, raising the possibility that NfGlck has only a limited role in parasite metabolism in this environment. However, the amoeboid trophozoite feeds on a variety of prey in the water, including bacteria, fungi, and other protists, which may generate substrates for NfGlck. The sediment at the bottom of freshwater sources is different, with free sugars, including glucose (1.1 mg/g dry weight), characterized in lake sediment samples (18, 19). Carbohydrates have also been characterized from lake surface mud samples, with glucose found at 13 to 191 mg/kg dry weight (20). These findings suggest that NfGlck plays a role in metabolism of nutrients acquired from the environment through either predation or directly from the sediment.
What role could NfGlck play during human infection? The process is usually initiated following forceful introduction of water contaminated with trophozoites into the nasal cavity. The presence of the trophozoites induces the host to secrete mucus, a response that serves as a barrier to invasion of the nearby tissue (21). The mucus contains an abundance of glycoproteins (including mucins), which compose the majority of its dry weight (22). To overcome this obstacle, the parasites release enzymes, including glycosidases, that degrade the mucin glycan components, potentially liberating substrates for NfGlck while aiding the parasite in accessing underlying tissue for ultimate invasion of the brain (23).
Degradation of host tissues during invasion provides access to glucose and glucose-bearing host macromolecules. As the parasite attacks the host cribriform plate, glucose availability is presumably enhanced once sufficient tissue damage has occurred to allow access to host blood, which has glucose levels that are maintained at ∼5 mM. The brain infection that follows exposes the parasite to lower (though still abundant) glucose concentrations, and the presence of parasites has been implicated in reducing the overall CSF glucose levels (5).
Whether NfGlck plays a central role in glucose catabolism in the diverse environments in which the parasite can reside, particularly the human brain, remains to be determined. However, because the enzyme also provides substrate to the pentose phosphate pathway (PPP) for the generation of sugar nucleotides, the activity is likely indispensable, a possibility that awaits the development of suitable molecular tools for genetic ablation of the enzyme activity in vivo. Nevertheless, the position of the enzyme at the head of glycolysis and the PPP suggests that interfering with its activity will yield potent antiparasitic compounds, and the differences in sequence and structure of NfGlck and HsGlck suggest inhibitor specificity can be established. Here, we have identified a number of enzyme inhibitors, but none that we tested impaired parasite growth at the levels tested (typically 10 μM). This may be for various reasons, including abundant substrate competitor molecules in the cytoplasm and insufficient access to the target in the cell. It is likely that much more potent inhibitors are required to see inhibition of parasite growth. Notably, however, the inhibitor ebselen (IC50 value of 0.88 μM) has been shown by others to be toxic to N. fowleri, with an EC50 value of 6.2 μM (24). It remains to be resolved if the toxicity is due to inhibition of the NfGlck or is the consequence of the known promiscuity of the reagent.
The NfGlck structure is highly similar to that of TcGlck. The T. cruzi enzyme was noted for having a structure consistent with that described for E. coli glucokinase (EcGlck), a group A Glck, which is a member of the group A ribokinase enzymes (10). Like other group A enzymes, NfGlck was limited to using ATP as a phosphoryl donor. However, kinetic differences between NfGlck and TcGlck raise the possibility that the Naegleria enzyme is an atypical group A member. This is primarily based on the observation that NfGlck, which can use glucose, glucosamine, or fructose as a substrate, lacks the glucose-limited substrate specificity that is a hallmark of group A enzymes. NfGlck and TcGlck also have distinct kinetic properties (7). NfGlck has an affinity for glucose that is ∼23-fold higher than that of TcGlck, perhaps explained by the additional hydrogen bonds provided by NfGlck His231 with the 1- and 2-hydroxyl groups of glucose, which are not present in TcGlck, where the equivalent residue is a Ser (Fig. 5A and B). Additionally, ADP inhibited NfGlck at much lower concentrations than those required for TcGlck inhibition (7).
The finding that NfGlck requires ATP for activity seems at odds with the presence of an N. fowleri PFK that is PPi dependent (4). Other pathogenic protists, like the anaerobes Entamoeba histolytica and Trichomonas vaginalis, have PPi-dependent glycolytic enzymes (4, 25), presumably because the alternative phosphoryl group donor allows optimized ATP production under energy-limiting conditions. The metabolic flexibility noted in Naegleria gruberi supports the notion that N. fowleri has a PPi-dependent PFK in order to support anaerobic fermentation (26). N. fowleri would likely encounter oxygen-poor environments, particularly when lake sediments are at higher temperatures during summer months. Alternatively, it is possible that an ATP-dependent PFK exists and has not yet been detected. It is known, for example, that some organisms can express either an ATP-dependent or PPi-dependent PFK in response to the types of environmental carbon sources available (27). Lastly, in some organisms a PPi-dependent PFK has been postulated to function as both a PFK in glycolysis and a fructose-bisphosphatase in gluconeogenesis (28). This is possible because thermodynamically (and under physiological conditions) PPi can be either consumed or regenerated. A similar role for the N. fowleri PPi-dependent PFK is unlikely, as parasites do not grow in the absence of glucose (Fig. 1). This suggests that any G6P generated by the PPi-dependent PFK is insufficient to meet the metabolic demands of the parasite and supports the notion that NfGlck is essential.
MATERIALS AND METHODS
Chemicals and reagents.
Glucose-6-phosphate dehydrogenase, β-NADP (NADP+), and phosphoenol pyruvate (PEP) were purchased from Alfa Aesar (Haverhill, MA). β-NAD (NADH), ATP, and dimethyl sulfoxide (DMSO) were from Fisher Scientific (Pittsburgh, PA), while 2-phenyl-1,2-benzisoselenazol-3(2H)-one (ebselen, [Eb]; PubChem SID856002) and glucosamine hydrochloride were obtained from VWR International (West Chester, PA). MMV688179 (2-[3-chloro-4-[5-[2-chloro-4-(diaminomethylideneamino)phenyl]furan-2-yl]phenyl]guanidine and MMV688271 (2-[2-chloro-4-[5-[3-chloro-4-(diaminomethylideneamino)phenyl]furan-2-yl]phenyl]guanidine) were provided by Medicines for Malaria Venture (Switzerland). The benzamidobenzoic acid-derived compounds (Table 1, entries 5 to 23) were provided by Jennifer E. Golden at the University of Wisconsin–Madison with >95% purity as determined by UV-mass spectrometry. See supporting information for synthetic details and characterization in Data File S1 in the supplemental material.
Growth inhibition assay.
N. fowleri (ATCC 30215) trophozoites were cultured axenically at 37°C in Nelson’s complete medium (NCM; 0.17% liver infusion broth [BD Difco, Franklin Lakes, NJ], 0.17% glucose, 0.012% sodium chloride, 0.0136% potassium phosphate monobasic, 0.0142% sodium phosphate dibasic, 0.0004% calcium chloride, 0.0002% magnesium sulfate, 10% heat-inactivated fetal bovine serum [FBS], 1% penicillin-streptomycin) in tissue culture-treated flasks (353135; Corning). Trophozoties were seeded at 2 × 106 cells/ml in 10 ml of NCM or NCM lacking glucose, and cultures were imaged daily on an EVOS imaging system (Thermo Fisher Scientific, Waltham, MA).
Recombinant NfGlck purification.
The open reading frame NF0035880 for the N. fowleri glucokinase (NfGlck) (http://amoebadb.org/amoeba/app/record/gene/NF0035880) was synthesized for codon optimization (Genescript, Piscataway, NJ), sequenced, and cloned into pQE-30 (Qiagen, Valencia, CA). Recombinant NfGlck, an ∼49-kDa protein, was purified by following a protocol developed for heterologous expression and purification of an HK from the African trypanosome. Briefly, an 8-ml bacterial culture of E. coli M15(pREP4) harboring pQE30-NfGlck was used to inoculate a 4-liter culture, which was grown to an optical density at 600 nm (OD600) of ∼0.6 and then induced for 24 h at room temperature with 500 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and purified using an approach modified from reference 29. After nickel column isolation (HisTrap FF; GE Healthcare Life Sciences, Pittsburgh, PA), active fractions were dialyzed into 20 mM Tris-HCl, pH 8, 50 mM NaCl and further purified by anion-exchange chromatography on a HiTrap Q XL (GE Healthcare Life Sciences, Pittsburgh, PA). The purified protein was stored in 25 mM HEPES-Na, pH 7.4, 150 mM NaCl, and 50% glycerol. Protein concentration was determined by Bradford assay using bovine serum albumin to generate a standard curve.
Glucokinase assay.
Enzyme was assayed in triplicate using a coupled reaction to measure enzyme activity. Briefly, enzyme (10 nM) in assay buffer (50 mM triethylamine hydrochloride [TEA-HCl], 3.3 mM MgCl2, pH 7.4) was added to clear 96-well plates and incubated for 15 min at room temperature. To start the reaction, 100 μl of substrate buffer (425 μM glucose, 1.5 mM ATP, 1 U/ml glucose-6-phosphate dehydrogenase, and 0.75 mM NADP+) was added to each well. The absorbance at 340 nm was then measured on a Biotek Synergy H1 microplate reader every 15 s for 2 min. Alternatively, enzyme activity was measured in a coupled reaction mixture containing pyruvate kinase and lactate dehydrogenase. Briefly, the 200-μl reaction mixture was placed in 96-well plates with NfGlck (10 nM) incubated in reaction buffer containing 50 mM TEA-HCl, pH 7.4, 3.3 mM MgCl2, 20 mM hexose, 2 mM ATP, 2.5 mM PEP, 0.6 mM NADH, and 1 μl pyruvate kinase-lactate dehydrogenase (Sigma, St. Louis, MO). This reaction measures the production of ADP through the coincident change in absorbance at 340 nm. Kinetic analyses were performed using KaleidaGraph 4.1 (Synergy Software, Reading, PA).
Expression and purification of NfGlck for crystallography.
The gene for NfGlck (AmoebaDB NF0035880) was amplified from N. fowleri RNA (the generous gift of Dennis Kyle, University of Georgia) and cloned into the expression vector pBG1861 using ligand-independent cloning (30). Expression from the vector yields a noncleavable 8-amino-acid tag (MAHHHHHH) immediately upstream of the start methionine of the gene (SSGCID target identifier [ID] NafoA.19900.a, SSGCID construct ID NafoA.19900.a.B11, and SSGCID batch NafoA.19900.a.B11.PW38443). NfGlck was expressed in E. coli Rosetta BL21(DE3)R3 by following standard SSGCID protocols as described previously (31). Purification was done using nickel-nitrilotriacetic acid affinity and size exclusion chromatography by following standard SSGCID protocols (32). The purified protein was concentrated to 13.78 mg/ml in its final buffer (25 mM HEPES, pH 7.0, 500 mM NaCl, 5% glycerol, 2 mM dithiothreitol, 0.025% NaN3), flash frozen in liquid nitrogen, and stored at −80°C.
Crystallization, data collection, and structure solution.
Crystallization set ups were performed using the apo protein and with the addition of 2 mM (each) MgCl2, adenylyl-imidodiphosphate (AMPPNP), and d-glucose and NfGlck at 13.78 mg/ml in 96-well XJR crystallization trays (Rigaku Reagents) with 0.4 μl protein mixed with a 0.4-μl reservoir, equilibrating against 80 μl reservoir solution. Crystallization conditions were searched for by using sparse matrix screens JCSG+ (Rigaku Reagents), MCSG1 (Microlytic/Anatrace), and Morpheus (Molecular Dimensions). Crystallization trays were incubated at 285 K. Several crystals were observed in the ligand trays, while no crystals grew in the apo trays.
Crystals from Morpheus condition C8 (12.5%, wt/vol, polyethylene glycol [PEG] 1000, 12.5%, wt/vol, PEG 3350, 12.5%, vol/vol, 2-methyl-2,4,-pentanediol, 30 mM each sodium nitrate, disodium hydrogen phosphate, ammonium sulfate, 100 mM morpholinepropanesulfonic acid-HEPES-Na, pH 7.5) were harvested and vitrified without adding additional cryoprotectant by plunging the crystals in liquid nitrogen. A diffraction data set was collected in house on a Rigaku FR-E+ SuperBright rotating anode equipped with Rigaku VariMax optics and a Saturn 944+ detector, using CuKα X-rays. The diffraction data were reduced with the XDS package (33) and are summarized in Table 1.
The structure was solved using the program MorDa (11) from the CCP4 package (34). The initial model was refined and extended using the phenix.ligand_pipeline script within Phenix (35). Manual model building was done with Coot (36), and the structure was refined in reciprocal space with Phenix.refine (35). Ligand restraints for AMPPNP and β-d-glucose were generated using the Grade web server (http://grade.globalphasing.org). The structure was validated using the built-in validation tools within Phenix.
Data availability.
The coordinates and structure factors were deposited in the PDB under accession code 6DA0.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christopher Rice from the laboratory of Dennis Kyle for providing the N. fowleri RNA library (to SSGCID) and for helpful comments on the manuscript. Work from the J.C.M. laboratory was supported in part by the U.S. National Institutes of Health Center for Biomedical Excellence (COBRE) grant under award number P20GM109094. This project has also been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN272201700059C. J.E.G. acknowledges institutional support from the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin–Madison with funding from the Wisconsin Alumni Research Foundation (WARF): UW2020225 infrastructure grant (J.E.G., principal invesigator). This work made use of the instrumentation at the UW−Madison Medicinal Chemistry Center, funded by the UW School of Pharmacy. M.K. was supported by the John & Jane Roudebush Distinguished Graduate Fellowship (School of Pharmacy Pharmaceutical Sciences Division), the Daniel H. Rich Distinguished Scholarship, and the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1256259.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02410-18.
REFERENCES
- 1.Yoder JS, Eddy BA, Visvesvara GS, Capewell L, Beach MJ. 2010. The epidemiology of primary amoebic meningoencephalitis in the USA, 1962-2008. Epidemiol Infect 138:968–975. doi: 10.1017/S0950268809991014. [DOI] [PubMed] [Google Scholar]
- 2.Seidel JS, Harmatz P, Visvesvara GS, Cohen A, Edwards J, Turner J. 1982. Successful treatment of primary amebic meningoencephalitis. N Engl J Med 306:346–348. doi: 10.1056/NEJM198202113060607. [DOI] [PubMed] [Google Scholar]
- 3.Linam WM, Ahmed M, Cope JR, Chu C, Visvesvara GS, da Silva AJ, Qvarnstrom Y, Green J. 2015. Successful treatment of an adolescent with Naegleria fowleri primary amebic meningoencephalitis. Pediatrics 135:e744–e748. doi: 10.1542/peds.2014-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mertens E, De Jonckheere J, Van Schaftingen E. 1993. Pyrophosphate-dependent phosphofructokinase from the amoeba Naegleria fowleri, an AMP-sensitive enzyme. Biochem J 292:797–803. doi: 10.1042/bj2920797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capewell LG, Harris AM, Yoder JS, Cope JR, Eddy BA, Roy SL, Visvesvara GS, Fox LM, Beach MJ. 2015. Diagnosis, clinical course, and treatment of primary amoebic meningoencephalitis in the United States, 1937-2013. J Pediatric Infect Dis Soc 4:e68–e75. doi: 10.1093/jpids/piu103. [DOI] [PubMed] [Google Scholar]
- 6.Kawai S, Mukai T, Mori S, Mikami B, Murata K. 2005. Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J Biosci Bioeng 99:320–330. doi: 10.1263/jbb.99.320. [DOI] [PubMed] [Google Scholar]
- 7.Caceres AJ, Quinones W, Gualdron M, Cordeiro A, Avilan L, Michels PA, Concepcion JL. 2007. Molecular and biochemical characterization of novel glucokinases from Trypanosoma cruzi and Leishmania spp. Mol Biochem Parasitol 156:235–245. doi: 10.1016/j.molbiopara.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Schuster FL. 2002. Cultivation of pathogenic and opportunistic free-living amebas. Clin Microbiol Rev 15:342–354. doi: 10.1128/CMR.15.3.342-354.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrke E, Arend C, Dringen R. 2015. 3-Bromopyruvate inhibits glycolysis, depletes cellular glutathione, and compromises the viability of cultured primary rat astrocytes. J Neurosci Res 93:1138–1146. doi: 10.1002/jnr.23474. [DOI] [PubMed] [Google Scholar]
- 10.D'Antonio EL, Deinema MS, Kearns SP, Frey TA, Tanghe S, Perry K, Roy TA, Gracz HS, Rodriguez A, D'Antonio J. 2015. Structure-based approach to the identification of a novel group of selective glucosamine analogue inhibitors of Trypanosoma cruzi glucokinase. Mol Biochem Parasitol 204:64–76. doi: 10.1016/j.molbiopara.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keegan RM, Long F, Fazio VJ, Winn MD, Murshudov GN, Vagin AA. 2011. Evaluating the solution from MrBUMP and BALBES. Acta Crystallogr D Biol Crystallogr 67:313–323. doi: 10.1107/S0907444911007530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence MC, Colman PM. 1993. Shape complementarity at protein/protein interfaces. J Mol Biol 234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 14.Sharlow ER, Lyda TA, Dodson HC, Mustata G, Morris MT, Leimgruber SS, Lee KH, Kashiwada Y, Close D, Lazo JS, Morris JC. 2010. A target-based high throughput screen yields Trypanosoma brucei hexokinase small molecule inhibitors with antiparasitic activity. PLoS Negl Trop Dis 4:e659. doi: 10.1371/journal.pntd.0000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duffy S, Sykes ML, Jones AJ, Shelper TB, Simpson M, Lang R, Poulsen SA, Sleebs BE, Avery VM. 2017. Screening the Medicines for Malaria Venture pathogen box across multiple pathogens reclassifies starting points for open-source drug discovery. Antimicrob Agents Chemother 61:e00379-17. doi: 10.1128/AAC.00379-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris MT, Walker DM, Drew ME, Mitchell WG, Dao K, Schroeder CE, Flaherty DP, Weiner WS, Golden JE, Morris JC. 2013. Interrogating a hexokinase-selected small-molecule library for inhibitors of Plasmodium falciparum hexokinase. Antimicrob Agents Chemother 57:3731–3737. doi: 10.1128/AAC.00662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flaherty DP, Harris MT, Schroeder CE, Khan H, Kahney EW, Hackler AL, Patrick SL, Weiner WS, Aube J, Sharlow ER, Morris JC, Golden JE. 2017. Optimization and evaluation of antiparasitic benzamidobenzoic acids as inhibitors of kinetoplastid hexokinase 1. ChemMedChem 12:1994–2005. doi: 10.1002/cmdc.201700592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers MA. 1965. Carbohydrates in aquatic plants and associated sediments from 2 Minnesota lakes. Geochim Cosmochim Acta 29:183. doi: 10.1016/0016-7037(65)90114-6. [DOI] [Google Scholar]
- 19.Whittaker JR, Vallentyne JR. 1957. On the occurrence of free sugars in lake sediment extracts. Limnol Oceanogr 2:98–110. doi: 10.4319/lo.1957.2.2.0098. [DOI] [Google Scholar]
- 20.Vallentyne JR, Bidwell RGS. 1956. The relation between free sugars and sedimentary chlorophyll in lake muds. Ecology 37:495–500. doi: 10.2307/1930172. [DOI] [Google Scholar]
- 21.Rojas-Hernández S, Jarillo-Luna A, Rodríguez-Monroy M, Moreno-Fierros L, Campos-Rodríguez R. 2004. Immunohistochemical characterization of the initial stages of Naegleria fowleri meningoencephalitis in mice. Parasitol Res 94:31–36. doi: 10.1007/s00436-004-1177-6. [DOI] [PubMed] [Google Scholar]
- 22.Bansil R, Stanley E, LaMont JT. 1995. Mucin biophysics. Annu Rev Physiol 57:635–657. doi: 10.1146/annurev.ph.57.030195.003223. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Castillo M, Cardenas-Guerra RE, Arroyo R, Debnath A, Rodriguez MA, Sabanero M, Flores-Sanchez F, Navarro-Garcia F, Serrano-Luna J, Shibayama M. 2017. Nf-GH, a glycosidase secreted by Naegleria fowleri, causes mucin degradation: an in vitro and in vivo study. Future Microbiol 12:781–799. doi: 10.2217/fmb-2016-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Debnath A, Nelson AT, Silva-Olivares A, Shibayama M, Siegel D, McKerrow JH. 2018. In vitro efficacy of ebselen and BAY 11-7082 against Naegleria fowleri. Front Microbiol 9:414. doi: 10.3389/fmicb.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mertens E, Ladror US, Lee JA, Miretsky A, Morris A, Rozario C, Kemp RG, Muller M. 1998. The pyrophosphate-dependent phosphofructokinase of the protist, Trichomonas vaginalis, and the evolutionary relationships of protist phosphofructokinases. J Mol Evol 47:739–750. doi: 10.1007/PL00006433. [DOI] [PubMed] [Google Scholar]
- 26.Fritz-Laylin LK, Prochnik SE, Ginger ML, Dacks JB, Carpenter ML, Field MC, Kuo A, Paredez A, Chapman J, Pham J, Shu S, Neupane R, Cipriano M, Mancuso J, Tu H, Salamov A, Lindquist E, Shapiro H, Lucas S, Grigoriev IV, Cande WZ, Fulton C, Rokhsar DS, Dawson SC. 2010. The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell 140:631–642. doi: 10.1016/j.cell.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 27.Alves AM, Euverink GJ, Santos H, Dijkhuizen L. 2001. Different physiological roles of ATP- and PP(i)-dependent phosphofructokinase isoenzymes in the methylotrophic actinomycete Amycolatopsis methanolica. J Bacteriol 183:7231–7240. doi: 10.1128/JB.183.24.7231-7240.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mertens E. 1991. Pyrophosphate-dependent phosphofructokinase, an anaerobic glycolytic enzyme? FEBS Lett 285:1–5. doi: 10.1016/0014-5793(91)80711-B. [DOI] [PubMed] [Google Scholar]
- 29.Morris MT, DeBruin C, Yang Z, Chambers JW, Smith KS, Morris JC. 2006. Activity of a second Trypanosoma brucei hexokinase is controlled by an 18-amino-acid C-terminal tail. Eukaryot Cell 5:2014–2023. doi: 10.1128/EC.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aslanidis C, de Jong PJ. 1990. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi R, Kelley A, Leibly D, Hewitt SN, Napuli A, Van Voorhis W. 2011. Immobilized metal-affinity chromatography protein-recovery screening is predictive of crystallographic structure success. Acta Crystallogr F Struct Biol Cryst Commun 67:998–1005. doi: 10.1107/S1744309111017374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryan CM, Bhandari J, Napuli AJ, Leibly DJ, Choi R, Kelley A, Van Voorhis WC, Edwards TE, Stewart LJ. 2011. High-throughput protein production and purification at the Seattle Structural Genomics Center for Infectious Disease. Acta Crystallogr F Struct Biol Cryst Commun 67:1010–1014. doi: 10.1107/S1744309111018367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project Network. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors were deposited in the PDB under accession code 6DA0.