

The increasing incidence of and high mortality rates associated with invasive fungal infections (IFIs) impose an enormous clinical, social, and economic burden on humankind. In addition to microbiological resistance to existing antifungal drugs, the large number of unexplained treatment failures is a serious concern.
KEYWORDS: Candida, Cdr1p, Tac1p, Upc2p, amphotericin B, antagonism, antifungal, azoles, echinocandins, resistance
ABSTRACT
The increasing incidence of and high mortality rates associated with invasive fungal infections (IFIs) impose an enormous clinical, social, and economic burden on humankind. In addition to microbiological resistance to existing antifungal drugs, the large number of unexplained treatment failures is a serious concern. Due to the extremely limited therapeutic options available, it is critical to identify and understand the various causes of treatment failure if patient outcomes are to improve. In this study, we examined one potential source of treatment failure: antagonistic drug interactions. Using a simple screen, we systematically identified currently approved medications that undermine the antifungal activity of three major antifungal drugs—fluconazole, caspofungin, and amphotericin B—on four prevalent human fungal pathogens—Candida albicans, Candida glabrata, Candida parapsilosis, and Candida tropicalis. This revealed that a diverse collection of structurally distinct drugs exhibit antagonistic interactions with fluconazole. Several antagonistic agents selected for follow-up studies induce azole resistance through a mechanism that depends on Tac1p/Pdr1p zinc-cluster transcription factors, which activate the expression of drug efflux pumps belonging to the ABC-type transporter family. Few antagonistic interactions were identified with caspofungin or amphotericin B, possibly reflecting their cell surface mode of action that should not be affected by drug efflux mechanisms. Given that patients at greatest risk of IFIs usually receive a multitude of drugs to treat various underlying conditions, these studies suggest that chemically inducible azole resistance may be much more common and important than previously realized.
INTRODUCTION
Invasive fungal infections (IFIs) are estimated to kill 1.5 million people each year (1), with mortality rates remaining disturbingly high, despite the availability of three major classes of antifungal drug (1). Millions more suffer debilitating infections of the mucosal membranes or destructive subcutaneous mycoses. Worryingly, the population of susceptible individuals and the incidences of both mucosal and systemic mycoses continue to rise due to the widespread use of broad-spectrum antibacterial agents, cancer chemotherapies, and the immune suppressive regimes necessary for organ transplantation, as well as the ongoing HIV and type 2 diabetes pandemics (2–6).
The azoles remain the most important and widely used class of antifungal treatment, acting through the inhibition of ergosterol biosynthesis. The polyenes, such as amphotericin B, damage the plasma membrane through direct interactions with ergosterol (7), while the members of the newest class of antifungals, the echinocandins, disrupt cell wall biosynthesis through inhibition of β1,3 glucan synthase (8). The increasing incidence of acquired fungal resistance, as well as the emergence of species with intrinsic resistance to each of these agents (9), undoubtedly contributes to the high frequency of treatment failure for IFIs. However, as many as two-thirds of clinical treatment failures occur in the absence of microbiological drug resistance (10–12). A variety of factors, such as inadequate drug distribution to the site of infection or the severity of a patient’s immune dysfunction, have been proposed to contribute to this phenomenon—however, little evidence is available in support of these arguments.
One factor that has been largely overlooked is the influence of other medications taken by the patient on the fungal pathogen itself. This is especially pertinent when one considers the fundamental similarity of fungal cells to those of their mammalian hosts. As eukaryotic organisms, fungi and mammals share similar cell biology and highly conserved signaling pathways. Accordingly, drugs designed to induce a physiological or biochemical response in humans may stimulate a similar response within fungal cells. These similarities have been previously exploited as the basis to identify existing drugs with antifungal activity or that enhance the activity of currently approved antifungals (13–16). However, there has not been a reciprocal effort to systematically identify pharmacotherapies that diminish the efficacy of antifungal drugs. Moreover, the influence of each coadministered medication on the clinical effectiveness of antifungal therapy remains largely unknown. This is important to consider given that the number of individuals within the population receiving more than one medication has grown dramatically in recent years (17, 18), and those at greatest risk of developing life-threatening fungal infections usually receive a multitude of drugs to treat a variety of underlying conditions (19). Recently, we screened a small collection of 129 oncology-related drugs and identified several that decreased the in vitro antifungal activity of the azoles on three of the most prevalent human fungal pathogens: Candida albicans, Candida glabrata, and Aspergillus fumigatus (20). Others have also noted individual drugs that oppose or antagonize the antifungal activity of the azoles (21, 22). The purpose of this study was to conduct a more comprehensive and systematic investigation of antifungal antagonism using a broader cross-section of approved medications in combination with all three antifungal drug classes. These screens revealed that a multitude of approved medications can induce antifungal resistance in C. albicans, C. glabrata, C. parapsilosis, and C. tropicalis. Finally, we have established one of the major mechanisms by which many of these antagonistic compounds exert their effect.
RESULTS
A systematic screen reveals that a variety of approved medications antagonize the antifungal activity of fluconazole.
To identify approved medications that undermine the efficacy of currently approved antifungal drugs, we conducted a simple screen to identify those which enhance fungal growth in the presence of inhibitory concentrations of fluconazole, caspofungin, or amphotericin B. The screens were conducted using the Prestwick collection of 1,280 compounds, ∼95% of which are off-patent drugs approved by the FDA or other agencies, and C. albicans SC5314 (23), C. glabrata ATCC 2001 (24), C. parapsilosis CDC317 (25), or C. tropicalis MYA3404 (26). The MIC of each antifungal drug was determined for each strain using the standard CLSI broth microdilution protocol (see Table S1 in the supplemental material) (27). The antagonism screens were then set up in 96-well plates with RPMI medium containing approximately 2× to 8× the MIC of fluconazole, caspofungin, or amphotericin B and 103 cells/well—conditions similar to those established by the CLSI. To ensure that the majority of drugs in the collection were provided at or above pharmacologically relevant concentrations, each library compound was added to a final concentration of 5 μM. Control wells contained either solvent alone (minus drug control) or the antifungal agent alone. After 24 h of incubation at 35°C, growth was quantified as optical density at 600 nm (OD600). Compounds that restored fungal growth in the presence of the antifungal drug (to ≥50% of the level seen with the untreated control) were called hits.
Thirteen compounds were identified that promoted C. albicans growth in the presence of 5 μM fluconazole (∼8-fold MIC) (Table 1). These hits encompass a wide array of therapeutic classes and do not appear to share significant structural or functional similarities. Seven compounds were selected for confirmation and follow-up analysis, based primarily on the prevalence of their clinical use and the likelihood of coadministration with fluconazole. This confirmed that the addition of any of the 7 compounds at a concentration of 5 μM resulted in an increase in the concentration of fluconazole required to inhibit C. albicans growth. The MIC of fluconazole increased approximately 8-fold in the presence of the antiemetic thiethylperazine and >128-fold in the presence of the nonsteroidal anti-inflammatory drug (NSAID) etofenamate (Fig. 1). The other 5 compounds, including the antipsychotic aripiprazole, the beta blocker penbutolol, and the corticosteroid mometasone, induced intermediate (16-fold to 32-fold) levels of resistance. Full checkerboard assays revealed that 5 of the 7 compounds tested antagonized fluconazole’s antifungal activity at concentrations of <1 μM (Fig. 2). Additional checkerboard assays revealed that all 7 compounds antagonized the antifungal activity of both voriconazole and itraconazole to similar extents (see Fig. S1 and S2 in the supplemental material), confirming that these interactions are not specific for fluconazole. Interestingly, when the same screen was conducted with 1 μM fluconazole (∼2-fold MIC), a total of 88 hits were found to restore C. albicans growth (Table S2). This suggests that a much larger number of approved medications may have a subtler effect on C. albicans susceptibility to fluconazole. The same screen was performed with C. parapsilosis and 25 μM fluconazole (∼4-fold MIC) as well as with C. tropicalis and 1.6 μM fluconazole (∼4-fold MIC), yielding 29 and 98 hits, respectively (Table S3 and Table S4). While the list of fluconazole antagonists identified was unique for each species, significant overlap was also observed (Fig. 3).
TABLE 1.
Agents from the Prestwick collection that suppress the antifungal activity of fluconazole on Candida albicans or Candida glabrataa
| Screen | Compound | Growth restoration (% of untreated control) |
|---|---|---|
| C. albicans, 5 μM fluconazole | Untreated | 100 |
| Fluconazole alone | 0 | |
| Fendiline hydrochloride | 132 | |
| Methiothepin maleate | 122 | |
| Hexetidine | 115 | |
| Prenylamine lactate | 83 | |
| Mometasone furoate | 121 | |
| Etofenamate | 102 | |
| Dimethisoquin hydrochloride | 58 | |
| Pinaverium bromide | 131 | |
| Halofantrine hydrochloride | 119 | |
| Penbutolol sulfate | 109 | |
| Thiethylperazine dimalate | 70 | |
| Toltrazuril | 63 | |
| Aripiprazole | 122 | |
| C. glabrata, 100 μM fluconazole | Untreated | 100 |
| Fluconazole alone | 0 | |
| Parthenolide | 61 | |
| Estramustine | 58 |
The Prestwick collection, containing a variety of FDA-approved drugs, was screened with each compound at a final concentration of 5 μM to identify those which restore the growth of either C. albicans or C. glabrata in the presence of inhibitory concentrations of fluconazole. Hits were defined as compounds that restored fungal growth to at least 50% of level seen with the untreated control wells.
FIG 1.
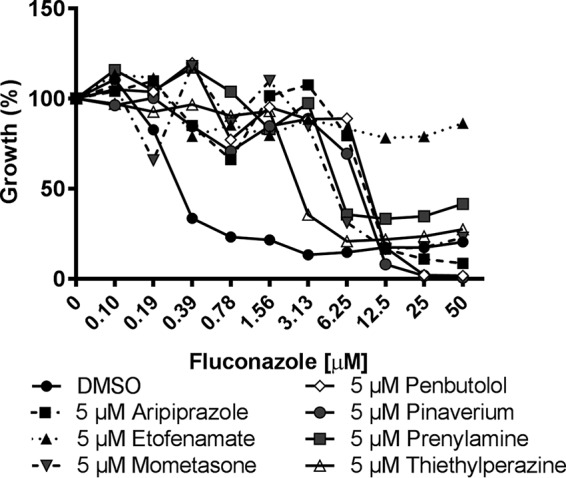
A variety of approved medications substantially diminish Candida albicans susceptibility to fluconazole. C. albicans (strain SC5314) susceptibility to fluconazole was tested in the absence (DMSO vehicle alone) or presence of 5 μM of the indicated compounds in RPMI medium according to the CLSI broth microdilution protocol. Growth was quantified as OD600 after 24 h of incubation at 35°C, and data are presented as percent growth relative to the drug-free control. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
FIG 2.

Several antagonistic compounds elevate Candida albicans fluconazole resistance at submicromolar concentrations. Checkerboard assays were performed in RPMI medium with C. albicans (strain SC5314) across a range of fluconazole doses and of (A) aripiprazole, (B) etofenamate, (C) mometasone, (D) penbutolol, (E) pinaverium, (F) prenylamine, and (G) thiethylperazine. After 24 h of incubation at 35°C, the growth in each well was quantified as OD600, normalized to the untreated control well, and data are presented as heat maps. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
FIG 3.
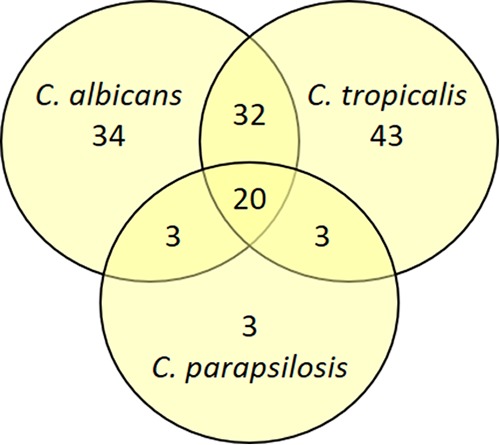
There is significant overlap in the azole antagonistic compounds from the C. albicans, C. parapsilosis, and C. tropicalis screens. The number of common and distinct compounds identified as hits from the fluconazole screens with these three species at 1, 25, and 1.6 μM, respectively, is illustrated in the Venn diagram.
We next conducted the same screen with C. glabrata in the presence of 100 μM fluconazole (∼4-fold MIC). Just two compounds were identified that restored growth to at least half the level observed in the untreated control—the steroidal estrogen and alkylating antineoplastic agent estramustine and parthenolide, which exhibits a variety of biological activities. Dose-response experiments confirmed that both compounds reduce fluconazole’s antifungal activity on C. glabrata, increasing the MIC by ∼8-fold (Fig. 4).
FIG 4.
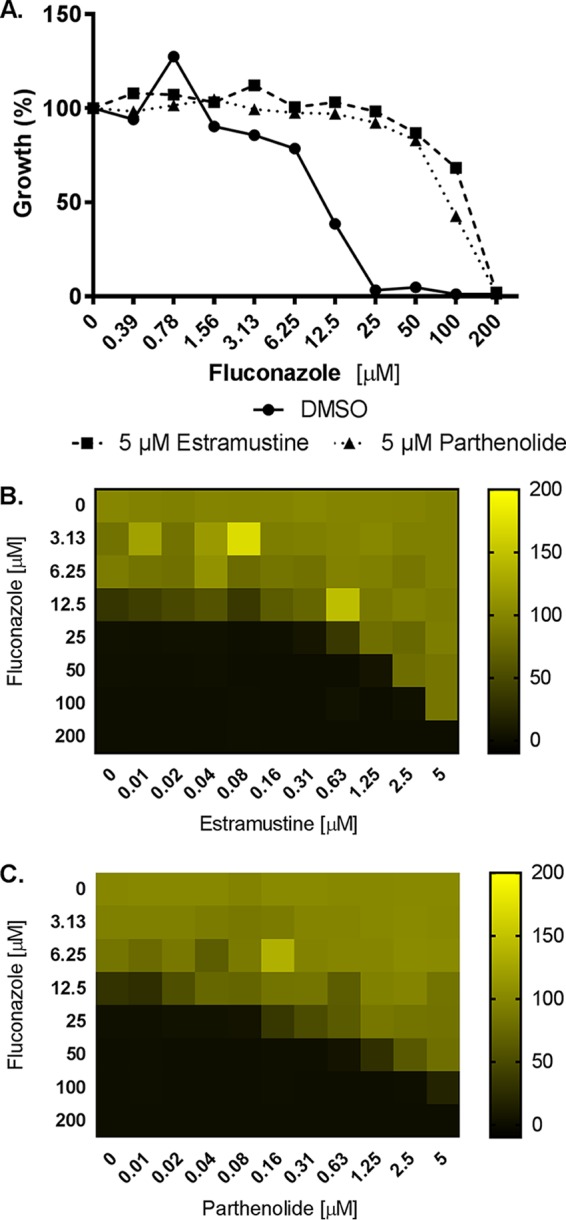
Estramustine and parthenolide elevate Candida glabrata fluconazole resistance. (A) The susceptibility of C. glabrata (strain ATCC 2001) to fluconazole was tested in the absence (DMSO vehicle alone) or presence of 5 μM estramustine or parthenolide in RPMI medium according to the CLSI broth microdilution protocol. Growth was quantified as OD600 after 24 h of incubation at 35°C, and data are presented as percent growth relative to the drug-free control. (B and C) Checkerboard assays were performed in RPMI medium with C. glabrata (ATCC 2001) across a range of fluconazole doses and of estramustine (B) and parthenolide (C). After 24 h of incubation at 35°C, the growth in each well was quantified as OD600, normalized to the untreated control well, and data are presented as heat maps. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
Similar antagonism screens were performed with the reference strains of each species in the presence of either 4-fold the MIC of caspofungin or 4-fold the MIC of amphotericin B. Just one compound—the topical antifungal amorolfine—restored C. albicans growth (to ≥50% that of the minus drug control) in the presence of caspofungin, while none restored growth of C. parapsilosis, C. tropicalis, or C. glabrata (Table 2). Two compounds—the antipsychotic haloperidol and ifenprodil—were identified as partially restoring the growth of C. albicans in the presence of amphotericin B (Table 2), but again, none restored the growth of C. parapsilosis, C. tropicalis, or C. glabrata. Dose-response experiments confirmed that amorolfine and haloperidol modestly increased the concentrations of caspofungin and amphotericin B, respectively, required to inhibit the growth of C. albicans (Fig. 5).
TABLE 2.
Agents from the Prestwick collection that suppress the antifungal activity of caspofungin or amphotericin B on Candida albicansa
| Screen | Compound | Growth restoration (% of untreated control) |
|---|---|---|
| 0.5 μM caspofungin | Untreated | 100 |
| Caspofungin alone | 0 | |
| Amorolfine | 58 | |
| 0.5 μM amphotericin B | Untreated | 100 |
| Fluconazole alone | 0 | |
| Haloperidol | 52 | |
| Ifenprodil tartrate | 63 |
The Prestwick collection, containing a variety of FDA-approved drugs was screened with each compound at a final concentration of 5 μM, to identify those which restore the growth of C. albicans in the presence of inhibitory concentrations of caspofungin or amphotericin B. Hits were defined as compounds that restore fungal growth to at least 50% of the untreated control wells.
FIG 5.
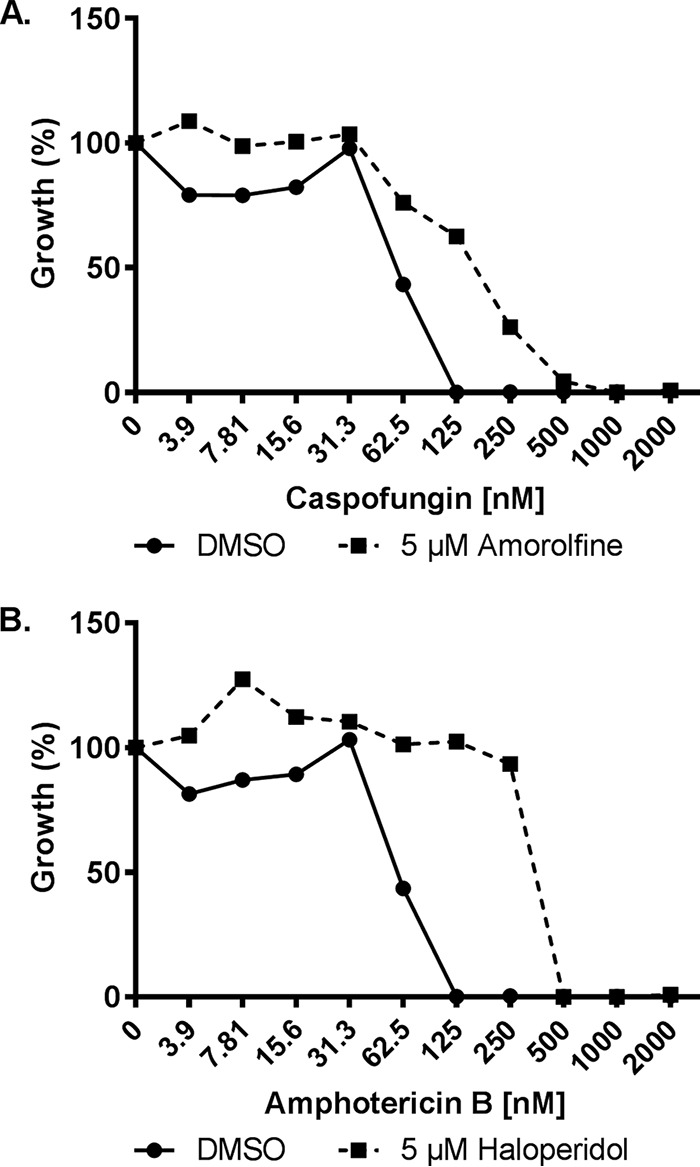
Amorolfine and haloperidol reduce Candida albicans susceptibility to caspofungin and amphotericin B, respectively. C. albicans (strain SC5314) susceptibility to caspofungin (A) or amphotericin B (B) were compared in the absence (DMSO vehicle alone) or presence of 5 μM amorolfine (A) and haloperidol (B) in RPMI medium according to the CLSI broth microdilution protocol. Growth was quantified as OD600 after 24 h of incubation at 35°C and is presented as percent growth relative to the drug-free control. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
Zinc-cluster transcription factors facilitate azole-antagonistic interactions in both C. albicans and C. glabrata.
In an effort to define the mechanisms underlying the antagonistic interactions observed with fluconazole, dose-response experiments were performed with a panel of C. albicans gene deletion strains lacking known mediators of antifungal resistance. This included three zinc-cluster transcription factors: Mrr1p, which activates expression of the major facilitator protein encoded by MDR1 (28); Tac1p, which activates the transcription of the CDR1 and CDR2 genes encoding ABC family multidrug transporters (29); and Upc2p, which activates the transcription of several genes in the ergosterol biosynthetic pathway (30). An mrr1Δ/Δ mutant exhibited the same patterns of fluconazole antagonism as those previously described for the wild-type strain, with all seven of the compounds tested (data not shown), indicating it has little or no relevant role. However, with the tac1Δ/Δ mutant, fluconazole antagonism was largely or completely lost for 6 of the 7 compounds. This indicates that the observed antagonism is dependent on Tac1p-mediated responses (Fig. 6 and Table 3). Only etofenamate was able to antagonize the activity of fluconazoles upon the tac1Δ/Δ mutant, suggesting that it acts through a distinct, Tac1p-independent mechanism. However, upon being tested with the upc2Δ/Δ mutant, the antagonism was completely abolished, indicating that etofenamate antagonizes fluconazole activity in a Upc2p-dependent manner (Fig. 6 and Table 3).
FIG 6.
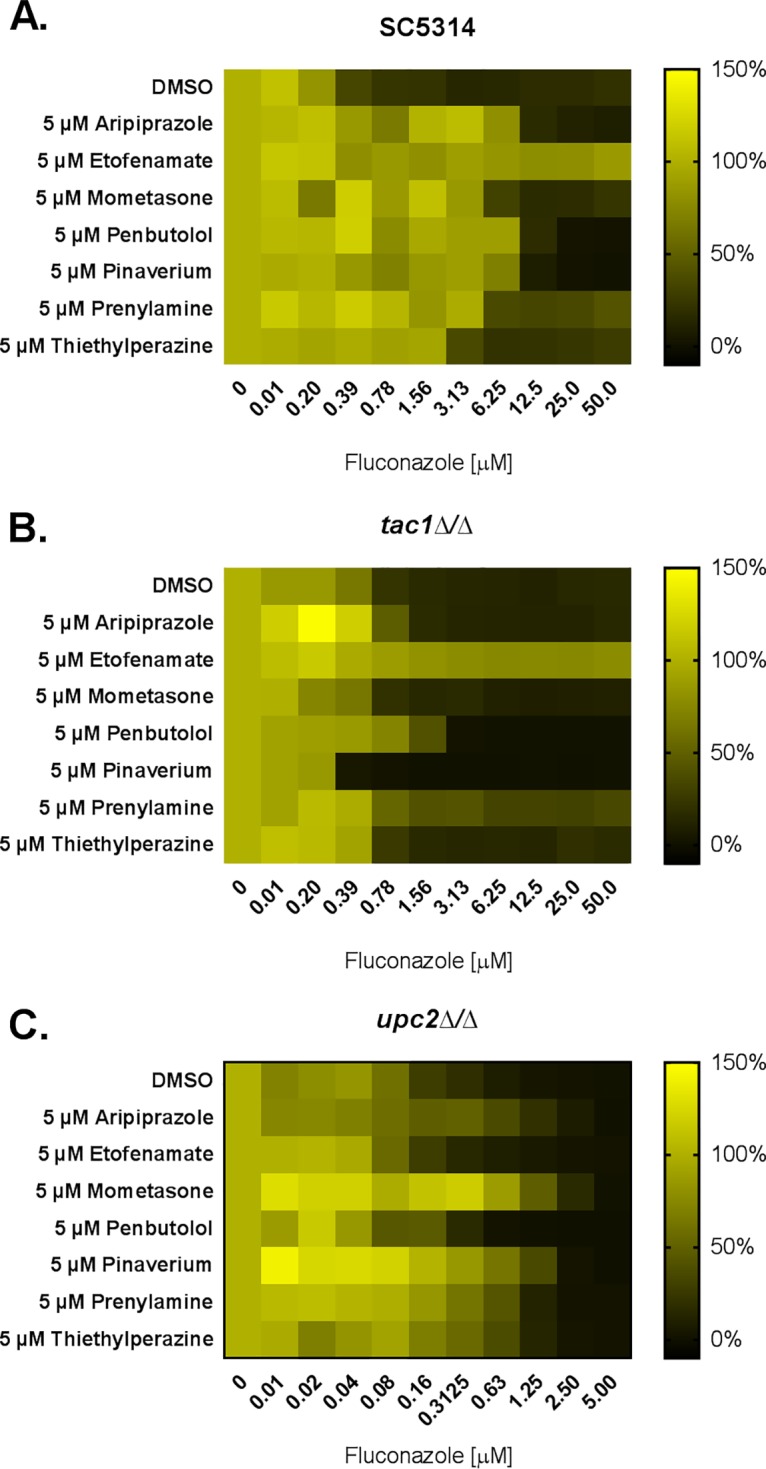
The Tac1p transcription factor is a major mediator of azole-antagonistic drug interactions. The susceptibility of wild-type (SC5314) (A), tac1Δ/Δ (B), and upc2Δ/Δ (C) C. albicans strains to fluconazole was tested in the absence (DMSO vehicle alone) or presence of 5 μM of the indicated compounds in RPMI medium according to the CLSI broth microdilution protocol. Growth was quantified as OD600 after 24 h of incubation at 35°C and expressed as a percentage of growth in the drug-free control, and data are presented as heat maps. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
TABLE 3.
Relative levels of fluconazole resistance induced in Candida albicans by selected antagonistic compoundsa
| Compound | Fluconazole resistance |
||
|---|---|---|---|
| Strain SC5314 | Strain tacΔ/Δ | Strain upc2Δ/Δ | |
| DMSO | 1× | 1× | 1× |
| Aripiprazole (5 μM) | 32× | 1× | >4× |
| Etofenamate (5 μM) | >256× | >128× | 1× |
| Mometasone (5 μM) | 16× | 1× | >4× |
| Penbutolol (5 μM) | 32× | 4× | >2× |
| Pinaverium (5 μM) | 32× | 1/2× | >4× |
| Prenylamine (5 μM) | 16× | 1× | >2× |
| Thiethylperazine (5 μM) | 8× | 1× | >2× |
Fluconazole susceptibility assays were performed using a modified version of the CLSI broth microdilution method for the indicated strains in the presence and absence of the seven indicated compounds at a final concentration of 5 μM. After 24 h of incubation, growth was quantified as OD600, and fluconazole MICs determined as the lowest concentration resulting in a 50% decrease in growth. The fluconazole MIC occurring with each drug treatment was then expressed relative to the fluconazole-alone control (DMSO).
To confirm that the Tac1p-dependent antagonistic interactions occur through activation of CDR1 and CDR2 transcription, wild-type C. albicans was grown with aripiprazole or etofenamate alone or in combination with fluconazole and the relative abundances of each gene’s transcripts were compared.
Treatment with 5 μM aripiprazole, either alone or in the presence of fluconazole, resulted in a dramatic induction of both CDR1 and CDR2, consistent with Tac1p activation. As expected, treatment with 5 μM etofenamate, which induces fluconazole resistance in a Tac1p-independent manner, did not elevate the levels of either transcript (Fig. 7).
FIG 7.
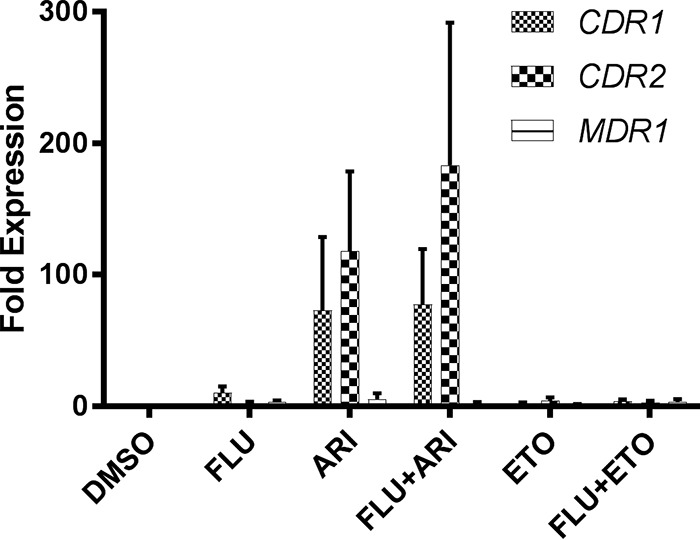
Aripiprazole induces transcription of the CDR1 and CDR2 genes in C. albicans. SC5314 was grown in YPD medium buffered with MOPS and adjusted to pH 7 in the presence of the indicated concentrations of drugs or of DMSO vehicle alone before RNA was isolated, and the relative abundance of the CDR1, CDR2, and MDR1 transcripts were determined by qRT-PCR analysis. The relative abundances of these transcripts in each sample were normalized to that of the ACT1 transcript and expressed relative to the level seen with the DMSO control (fold expression). Flu, fluconazole; Ari, aripiprazole; Eto, etofenamate. Mean and standard deviations of results from three separate experiments, each performed with three technical replicates, are shown.
In C. glabrata, Pdr1p—the functional paralog of Tac1p—activates the transcription of PDR5, a gene encoding an ABC type multidrug transporter that is a major driver of azole resistance in this species (31). Fluconazole dose-response experiments were performed with a C. glabrata pdr1Δ mutant in the presence or absence of both parthenolide or estramustine. Antagonism was significantly reduced compared to the levels seen with the wild-type strain for both compounds (Fig. 8 and Table 4), indicating that the Pdr1p transcription factor is required. In contrast, both estramustine and parthenolide antagonized the effect of fluconazole’s antifungal activity on a C. glabrata upc2aΔ mutant, indicating that these interactions were not facilitated by the Upc2a transcription factor.
FIG 8.
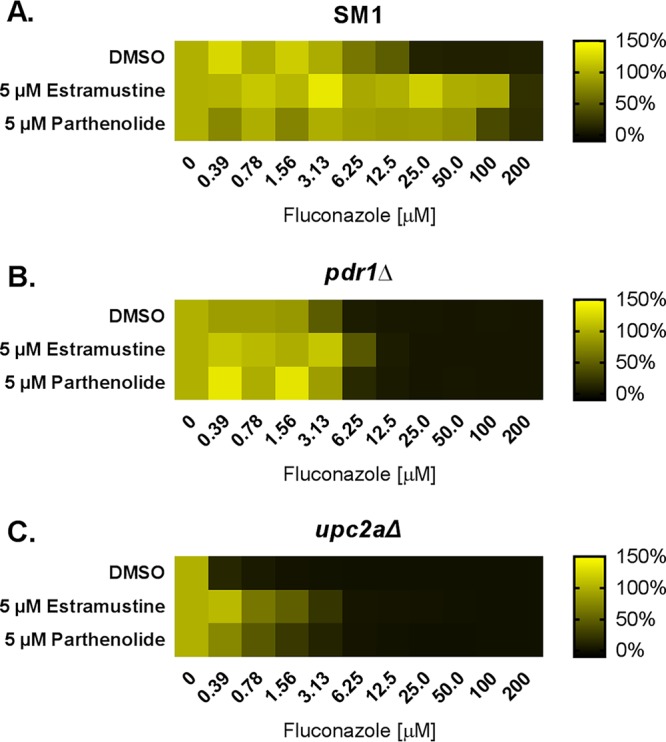
The Pdr1p transcription factor facilitates azole-antagonistic drug interactions in Candida glabrata. The susceptibility of wild-type (SM1) (A), pdr1Δ (B), and upc2aΔ (C) strains of C. glabrata to fluconazole was tested in the absence (DMSO vehicle alone) or presence of 5 μM of the indicated compounds in RPMI medium according to the CLSI broth microdilution protocol. Growth was quantified as OD600 after 24 h of incubation at 35°C, expressed as a percentage of growth in the drug-free control, and data are presented as heat maps. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
TABLE 4.
Relative levels of fluconazole resistance induced in Candida glabrata by selected antagonistic compoundsa
| Compound | Strain SM1 | Strain pdr1Δ |
|---|---|---|
| DMSO | 1× | 1× |
| Estramustine (5 μM) | 8× | 2× |
| Parthenolide (5 μM) | 8× | 1× |
Fluconazole susceptibility assays were performed using a modified version of the CLSI broth microdilution method for the indicated strains in the presence and absence of the two indicated compounds at a final concentration of 5 μM. After 24 h of incubation, growth was quantified as OD600, and fluconazole MICs determined as the lowest concentration resulting in a 50% decrease in growth. The fluconazole MIC occurring with each drug treatment was then expressed relative to the fluconazole-alone control (DMSO).
Induced and genetically encoded mechanisms can act in concert to elevate azole resistance.
Azole resistance in Candida is usually a multifactorial trait, with multiple genetically encoded mechanisms required to confer clinically relevant levels of azole resistance (32, 33). We therefore determined if the increase in fluconazole MIC induced by the aforementioned medications would be able to further elevate resistance in strains possessing a single, genetically encoded mechanism of resistance. We first compared the effects of the different drugs on a strain harboring an activated allele of UPC2 (34). As expected, the UPC2G648D allele elevated the MIC of fluconazole by 8-fold compared to the isogenic control (Fig. 9). The addition of any of the 7 azole-antagonistic compounds tested further elevated the MIC of the UPC2G648Dstrain (Fig. 9), demonstrating that they can make an independent and additive contribution to azole resistance. A similar pattern of results was observed with a strain harboring an activated allele of MRR1 (35). However, as expected, the effects of the six Tac1p-dependent antagonistic drugs were much less pronounced in a strain harboring an activated allele of TAC1 (Fig. 9). These data are again consistent with these six compounds acting through activation of the Tac1p transcription factor.
FIG 9.

Drug-induced and genetically encoded mechanisms can act in an additive manner to confer azole resistance in Candida albicans. The susceptibility of wild-type C. albicans (SC5314) (A), of strains harboring activated (gain of function—GOF) alleles of MRR1 (B), TAC1 (D), or UPC2 (F), or of isogenic control strains (C, E, and G) to fluconazole was tested in the absence (DMSO vehicle alone) or presence of 5 μM of the indicated compounds in RPMI medium according to the CLSI broth microdilution protocol. Growth was quantified as OD600 after 24 h of incubation at 35°C, expressed as a percentage of growth in the drug-free control, and presented as heat maps. Data represent averages of results from technical duplicates from a single experiment representative of two biological replicates.
DISCUSSION
Reports have suggested that as many as one-third of all patients with disseminated Candida infections fail to respond to antifungal therapy (10, 36–39). Both intrinsic and acquired mechanisms of antifungal drug resistance are known to reduce the chances of successful treatment of IFIs (11). However, on the basis of in vitro susceptibility testing, a substantial proportion of treatment failures occur in patients infected with antifungal-sensitive isolates. In addition, many infections involving apparently drug-resistant fungi do in fact respond to treatment (40). In fact, data from clinical trials indicate response rates of 60% to 81% with fluconazole for patients with disseminated Candida infections caused by isolates with in vitro MICs of ≤8 μg/ml, i.e., that are deemed susceptible according to current clinical breakpoints (10). Several host-related factors have been proposed to explain the discordance between the results of in vitro susceptibility tests and patient outcomes (10).
First, any factor that impacts an antifungal drug pharmacokinetics, including the efficiency of drug distribution and peak concentrations at the site of infection as well as the duration over which supratherapeutic concentrations are maintained, is likely to be a critical determinant of a patient’s response to therapy (41, 42). Certainly, subtherapeutic concentrations of the antifungal drug at the site of infection could explain unexpected treatment failures in those afflicted by a susceptible fungus. To some extent, these factors may be controlled through dosing schedules and potentially the route of antifungal administration. Nonetheless, some tissues pose specific challenges for a given class of antimicrobial, and pathology induced by the invading microbe (e.g., necrosis) may also restrict drug penetration to foci of infection. Additionally, the host’s genetics, age, physiological status (e.g., renal or hepatic function), underlying comorbidities, and drug-drug interactions may directly or indirectly impinge upon the pharmacokinetics of an antimicrobial drug and thus, ultimately, its clinical efficacy.
The severity of patient immunosuppression may also have an impact upon the response to antifungal therapy. Patients experiencing severe immunosuppression may be less likely to respond to treatment, especially with fungistatic drugs such as the azoles, and the response may depend in part upon host immune system-mediated clearance. However, it has been difficult to find unequivocal evidence in support of this argument since immunosuppressed mice and immunocompetent mice with a disseminated C. albicans infection respond similarly to azole treatment (43). Other factors that might account for variable patient outcomes relate to the size of the infectious dose and/or the time elapsed before an appropriate antifungal is administered. Intuitively, a more severe infection with a heavier fungal burden would be expected to have a reduced chance of therapeutic success. Similarly, more-advanced infections that have already caused significant damage to the host may also be associated with worse therapeutic outcomes. Certainly, rapid diagnosis, identification of the etiological agent, and administration of an appropriate antifungal agent can dramatically reduce the mortality rates of IFIs. Thus, while standardized antifungal susceptibility tests can be a useful predictive tool to facilitate selection of the antimicrobial agent with the greatest chance of therapeutic success, there are clearly a variety of additional factors that determine the likelihood of an effective intervention.
The results of this study have highlighted another factor that could potentially have a substantive impact upon a patient’s response to antifungal therapy and ultimately help explain poor outcomes—the other medications that a patient receives. While there are a number of previous reports of antifungal antagonism, these individual interactions were usually found inadvertently and/or were restricted to testing of specific drug combinations (22, 44, 45). Notably, Henry et al. tested a small set of 16 anti-infective agents and found that both sulfadiazine and albendazole antagonized the in vitro activity of fluconazole on C. albicans to a modest degree (21). As such, the current study and our previous one (20) appear to be the only comprehensive and systematic efforts to identify this type of interaction using multiple classes of antifungal drug and with multiple pathogenic species. Our most striking finding is that antagonistic interactions are much more common than previously appreciated. A staggering 139 of the 1,280 compounds in the Prestwick collection were found to induce fluconazole resistance in at least one of the four Candida species tested. Given that the collection used in this study is far from a complete representation of the current pharmacopeia, we anticipate that a more comprehensive screen of all approved medications would reveal an even larger number of antagonistic interactions. Thus, on the basis of the prevalence of this phenomenon, it is highly likely that one or more of the antagonistic interactions reported here are clinically relevant and reduce the efficacy of antifungal therapy.
The results of this study have provided several additional interesting insights, each of which has potentially important clinical implications. First, the compounds identified as antifungal antagonists were not obviously enriched for any one mechanistic class or indication and no common chemical features were apparent. Given the often complex and patient-specific drug regimens provided to those with IFIs, as well as the species specificity of the antagonistic medications, it would likely be extremely difficult to detect the negative consequences of such interactions through clinical observation alone. Second, notably fewer drugs were identified that antagonized either caspofungin or amphotericin B activity compared to fluconazole. This could reflect the fact that both amphotericin and caspofungin are fungicidal against Candida species, whereas the azoles are only fungistatic. The static nature of fluconazole’s activity may afford a greater opportunity for antagonistic compounds to induce protective physiological responses in the fungus than is the case for antifungals with a rapidly cidal mode of action. In our straightforward screening protocol, the antifungal drug was essentially coadministered with the test compound. A greater number of antagonistic agents might have been detected if the fungi had been precultured with each library compound before either amphotericin or caspofungin was introduced, as this might have provided an opportunity to induce or activate protective responses that oppose these agents. Perhaps a more relevant explanation for the different frequencies with which antagonistic compounds were identified with the three classes of antifungal drug relates to the cellular location of their respective targets. Lanosterol demethylase (Erg11p) is anchored to the endoplasmic reticulum within the cell, whereas both ergosterol and β-(1,3)-d-glucan synthase, targets of amphotericin and caspofungin, respectively, are partially exposed at the cell surface. Furthermore, the echinocandins are believed to inhibit β-(1,3)-d-glucan synthase through binding to an extracellular domain (46). As such, in contrast to the azoles, neither amphotericin B nor caspofungin needs to enter the cell to engage their target. Either way, the echinocandins are not substrates of the same drug efflux pumps that confer azole resistance (47). This is significant based on our finding that the majority of the fluconazole-antagonistic compounds act through inducing the expression of drug efflux pumps that transport the antifungal out of the cell. These mechanisms are not likely to affect sensitivity to antifungals that act at the cell surface, as they would not remove these agents from their site of action. As such, we might expect antifungals which act upon intracellular targets to be more prone to antagonistic drug interactions than those acting upon cell surface targets.
Traditionally, antifungal resistance has been considered to result from either the selection of intrinsically resistant species or the acquisition of mutations that confer resistance in a previously susceptible isolate. In both instances, antifungal resistance is a genetically encoded trait that is stably inherited from one generation to the next. Several genetic mechanisms can confer azole resistance to Candida spp., including those which alter the expression or function of enzymes in the ergosterol biosynthesis pathway. However, the most common involve mutations that result in increased expression of the efflux pumps that actively transport the azoles out of the cytoplasm (48, 49). In C. albicans, gain-of-function mutations in the zinc-cluster transcription factors Mrr1p and Tac1p trigger the transcription of MDR1 and of CDR1 and CDR2, respectively, each of which encode azole efflux pumps. The results of this and previous studies (21) challenge the perception of antifungal susceptibility as a relatively fixed and genetically encoded trait and suggest that the incidence of inducible antifungal resistance may have been grossly underestimated. This may help to explain the often-poor correlation between the results of in vitro antifungal susceptibility testing and patient outcomes. In the simplest case, a patient receiving medications that sensitize an invading fungal pathogen to an antifungal is likely to be more responsive to therapy than a patient receiving an antagonistic medication. Thus, all medications received by the patient could potentially have a pronounced impact upon the chances of therapeutic success. It is interesting to note that many of the antagonistic drugs act through the same mechanisms as those previously described as genetically encoded determinants, i.e., zinc-cluster transcription factors. However, this does not rule out the possibility that other antagonistic compounds may act through unrelated or distinct mechanisms.
Our results are consistent with a previous study by Thakur et al. that demonstrated that C. glabrata Pdr1p plays a direct role in sensing compounds through a xenobiotic binding domain, which leads to the activation of the transcription factor and the upregulation of a major efflux pump (50). That there were relatively few azole antagonistic hits common to C. glabrata and the other Candida species is somewhat surprising, especially given the significant overlap of the other three species. This suggests that there may be divergence in the specific xenobiotic detection mechanisms and/or associated substrate specificities responsible for the induction of the multidrug resistance pumps. Either way, our findings leave several unanswered questions. How can such a diverse group of xenobiotics be sensed by a single pathway? Does Tac1p/Pdr1p directly interact with and sense the antagonistic drugs, or are other ancillary proteins required? Additionally, do variants exist in genes encoding the xenobiotic-sensing proteins that alter sensitivity or substrate specificity and hence select for the medications that induce resistance for a given fungal species?
While the results described above are interesting from a biological perspective, the fundamentally important question remains—do the antifungal antagonistic interactions observed in vitro occur within patients, and if so, are they sufficient to affect clinical efficacy or outcomes? Since a single concentration of 5 μM was selected for screening purposes, it is unlikely that all of the compounds identified through our screens would be antagonistic at pharmacologically relevant concentrations or under physiological conditions. However, checkerboard assays revealed that four of the seven antagonistic compounds selected for follow-up analysis were able to elevate C. albicans fluconazole resistance at, or close to, pharmacologically relevant concentrations. For example, plasma concentrations of ∼1 μM aripiprazole, a widely used antipsychotic and mood stabilizer, can be achieved in patients, a concentration sufficient to increase the fluconazole MIC of C. albicans by 4-fold to 8-fold. Thus, it seems highly likely that at least a subset of the described antagonistic drugs would affect susceptibility in patients and, by extension, the chances for a successful intervention.
While drug-drug interactions have been a serious concern from the perspective of patient toxicity, the effects of most medications on fungal physiology and antifungal susceptibility have not been considered to date. A more comprehensive assessment of all approved medications with a broader variety of infectious fungi is needed to determine the full extent of this phenomenon. Furthermore, the potential clinical implications of antifungal antagonism should be thoroughly investigated using animal models of invasive fungal disease, as well as careful analysis of clinical outcomes from large patient cohorts that are appropriately controlled for and stratified according to their underlying risk factors.
MATERIALS AND METHODS
Growth conditions.
Strains were routinely grown on YPD medium (1% yeast extract, 2% peptone, 2% dextrose) at 30°C. The RPMI 1640 medium (Sigma-Aldrich) used for antifungal susceptibility testing as well as for the antagonism screens was prepared according to the CLSI M27-A3 document (51) and was buffered with morpholinepropanesulfonic acid (MOPS), with pH adjusted to 7.0 using NaOH and HCl.
Fungal strains.
C. albicans strain SC5314 (23), C. parapsilosis strain CDC317 (25), and C. tropicalis strain MYA-3404 (26) and isolate Ct8 and C. glabrata strain ATCC 2001 (24) were used throughout this study. Mutant strains used to determine the mechanism(s) of these interactions are listed in Table S5 in the supplemental material.
Compounds.
Stock solutions of fluconazole, itraconazole, voriconazole, amphotericin B, and caspofungin (Sigma-Aldrich) and each hit compound (Selleckchem and Prestwick) were prepared at 10 mM in dimethyl sulfoxide (DMSO) and diluted as needed in the same solvent. The Prestwick library was purchased from Prestwick Chemical, with all 1,280 compounds dissolved in DMSO at 10 mM in 96-well plates. Each was further diluted in DMSO to 1 mM, and 1-μl volumes were dispensed into the flat-bottomed 96-well polystyrene plates that were used for the chemical screens.
Antifungal susceptibility testing.
Antifungal susceptibility testing was performed using the broth microdilution method essentially as described in CLSI document M27-A3 (51), with minor modifications. RPMI 1640 medium (Sigma-Aldrich) was prepared according to the CLSI document. Each antifungal drug (fluconazole, caspofungin, or amphotericin B) was diluted in DMSO to a final 200× concentration before dilution in buffered RPMI medium at a final 2× concentration. Candida spp. were grown overnight in YPD at 30°C and resuspended at 1 × 104 cells/ml in RPMI medium (pH 7.0). Finally, 100 μl of the RPMI medium containing a final 2× drug concentration was dispensed into the wells of a flat-bottomed polystyrene 96-well plate, and 100 μl of the cell suspension was added to each well. The final concentration of DMSO was 0.5% for all treatments, with drug-free control wells having DMSO alone. Plates were incubated without shaking for 24 h at 35°C, and then the content of each well was carefully resuspended by pipetting up and down using a multichannel pipette. Fungal growth in each well was then quantified by measuring OD600 using a Cytation 5 plate reader (Bio-Tek Instruments, Inc.), and the results were expressed as a percentage of growth in the drug-free control well.
To test the extent of antifungal antagonism of hit compounds identified from the screen, antifungal susceptibility assays were performed as described above but in the absence or presence of the hit compound at a final concentration of 5 μM.
Antifungal antagonism screens.
The wells of the 96-well flat-bottom assay plates were seeded with 1 μl of the 1 mM stock solutions of each library compound in DMSO or with DMSO alone (control). Candida spp. were grown overnight in YPD medium at 30°C, and the cells were harvested by centrifugation, washed twice in phosphate-buffered saline (PBS), and resuspended at ∼5 × 103 cells/ml in RPMI medium supplemented with the indicated concentration of fluconazole, amphotericin B, or caspofungin. A 199-μl volume of the cell-plus-antifungal drug suspension was then dispensed into each well of the drugged 96-well plates. Control wells had antifungal drug but no test/library compound (antifungal alone) or had neither the antifungal nor the library compound (minus drug/untreated control). The final concentration of DMSO was 0.55% in all wells, with each library compound supplied at a final concentration of 5 μM. After 24 h of incubation at 35°C, the fungal cells in each well were resuspended using a multichannel pipette and growth was measured as OD600. Wells with visible precipitation were excluded from analysis.
RNA isolation and reverse transcription-quantitative PCR (qRT-PCR) analysis.
C. albicans strain SC5314 was grown in YPD medium at 30°C overnight before being subcultured into fresh YPD medium supplemented with the indicated concentrations of fluconazole, aripiprazole, and/or etofenamate or with DMSO (minus drug control) to an OD600 of 0.1, incubated at 30°C with shaking for 6 h, harvested by centrifugation, and frozen at −80°C on three separate days. Total cellular RNA was extracted using the hot phenol method (52). cDNA was synthesized from total RNA using a Verso cDNA synthesis kit (Thermo Scientific), in accordance with the manufacturer's instructions, and was used as the template for amplification of ACT1, MDR1, CDR1, or CDR2 transcripts using Maxima SYBR green PCR master mix (ThermoScientific) in technical triplicate. Gene-specific primers were designed using the PrimerQuest Tool from Integrated DNA Technologies and are listed in Table S6. The threshold cycle (2−ΔΔCT) method was used to calculate changes in gene expression among the strains (53).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Daniel Diekema for providing the clinical isolate used in this study.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI099080.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00054-19.
REFERENCES
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Maertens J, Vrebos M, Boogaerts M. 2001. Assessing risk factors for systemic fungal infections. Eur J Cancer Care (Engl) 10:56–62. doi: 10.1046/j.1365-2354.2001.00241.x. [DOI] [PubMed] [Google Scholar]
- 3.Sobel JD. 2007. Vulvovaginal candidosis. Lancet 369:1961–1971. doi: 10.1016/S0140-6736(07)60917-9. [DOI] [PubMed] [Google Scholar]
- 4.Vincent JL, Anaissie E, Bruining H, Demajo W, el-Ebiary M, Haber J, Hiramatsu Y, Nitenberg G, Nystrom PO, Pittet D, Rogers T, Sandven P, Sganga G, Schaller MD, Solomkin J. 1998. Epidemiology, diagnosis and treatment of systemic Candida infection in surgical patients under intensive care. Intensive Care Med 24:206–216. doi: 10.1007/s001340050552. [DOI] [PubMed] [Google Scholar]
- 5.Wey SB, Mori M, Pfaller MA, Woolson RF, Wenzel RP. 1989. Risk factors for hospital-acquired candidemia. A matched case-control study. Arch Intern Med 149:2349–2353. doi: 10.1001/archinte.1989.00390100145030. [DOI] [PubMed] [Google Scholar]
- 6.Pfaller MA, Diekema DJ. 2010. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- 7.Tiphine M, Letscher-Bru V, Herbrecht R. 1999. Amphotericin B and its new formulations: pharmacologic characteristics, clinical efficacy, and tolerability. Transpl Infect Dis 1:273–283. doi: 10.1034/j.1399-3062.1999.010406.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen SC, Slavin MA, Sorrell TC. 2011. Echinocandin antifungal drugs in fungal infections: a comparison. Drugs 71:11–41. doi: 10.2165/11585270-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Pfaller MA. 2012. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am J Med 125:S3–S13. doi: 10.1016/j.amjmed.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Rex JH, Pfaller MA, Galgiani JN, Bartlett MS, Espinel-Ingroff A, Ghannoum MA, Lancaster M, Odds FC, Rinaldi MG, Walsh TJ, Barry AL. 1997. Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro-in vivo correlation data for fluconazole, itraconazole, and candida infections. Subcommittee on Antifungal Susceptibility Testing of the National Committee for Clinical Laboratory Standards. Clin Infect Dis 24:235–247. doi: 10.1093/clinids/24.2.235. [DOI] [PubMed] [Google Scholar]
- 11.Sanglard D, Odds FC. 2002. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2:73–85. doi: 10.1016/S1473-3099(02)00181-0. [DOI] [PubMed] [Google Scholar]
- 12.Jessup CJ, Pfaller MA, Messer SA, Zhang J, Tumberland M, Mbidde EK, Ghannoum MA. 1998. Fluconazole susceptibility testing of Cryptococcus neoformans: comparison of two broth microdilution methods and clinical correlates among isolates from Ugandan AIDS patients. J Clin Microbiol 36:2874–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins N, Spitzer M, Yu T, Cerone RP, Averette AK, Bahn Y-S, Heitman J, Sheppard DC, Tyers M, Wright GD. 2015. An antifungal combination matrix identifies a rich pool of adjuvant molecules that enhance drug activity against diverse fungal pathogens. Cell Rep 13:1481–1492. doi: 10.1016/j.celrep.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui J, Ren B, Tong Y, Dai H, Zhang L. 2015. Synergistic combinations of antifungals and anti-virulence agents to fight against Candida albicans. Virulence 6:362–371. doi: 10.1080/21505594.2015.1039885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spitzer M, Griffiths E, Blakely KM, Wildenhain J, Ejim L, Rossi L, De Pascale G, Curak J, Brown E, Tyers M, Wright GD. 2011. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Mol Syst Biol 7:499. doi: 10.1038/msb.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Yan K, Zhang Y, Huang R, Bian J, Zheng C, Sun H, Chen Z, Sun N, An R, Min F, Zhao W, Zhuo Y, You J, Song Y, Yu Z, Liu Z, Yang K, Gao H, Dai H, Zhang X, Wang J, Fu C, Pei G, Liu J, Zhang S, Goodfellow M, Jiang Y, Kuai J, Zhou G, Chen X. 2007. High-throughput synergy screening identifies microbial metabolites as combination agents for the treatment of fungal infections. Proc Natl Acad Sci U S A 104:4606–4611. doi: 10.1073/pnas.0609370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hovstadius B, Hovstadius K, Astrand B, Petersson G. 2010. Increasing polypharmacy—an individual-based study of the Swedish population 2005–2008. BMC Clin Pharmacol 10:16. doi: 10.1186/1472-6904-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong W, Maradit-Kremers H, St Sauver JL, Yawn BP, Ebbert JO, Roger VL, Jacobson DJ, McGree ME, Brue SM, Rocca WA. 2013. Age and sex patterns of drug prescribing in a defined American population. Mayo Clin Proc 88:697–707. doi: 10.1016/j.mayocp.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badiee P, Hashemizadeh Z. 2014. Opportunistic invasive fungal infections: diagnosis & clinical management. Indian J Med Res 139:195–204. [PMC free article] [PubMed] [Google Scholar]
- 20.Butts A, Reitler P, Ge W, Fortwendel JR, Palmer GE. 26 June 2018. Commonly used oncology drugs decrease antifungal effectiveness against Candida and Aspergillus species. Antimicrob Agents Chemother doi: 10.1128/AAC.00504-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry KW, Cruz MC, Katiyar SK, Edlind TD. 1999. Antagonism of azole activity against Candida albicans following induction of multidrug resistance genes by selected antimicrobial agents. Antimicrob Agents Chemother 43:1968–1974. doi: 10.1128/AAC.43.8.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tome M, Zupan J, Tomicic Z, Matos T, Raspor P. 2018. Synergistic and antagonistic effects of immunomodulatory drugs on the action of antifungals against Candida glabrata and Saccharomyces cerevisiae. PeerJ 6:e4999. doi: 10.7717/peerj.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odds FC, Brown AJP, Gow N. 2004. Candida albicans genome sequence: a platform for genomics in the absence of genetics. Genome Biol 5:230. doi: 10.1186/gb-2004-5-7-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koszul R, Malpertuy A, Frangeul L, Bouchier C, Wincker P, Thierry A, Duthoy S, Ferris S, Hennequin C, Dujon B. 2003. The complete mitochondrial genome sequence of the pathogenic yeast Candida (Torulopsis) glabrata. FEBS Lett 534:39–48. doi: 10.1016/S0014-5793(02)03749-3. [DOI] [PubMed] [Google Scholar]
- 25.Kuhn DM, Mukherjee PK, Clark TA, Pujol C, Chandra J, Hajjeh RA, Warnock DW, Soll DR, Ghannoum MA. 2004. Candida parapsilosis characterization in an outbreak setting. Emerg Infect Dis 10:1074–1081. doi: 10.3201/eid1006.030873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joly S, Pujol C, Schroppel K, Soll DR. 1996. Development of two species-specific fingerprinting probes for broad computer-assisted epidemiological studies of Candida tropicalis. J Clin Microbiol 34:3063–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rex JH, Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts: approved standard. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 28.Morschhauser J, Barker KS, Liu TT, Bla BWJ, Homayouni R, Rogers PD. 2007. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog 3:e164. doi: 10.1371/journal.ppat.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother 49:1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 32.Rogers PD, Barker KS. 2003. Genome-wide expression profile analysis reveals coordinately regulated genes associated with stepwise acquisition of azole resistance in Candida albicans clinical isolates. Antimicrob Agents Chemother 47:1220–1227. doi: 10.1128/AAC.47.4.1220-1227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whaley SG, Berkow EL, Rybak JM, Nishimoto AT, Barker KS, Rogers PD. 12 January 2017. Azole antifungal resistance in Candida albicans and emerging non-albicans Candida species. Front Microbiol doi: 10.3389/fmicb.2016.02173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heilmann CJ, Schneider S, Barker KS, Rogers PD, Morschhauser J. 2010. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob Agents Chemother 54:353–359. doi: 10.1128/AAC.01102-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schubert S, Barker KS, Znaidi S, Schneider S, Dierolf F, Dunkel N, Aid M, Boucher G, Rogers PD, Raymond M, Morschhauser J. 2011. Regulation of efflux pump expression and drug resistance by the transcription factors Mrr1, Upc2, and Cap1 in Candida albicans. Antimicrob Agents Chemother 55:2212–2223. doi: 10.1128/AAC.01343-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reboli AC, Rotstein C, Pappas PG, Chapman SW, Kett DH, Kumar D, Betts R, Wible M, Goldstein BP, Schranz J, Krause DS, Walsh TJ; Anidulafungin Study Group. 2007. Anidulafungin versus fluconazole for invasive candidiasis. N Engl J Med 356:2472–2482. doi: 10.1056/NEJMoa066906. [DOI] [PubMed] [Google Scholar]
- 37.Reboli AC, Shorr AF, Rotstein C, Pappas PG, Kett DH, Schlamm HT, Reisman AL, Biswas P, Walsh TJ. 2011. Anidulafungin compared with fluconazole for treatment of candidemia and other forms of invasive candidiasis caused by Candida albicans: a multivariate analysis of factors associated with improved outcome. BMC Infect Dis 11:261. doi: 10.1186/1471-2334-11-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kullberg BJ, Viscoli C, Pappas PG, Vazquez J, Ostrosky-Zeichner L, Rotstein C, Sobel JD, Herbrecht R, Rahav G, Jaruratanasirikul S, Chetchotisakd P, Van Wijngaerden E, De Waele J, Lademacher C, Engelhardt M, Kovanda LL, Croos-Dabrera R, Fredericks C, Thompson GR III.. 5 October 2018. Isavuconazole versus caspofungin in the treatment of candidemia and other invasive candida infections: the ACTIVE trial. Clin Infect Dis doi: 10.1093/cid/ciy827. [DOI] [PubMed] [Google Scholar]
- 39.Andes D. 5 October 2018. Has the optimal therapy for invasive candidiasis now been defined? Clin Infect Dis doi: 10.1093/cid/ciy830. [DOI] [PubMed] [Google Scholar]
- 40.Rex JH, Pfaller MA. 2002. Has antifungal susceptibility testing come of age? Clin Infect Dis 35:982–989. doi: 10.1086/342384. [DOI] [PubMed] [Google Scholar]
- 41.Andes D, van Ogtrop M. 1999. Characterization and quantitation of the pharmacodynamics of fluconazole in a neutropenic murine disseminated candidiasis infection model. Antimicrob Agents Chemother 43:2116–2120. doi: 10.1128/AAC.43.9.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drusano GL. 1988. Role of pharmacokinetics in the outcome of infections. Antimicrob Agents Chemother 32:289–297. doi: 10.1128/AAC.32.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van T Wout JW, Mattie H, van Furth R. 1989. Comparison of the efficacies of amphotericin B, fluconazole, and itraconazole against a systemic Candida albicans infection in normal and neutropenic mice. Antimicrob Agents Chemother 33:147–151. doi: 10.1128/AAC.33.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai Y-W, Campbell LT, Wilkins MR, Pang CNI, Chen S, Carter DA. 2016. Synergy and antagonism between iron chelators and antifungal drugs in Cryptococcus. Int J Antimicrob Agents 48:388–394. doi: 10.1016/j.ijantimicag.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Cokol M, Weinstein ZB, Yilancioglu K, Tasan M, Doak A, Cansever D, Mutlu B, Li S, Rodriguez-Esteban R, Akhmedov M, Guvenek A, Cokol M, Cetiner S, Giaever G, Iossifov I, Nislow C, Shoichet B, Roth FP. 2014. Large-scale identification and analysis of suppressive drug interactions. Chem Biol 21:541–551. doi: 10.1016/j.chembiol.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson ME, Edlind TD. 2012. Topological and mutational analysis of Saccharomyces cerevisiae Fks1. Eukaryot Cell 11:952–960. doi: 10.1128/EC.00082-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niimi K, Maki K, Ikeda F, Holmes AR, Lamping E, Niimi M, Monk BC, Cannon RD. 2006. Overexpression of Candida albicans CDR1, CDR2, or MDR1 does not produce significant changes in echinocandin susceptibility. Antimicrob Agents Chemother 50:1148–1155. doi: 10.1128/AAC.50.4.1148-1155.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perea S, Lopez-Ribot JL, Kirkpatrick WR, McAtee RK, Santillan RA, Martinez M, Calabrese D, Sanglard D, Patterson TF. 2001. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 45:2676–2684. doi: 10.1128/AAC.45.10.2676-2684.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White TC. 1997. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob Agents Chemother 41:1482–1487. doi: 10.1128/AAC.41.7.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thakur JK, Arthanari H, Yang F, Pan S-J, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Näär AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604. doi: 10.1038/nature06836. [DOI] [PubMed] [Google Scholar]
- 51.CLSI. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard-3rd ed CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 52.Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 18:3091–3092. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.