

Abstract
Abstract. Arrangement of the intestinal cell lining, as it is, into distinct anatomically defined zones where proliferation is confined to the crypts, makes it an ideal tissue to study growth control mechanisms. While many methods have been used to quantify cell proliferation in the gut, several of them have severe limitations and others (although potentially better) have been misused and misinterpreted. Here, correct use and interpretation of labelling studies will be described as will a well established alternative method that provides equivalent results for one‐sixth of the effort.
INTRODUCTION
While much basic information on cell cycle parameters and molecular control of cell proliferation has been derived from cultured‐cell studies, often immortalized cell lines (widely used for such investigations) have been produced by transformation of cells from a normal phenotype and/or use of those that initially had malignant cell proliferation properties. Results obtained thus cannot necessarily be applied to normal cell and tissue counterparts. The gastrointestinal epithelium is part of a complex multilayered defence system that interacts with many other tissue systems and the luminal contents – especially commensal bacteria. Furthermore, in situ enterocytes are normally protected from extracellular chemicals rather than being immersed in them. A good example of difference in properties between in vitro and in vivo environments is provided by the ‘butyrate paradox’. Butyrate is a product of fibre breakdown by bacteria in the hindgut; it is a powerful stimulus for differentiation and apoptosis in vitro, but in vivo is a potent stimulator of cell division (Hass et al. 1997; Goodlad 2001).
While in vitro studies have a place, they cannot reflect the cornucopia of responses of complex tissues to stimulation, thus, animal studies are essential. Widely, workers in the field of gastroenterology have only a very limited appreciation of the problems and pitfalls associated with cell proliferation assays, or in vitro versus in vivo metabolic comparisons, and this can lead to inappropriate conclusions being drawn. Latterly, there has been a substantial increase in studies in the mouse gut following the introduction of methods for generating genetically engineered animals, and measurement of cell proliferation status in a variety of tissues is often one of the endpoints in these studies.
Increased levels of cell proliferation is generally considered to be one of the earliest events in the multistage model of cancer development (Fearon & Vogelstein 1990) and indicates the requirement of a promoter of carcinogenesis to ‘fix’ mutations before ‘genetic housekeeping’ repairs defects. There are reports that currently the use of measures of cell proliferation in human cancer studies has somewhat fallen out of favour (Sanderson et al. 2004), but this may be a failing as the use of misappropriate methods and/or the misuse of better methods has been the order of the day. However, the importance of cell proliferation studies in cancer has recently been reaffirmed by the finding that the biggest changes in gene expression between normal and tumour tissue samples occurs in those genes involved in the control of cell proliferation (Whitfield et al. 2006).
This article will attempt to provide an overview of the methods available for quantifying cell proliferation in animals and also in humans. Some of the widely used methods are inherently flawed and little credence can be given to the conclusions generated by them. Others are basically sound, but can yield false conclusions if the three‐dimensional nature of the gut is not taken into account.
MEASURES AVAILABLE
Gross measures of cell proliferation
The end result of an altered level of cell production in a tissue is ultimate increased or decreased tissue mass; experimentally, simply weighing the various sections of the gastrointestinal tract can give valuable information and a quick indication of the difference between average control guts and average experimental guts. First, tissue must be rinsed with cold‐buffered saline to clean the segments, which must then be blotted on folded paper towel.
In the laboratory, it must be borne in mind though that gut weight change takes a few days to manifest itself and does not distinguish between epithelial, loose connective tissue or muscle layer effects. Inflammation and oedema may also influence mass measurement results therefore further measures are desirable.
Dividing cells in the S‐phase (DNA synthesis) of cell division incorporates thymidine into their DNA and this is easily detected in tissues by using a scintillation counter. The method is quick and easy, but unfortunately the results generated are of questionable value. One problem is that tritiated thymidine is readily broken down resulting in cytoplasm widely contaminated with tritium. The problem can be overcome if the DNA is extracted, but metabolism of thymidine by salvage pathways is such that agents or interventions can alter the activity of the thymidine kinase enzyme involved so that tritiated thymidine uptake does not always reflect DNA synthesis (Maurer 1981). In addition, tissue digests cannot distinguish between uptakes into the various components of the tissue. The main flaw of the method however, is that one cannot decide if 100 beta radiation counts is the result of 10 cells each providing 10 counts or of 100 cells each providing one count. Most of these areas of potential inaccuracy with a tritiated thymidine uptake technique, however, can be overcome by performing autoradiography.
The value of the latter approach and the limitations of the former were dramatically demonstrated when our group performed an investigation on the action of a prostaglandin analogue whose activity increased stomach mass. The study originally planned to use tritiated thymidine per unit DNA as the assessment of cell proliferation endpoint; however, in the event, autoradiograph tritium‐positive cell scoring was also used. When the analyses were published, the gross thymidine measures showed no differences between groups (Goodlad et al. 1990), while the careful study of autoradiographs demonstrated significant changes in both cell proliferation (Goodlad et al. 1989) and cell migration (Goodlad et al. 1990). Several groups still use gross thymidine levels as an assay of cell proliferation, especially in vitro where there is less complexity, yet the problems still apply and this should always be taken into consideration. Use of whole tissue thymidine incorporation because of its simplicity is no longer necessary, as there are easy to use fluorimetric assays available (Otto 2005) that quickly measure DNA content to assay cell number.
Fluorescence‐activated cell sorters analysis
Fluorescence‐activated cell sorters can be used to determine proportions of cells in the stages of the cell cycle, however, flow cytometers require the test sample provided as a cell suspension; whole tissues are difficult to prepare accurately in this way and the cell type subpopulations are mixed. In the intestine, epithelial tissue can be separated from the other tissue components (Weiser 1973, Kearsey & Stadnyk 1996) but this is time consuming, and numbers of samples involving the many animals required to achieve statistical power become difficult to provide. In addition, it is vital to ensure for these protocols that there is no clumping of cells.
Intrinsic markers of cell proliferation
As cells pass through the cell division cycle, many different proteins are expressed and several of them are specific enough for them to be detected to demonstrate cycling cells or cells about to enter DNA synthesis. Between the S‐phase and mitosis there is a short gap (G2) then the chromosomes separate in mitosis (M‐phase). Cells in mitosis are easy to identify histologically and counting them can provide an excellent indication of proliferative status in the tissue without the problems and pitfalls associated with immunohistochemical identification of cell cycle antigens. The traditional stages of the cell cycle vary somewhat between conditions/tissues/species, yet mitosis is often 0.5 h to 1 h long while the S‐phase takes around 8 h (Wright & Alison 1984). Counting mitotic cells, down the microscope, means that there are few events available to score; however, a squash preparation technique is described below in which whole crypts are examined at once so that as many mitoses can be seen per crypt as S‐phase cells are seen in section.
In histological sections, mitotic figures (cells in which chromosomes are in their condensed mode, thus easily visible) are usually counted from routine haematoxylin‐ and eosin‐stained sections. However, they are more readily identified if the Feulgen technique is used to stain their DNA. Careful examination is required to positively identify mitotic figures and criteria need to be set so that different quality of preparations can be scored; for example in a well‐prepared section early prophases and very late telophases can often be seen, but on other occasions this may not be the case, and these are best excluded. Monoclonal antibodies directed against histone H3 phosphoserine have been used to visualize mitotic chromosomal condensation (Hirata et al. 2004) and provide staining indices that correlate well with simple mitotic indices, but are nearly twice as numerous (to provide indices, the number of ‘positive’ cells is expressed as a percentage of the total cell number counted). The use of such antibodies may help distinguish mitotic figures and apoptotic figures; however, an alternative would be to cut ‘semithin’ (around one micron thick) sections, for extra clarity. Both approaches may be useful in the initial stages of a study to help the observer to be come familiar with the tissue thus to be able to score accurately. It is accepted that there will always be some inter‐observer variation in accuracy, thus, it is important not to mix observers when comparing groups or sites as this can be a danger, especially when samples from large studies are randomised for ‘blind scoring’.
While there are several antibodies that can detect cell cycle associated proteins only a few are in regular use, and some of these should definitely not be! This includes the so‐called proliferative cell nuclear antigen (PCNA) that is still widely employed. Results obtained after PCNA staining must be viewed with suspicion as the number of positive cells is greatly dependant on the time passed in tissue fixation and pre‐treatment of the tissue. Even if these can be standardized, PCNA labelling is not reliable as very large amounts of PCNA expression can be seen near tumours or in the tissue of animals given growth factors, despite a complete lack of tritiated thymidine uptake (Hall et al. 1994). PCNA is a known DNA clamp and is also active in DNA repair; however, this does not explain its anomalous expression in the above study.
For cell proliferation studies, there is no need to use PCNA as other, far better, intrinsic markers of proliferation exist. One of the best of these is the antibody to the Ki67 protein, also known as MIB1 (murine version) (Gerdes et al. 1992; McCormick et al. 1993). The antigen is expressed in cells throughout the cell cycle from early in gap 1 (G1) whereas PCNA expression is, perhaps, even more comprehensively through the whole cell cycle.
Intrinsic markers of proliferation can be used in a manner similar to that of extrinsic markers described below.
Extrinsic measures of cell proliferation
Intrinsic measures are useful when retrospective analysis needs to be performed or if human tissue is to be studied; however, if the proliferation analysis is planned at the start of an investigation, it is far better to provide uptake agents ahead of the experiment, to be able to label the S‐phase or cells accumulated in metaphase. The classical S‐phase label is tritiated thymidine. While the gross uptake of tritiated thymidine is not recommended (see above), measuring uptake on an all or none (per cell) basis is very useful, and most of the basic information on intestinal epithelial kinetics has been obtained from the study of autoradiographs. The results are very clean, as unincorporated label is washed out during histological dehydration and embedding. Originally, the standard dose of tritiated thymidine used was 1 mCi/kg, but later this was reduced to 0.5 mCi/kg, and if administered 1 h before death, all dividing cells will be ‘flash’ labelled. Tissue is sampled as required and is processed by fixation, dehydration and embedding in paraffin wax in the routine manner. Sections are cut and collected on glass slides. Under darkroom conditions, each slide is dipped into liquid photographic emulsion so that a layer covers the histological section. In this manner, the tritiated thymidine's β particles react with the silver salts of the photographic emulsion. Latent images are created which are developed photographically. The short path length of tritium β particle emission means that the resultant silver grains are virtually exactly over the source of radiation. Moreover, the effect of background reactions (grains attributable to other sources) can be measured and corrected for, using the Poisson distribution method; however, after performing such calculations, ad hoc threshold of three to five grains per nucleus to be considered as labelled, was usually found to apply. The use of tritiated thymidine has unfortunately fallen out of favour and been superseded by another nucleotide recursion namely bromodeoxyuridine (BrdU). The reason for this is not that BrdU gives better results, but that its use does not involve the attention of the radiation protection officer. BrdU is readily detected by using an immunohistochemical method and provides equivalent results; however, as with all immunohistochemical methods, results can vary and one needs to decide at what point brownish nuclei are scored as brown (labelled) or not. BrdU is usually administered 1 h before killing the target, at a dose of 50 mg/kg. Tritiated thymidine and BrdU can be given at different times to the same animal(s) to study cell migration, in which case the thymidine is administered 17 h before killing and BrdU 1 h before (Marchbank et al. 2001) or to determine the rates of entry and exit from the S‐phase (Chwalinski et al. 1988).
Use of labels to determine cell proliferation parameters
The markers described above are used to measure cell proliferation, usually expressed as a proliferative index (as described above). While this can often give a useful measure, two fundamental problems exist. The first is that all these measures are what are known as ‘state’ parameters. This means that they cannot account for the dynamics of the system. The analogy is that simply taking an aerial photograph of a motorway cannot measure the flux of vehicles, as the 100 vehicles between junction 1 and junction 2 (state) may be going at 10 mph or 100 mph, thus unless some measure of time is also included the rate of flow cannot be calculated. The most straightforward method to histologically measure rate of cell proliferation is to determine the accumulation of arrested metaphases in a tissue as time passes; the other way involves double labelling to determine the rate of S‐phase entry (and exit) (Chwalinski et al. 1988).
In the intestine, cell efflux from a crypt is the product of the proportion of crypt cells involved in proliferation (the growth fraction), crypt size and the cell cycle time. In most cases, cell cycle time does not appear to vary greatly. Crypt size, however, often alters so that reliance on a simple labelling index can be misleading. If labelling indices are seen to change it is likely that something has happened, but if no changes are seen a more careful analysis is required. This is because it is possible that both sides of the fraction (labelled cells/total cell number) may have changed concurrently (Goodlad & Alferez 2005).
In our laboratory, we have found that cell production is very dependant on crypt size (denominator). This can have profound implications when interpreting labelling indices. One of the most dramatic examples of this was seen in our studies of epidermal growth factor (EGF) in intravenously fed rats. The weight of the gut halved when rats were starved of luminal content when fed intravenously, which was reversed by EGF treatment (Goodlad et al. 1987). Labelling indices could detect response to EGF administration, but not the profound atrophy of the gut in response to intravenous feeding. The effect of intravenous feeding only became apparent when changes in denominator were taken into account by expressing the results as labelling (or mitoses) per crypt (1989, 1992). This conversion involved several measures and some calculation to ultimately provide the same results obtained by using the far faster and simpler crypt ‘microdissection’ method (described below) that uses whole crypts as the denominator (1987, 1991a; Goodlad 1994).
A second example of the denominator effect was seen in our studies of the prostaglandin analogue misoprostol, on gastric glands; labelling index remained unchanged (despite a large effect on stomach weight). Results of the drug treatment only became apparent when they were expressed as labelling index per gastric gland (Goodlad et al. 1991b). Failure to appreciate this denominator effect is perhaps the most common proliferation‐related error in the literature (Goodlad 1995; Goodlad & Alferez 2005).
The distribution of labelled (and mitotic) cells within the crypt
There is some evidence that upward migration of the proliferative cell compartment is associated with malignant conditions (Gerdes et al. 1993). This can be measured by recording the position of labelled cells within the crypt. Microscopically, one must first find crypts that have been cut down the middle (axially sectioned) and call the lowermost cell position 0. One then ascends the crypt recording all positions that have labelled cells (and mitotic cells) and keep counting until one reaches the top of the crypt. Once 30 half‐crypts have been scored, one needs to add up the values per position and generate labelling per crypt position graphs. We determine the position of maximum labelling then half maximum labelling that is interpreted as the growth fraction. Alternatively, this position can be derived assuming a Gaussian distribution where the growth fraction (Ip) can be defined by the half‐maximum labelling point, which is the mean +0.833 times the standard deviation. One problem with such scores occurs if the crypts are of different lengths. Computer programs exist for the ‘standardization’ of crypts, but these tend to be not user friendly; the principal is to expand all crypts to, say 1000 cells, and then shrink back to the average crypt length.
A far simpler means of accounting for this is to ‘standardize’ the crypts under the microscope. In our laboratory, we have a five‐step grid eye‐piece graticule for a microscope fitted with an optical zoom, which is then used to fit the crypt into the grid. Labelled cells in each of the five zones are then recorded and are thus automatically standardized. We also have used a drawing tube (which projects an image into the microscope field of view) to project an image of a grid that can be altered in size by moving it up or down on a scissor jack.
The metaphase arrest technique
The ‘most accurate and elegant in vivo method’ (Seitz et al. 1998) is the metaphase arrest technique. Administration to an animal, of vincristine sulphate at a dose 1 mg/kg, arrests cells as they enter mitosis. This can be either used to provide an augmented mitotic index (2 h after vincristine) or it can be used to determine the rate of entry of cells into mitosis or the crypt cell production rate (CCPR) (Goodlad 1994). This is a dynamic ‘rate’ measurement, as opposed to the passive ‘state’ measures described above. Animals are killed at timed intervals, 30–180 min after vincristine. The slope of the curve of metaphases over time will then provide the CCPR ± its standard error. The CCPR can be determined in histological sections but it is far easier to use the so‐called ‘crypt microdissection’ technique (Wimber & Lamerton 1963; Goodlad 1994). If a change in proliferation parameters is detected using the CCPR method, there is no other explanation that can be put forward as it accounts for all the factors determining crypt cell efflux, namely crypt size, growth fraction and cell cycle time.
Crypt microdissection
People are often put off by the name of the technique, but in reality it is quite simple. Tissue must be fixed in Carnoy's fluid and stored in 70% ethanol. When it is to be scored it is taken through a descending series of alcohols and hydrolysed at 60 °C for 10 min, then stained in Schiff's reagent (the Feulgen reaction). After 45 min, a small piece of the tissue can be cut off and placed on a glass microscope slide with a drop of 45% acetic acid. Mounted needles or disposable ophthalmic knives are then used to gently tease the tissue apart under a stereomicroscope. A coverslip can determine the correct amount of tissue, as if there is too much the coverslip will crack and if there is not enough when it is applied it is possible to generate a single cell suspension!
The preparation is then taken to a standard microscope and the number of metaphases per crypt scored. As we express our results as metaphases per (microdissected) crypt we avoid the problems associated with changes in denominator referred to above and greatly speed up the counting process so that the method is about six times faster than scoring traditional histological sections. In addition, the results are ready for statistical analysis, unlike histological scores that require several subsequent calculations.
The microdissection technique can still provide valuable results even without vincristine treatment, as native mitoses can be counted (Chaudhary et al. 2000). These are not as numerous as S‐phase cells, but as the entire crypt is scored, a similar number of events can be obtained. The technique is ideal for studies of human biopsies, as unlike biopsy sections, hundreds of scorable crypts can be obtained (Goodlad et al. 1991a; Wong et al. 2002). If there are many figures scoring is facilitated by using a 10 × 10 square eye‐piece graticule to subdivide the field of view. A convention needs to be established so that only figures on two sides of each square are counted. Microdissected crypts can also be scored to determine the distribution of mitoses within zones as referred to above. In addition, a drawing tube can be used to trace crypts (and villi) to determine their area. For such measures, the drawing tube must be calibrated so that a line on the drawing paper corresponds to a known distance on a stage graticule. The drawings can then be scanned and quantified using the free programme National Institutes of Health image (http://rsb.info.nih.gov/nih‐image/). Apoptotic figures can also be identified in this way and scoring per crypt greatly simplifies the assessment, especially as these are normally rare (in normal tissue, approximately one per 10 crypts). The microdissection technique allows hundreds of crypts to be scored and there is no need to count the number of cells.
Specimen site
There is a marked proximal to distal gradient in villus size in the small intestine (Wright & Alison 1984) but this is far less pronounced for crypt size. The explanation for this is that there is also a gradient in the number of crypts supplying a villus. In the rat, there are 27 crypts per villus in the duodenum and 10 per villus in the terminal ileum. In the mouse, the ratio is 14 : 1 for the duodenum and 6 : 1 for the ileum, and in man the duodenal crypt villus ratio is 7 : 1 (Wright & Alison 1984). Differences in proliferative rates of the different sections of colon are equivocal with some groups reporting no changes in labelling index in the (human) colon (Liu et al. 1999), others (using the more reliable crypt cell microdissection method) having found a significant reduction in proliferation as one descends the colon (Goodlad et al. 1991; Mills et al. 2001). In humans, the small intestinal crypts are considerably smaller and have less mitotic figures per crypt, but if expressed as mitoses per unit area the values are remarkably constant (Goodlad et al. 1991a).
Productivity of the microdissection method allows one to score sites from the proximal, mid and distal small intestine and colon, usually defined in terms of their percentage length, as the anatomical and histological terms used in humans do not apply to rodents.
Crypt fission
The gut can also increase its mass by the process of crypt fission in addition to cell proliferation (St Clair & Osborne 1985; Cheng et al. 1986). An indentation in the base of the crypt is first seen, which may then enlarge to ‘unzip’ the crypt into two new crypts (Fig. 1). This is the main means of increasing crypt number in the young animal (St Clair & Osborne 1985) but is still seen in the adult and can be increased following damage (Cairnie & Millen 1975), colitis (Cheng et al. 2000; Chen et al. 2005) and chemical carcinogens (Park et al. 1997; Wasan 1998). Crypt fission provides a mechanism for the evolution of clonal crypt populations in the neonate (Park et al. 1995) and in cancer (Wong et al. 2002). Crypt fission is not very apparent in tissue sections but is readily observed in microdissected crypts. The technique is similar to that described above but more care is required in the dissection.
Figure 1.
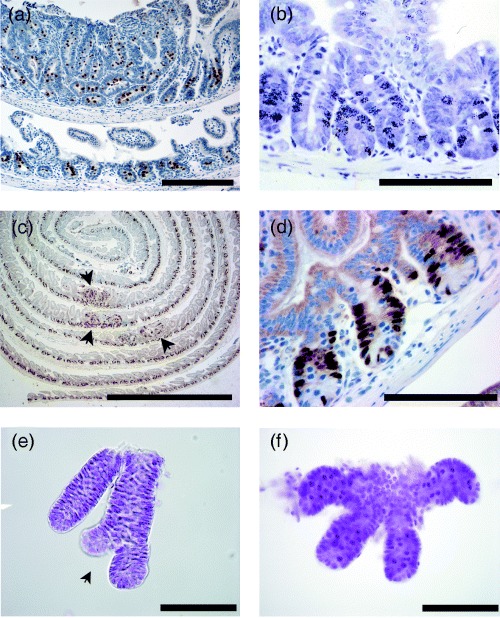
(a) Histone H3 phosphoserine staining of mitotic chromosomal condensation in ‘Swiss roll’ of mouse small intestine. Top segment shows a polyp from an ApcMin/+ mouse (of intestinal cancer) with enlarged crypts. (b) Autoradiograph of mouse small intestine showing developed silver grains overlying nuclei that have incorporated tritiated thymidine. (c) ‘Swiss roll’ of bromodeoxyuridine (BrdU)‐stained small intestine from an ApcMin/+ mouse, note that the label delineates the proliferative zone. Arrows indicate polyps with disorganized cell proliferation. (d) High power view of BrdU labelled small bowel crypts. (e) Microdissected crypts showing crypt fission, note that the microscope condenser has been lowered to increase contrast and help visualise the crypt edges. (f) Microdissected crypts showing dark staining arrested metaphases. Scale bar: a = 200 µm, c = 500 µm and 100 µm for b, d, e and f.
Gut preparation
The gut is removed and rinsed with cold saline at autopsy using a ‘Gilson’ pipette tip that has been cut short, to fit on a standard luer syringe. Samples of tissue can be fixed in appropriate containers or whole mounts prepared. Whole mounts are very useful if other measures, such as searching for aberrant crypt foci, for polyps or ‘micropolyp’ formation are intended. We have recently described the construction and use of a device to aid the process (Rudling et al. 2006). In brief, stainless steel rods are threaded through the segments of intestine and a cutting guide is used to guide a scalpel and dissect the segments longitudinally. The rods are then rolled sideways to gently flatten the tissue onto a piece of filter paper or card.
Fixation
We routinely use Carnoy's fluid for fixing our tissues as nuclear detail is enhanced and this fixative is required for the crypt cell microdissection technique. Formalin is usually used if in situ hybridization or certain immunohistochemical techniques are required. Degree of immunoreactivity is dependent on the time of fixation and can be reduced dramatically by delay in fixation or by over fixation (Hirata et al. 2004).
Suppliers
Bromodeoxyuridine is available from Sigma‐Aldrich Ltd., the Old Brickyard, New Road, Gillingham, Dorset, SP8 4XT, UK. Vincristine sulphate can be obtained as a powder, but it is safer to order a 1 mg/ml solution in sterile vials with a needle septum (it is also considerably cheaper this way) and can be ordered from Mayne Pharma Plc, Queensway, Royal Leamington Spa, Warwickshire, CV31 3RW, UK. Schiff's reagent can be ordered from VWR International Ltd., Merck House Poole Dorset BH15 1TD, UK. Ophthalmic knives can be supplied by Meddis Ltd., Henderson House Hithercroft Road, Wallingford, OX10 9DG, UK. A wide range of eyepiece and stage graticules are available from Pyser SGI Ltd., Fircroft Way, Edenbridge, Kent, TN8 6HA, UK.
REFERENCES
- Cairnie AB, Millen BH (1975) Fission of crypts in the small intestine of the irradiated mouse. Cell Tissue Kinet. 8, 189–196. [DOI] [PubMed] [Google Scholar]
- Chaudhary M, Mandir N, Fitzgerald AJ, Howard JK, Lord GM, Ghatei MA, Bloom SR, Goodlad RA (2000) Starvation, leptin and epithelial cell proliferation in the gastrointestinal tract of the mouse. Digestion 61, 223–229. [DOI] [PubMed] [Google Scholar]
- Chen R, Rabinovitch PS, Crispin DA, Emond MJ, Bronner MP, Brentnall TA (2005) The initiation of colon cancer in a chronic inflammatory setting. Carcinogenesis 26, 1513–1519. [DOI] [PubMed] [Google Scholar]
- Cheng H, Bjerknes M, Amar J, Gardiner G (1986) Crypt production in normal and diseased human colonic epithelium. Anat. Rec. 216, 44–48. [DOI] [PubMed] [Google Scholar]
- Cheng L, Araki K, Furuya Y, Matsuoka T, Mashima K, Kobayashi M, Matsuura K (2000) Morphological study of the regeneration mechanism of acetic acid‐injured colon crypts in the rat. Med. Electron Microsc. 33, 165–171. [DOI] [PubMed] [Google Scholar]
- Chwalinski S, Potten CS, Evans G (1988) Double labelling with bromodeoxyuridine and 3H‐thymidine of proliferating cells in small intestinal epithelium in steady state and after irradiation. Cell Tissue Kinet. 21, 317–329. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Becker MHG, Key G, Cattoretti G (1992) Immunohistochemical detection of tumour growth fraction (Ki67 antigen) in formalin fixed and routinely processed tissues. J. Pathol. 168, 85–87. [DOI] [PubMed] [Google Scholar]
- Gerdes H, Gillin JS, Zimbalist E, Urmacher C, Lipkin M, Winawer SJ (1993) Expansion of the epithelial‐cell proliferative compartment and frequency of adenomatous polyps in the colon correlate with the strength of family history of colorectal‐cancer. Cancer Res. 53, 279–282. [PubMed] [Google Scholar]
- Goodlad RA (1994) Microdissection‐based techniques for the determination of cell proliferation in gastrointestinal epithelium: application to animal and human studies In: Celis JE, ed. Cell Biology: a Laboratory Handbook, pp. 205–216. San Diego, CA: Academic Press. [Google Scholar]
- Goodlad RA (1995) Defective denominators, or will people never learn? Gastroenterology 108, 1963. [DOI] [PubMed] [Google Scholar]
- Goodlad RA (2001) Dietary fibre and the risk of colorectal cancer. Gut 48, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlad RA, Alferez D (2005) Defective denominators. Gut 54, 1502–1503. [PMC free article] [PubMed] [Google Scholar]
- Goodlad RA, Wilson TG, Lenton W, Wright NA, Gregory H, McCullagh KG (1987) Intravenous but not intragastric urogastrone‐EGF is trophic to the intestinal epithelium of parenterally fed rats. Gut 28, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlad RA, Madgwick AJ, Moffatt MR, Levin S, Allen JL, Wright NA (1989) Prostaglandins and the gastric epithelium: effects of misoprostol on gastric epithelial cell proliferation in the dog. Gut 30, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlad RA, Madgwick AJ, Moffatt MR, Levin S, Allen JL, Wright NA (1990) Effects of misoprostol on cell migration and transit in the dog stomach. Gastroenterology 98, 90–95. [DOI] [PubMed] [Google Scholar]
- Goodlad RA, Levi S, Lee CY, Mandir N, Hodgeson H, Wright NA (1991a) Morphometry and cell proliferation in endoscopic biopsies: evaluation of a technique. Gastroenterology 101, 1235–1241. [DOI] [PubMed] [Google Scholar]
- Goodlad RA, Mandir N, Levin S, Allen JL, Wright NA (1991b) Prostaglandins and the colonic epithelium. Effects of misoprostol on crypt size, cell production, and cell migration in the dog. Gastroenterology 101, 1229–1234. [PubMed] [Google Scholar]
- Goodlad RA, Lee CY, Wright NA (1992) Cell proliferation in the small intestine and colon of intravenously fed rats: effects of urogastrone‐epidermal growth factor. Cell Prolif. 25, 393–404. [DOI] [PubMed] [Google Scholar]
- Hall PA, Coates PJ, Goodlad RA, Hart IR, Lane DP (1994) Proliferating cell nuclear antigen expression in non‐cycling cells may be induced by growth factors in vivo . Br. J. Cancer 70, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass R, Busche R, Luciano L, Reale E, Engelhardt WV (1997) Lack of butyrate is associated with induction of Bax and subsequent apoptosis in the proximal colon of guinea pig. Gastroenterology 112, 875–881. [DOI] [PubMed] [Google Scholar]
- Hirata A, Inada K, Tsukamoto T, Sakai H, Mizoshita T, Yanai T, Masegi T, Goto H, Inagaki M, Tatematsu M (2004) Characterization of a monoclonal antibody, HTA28, recognizing a histone H3 phosphorylation site as a useful marker of M‐phase cells. J. Histochem. Cytochem. 52, 1503–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey JA, Stadnyk AW (1996) Isolation and characterization of highly purified rat intestinal intraepithelial lymphocytes. J. Immunol. Methods 194, 35–48. [DOI] [PubMed] [Google Scholar]
- Liu LU, Holt PR, Krivosheyev V, Moss SF (1999) Human right and left colon differ in epithelial cell apoptosis and in expression of Bak, a pro‐apoptotic Bcl‐2 homologue. Gut 45, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchbank T, Cox HM, Goodlad RA, Giraud AS, Moss SF, Poulsom R, Wright NA, Jankowski J, Playford RJ (2001) Effect of ectopic expression of rat trefoil factor family 3 (rTFF3, intestinal trefoil factor) in the jejunum of transgenic mice. J. Biol. Chem. 276, 24 088–24 096. [DOI] [PubMed] [Google Scholar]
- Maurer HR (1981) Potential pitfalls of 3H‐thymidine technique to measure cell proliferation. Cell Tissue Kinet. 14, 111–120. [DOI] [PubMed] [Google Scholar]
- McCormick D, Chong H, Hobbs C, Hall PA (1993) Reliable detection of the Ki67 antigen in fixed and wax embeddded sections with the noverl monoclonal antibody MIB1. J. Pathol. 169, 175A. [DOI] [PubMed] [Google Scholar]
- Mills SJ, Mathers JC, Chapman PD, Burn J, Gunn A (2001) Colonic crypt cell proliferation state assessed by whole crypt microdissection in sporadic neoplasia and familial adenomatous polyposis. Gut 48, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto WR (2005) Fluorimetric DNA assay of cell number. Methods Mol. Biol. 289, 251–262. [DOI] [PubMed] [Google Scholar]
- Park HS, Goodlad RA, Wright NA (1995) Crypt fission in the small intestine and colon. A mechanism for the emergence of G6PD locus‐mutated crypts after treatment with mutagens. Am. J. Pathol. 147, 1416–1427. [PMC free article] [PubMed] [Google Scholar]
- Park HS, Goodlad RA, Wright NA (1997) The incidence of aberrant crypt foci and colonic carcinoma in dimethylhydrazine‐treated rats varies in a site‐specific manner and depends on tumor histology. Cancer Res. 57, 4507–4510. [PubMed] [Google Scholar]
- Rudling R, Kitau J, Mandir N, Goodlad RA (2006) A simple device to rapidly prepare whole mounts of murine intestine. Cell Prolif. 39, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson P, Johnson IT, Mathers JC, Powers HJ, Downes CS, McGlynn AP, Dare R, Kampman E, Pool‐Zobel BL, Bingham SA, Rafter JJ (2004) Emerging diet‐related surrogate end points for colorectal cancer: UK Food Standards Agency diet and colonic health workshop report. Br. J. Nutr. 91, 315–323. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Simanowski UA, Homann N, Waldherr R (1998) Cell proliferation and its evaluation in the colorectal mucosa: effect of ethanol. Z. Gastroenterol. 36, 645–655. [PubMed] [Google Scholar]
- St Clair WH, Osborne JW (1985) Crypt fission and crypt number in the small and large bowel of postnatal rats. Cell Tissue Kinet. 18, 255–262. [DOI] [PubMed] [Google Scholar]
- Wasan HS, Park HS, Liu KC, Mandir NK, Winnett A, Saseni P, Bodmer WF, Goodlad RA, Wright NA (1998) APC in the regulation of intestinal crypt fission. J. Pathol. 185, 246–255. [DOI] [PubMed] [Google Scholar]
- Weiser MM (1973) Intestinal epithelial cell surface membrane glycoprotein synthesis. I. An indicator of cellular differentiation. J. Biol. Chem. 248, 2536–2541. [PubMed] [Google Scholar]
- Whitfield ML, George LK, Grant GD, Perou CM (2006) Common markers of proliferation. Nat. Rev. Cancer 6, 99–106. [DOI] [PubMed] [Google Scholar]
- Wimber DR, Lamerton L (1963) Cell population studies on the intestine of continuously irradiated rats. Radiat. Res. 18, 137–146. [PubMed] [Google Scholar]
- Wong WM, Mandir N, Goodlad RA, Wong BC, Garcia SB, Lam SK, Wright NA (2002) Histogenesis of human colorectal adenomas and hyperplastic polyps: the role of cell proliferation and crypt fission. Gut 50, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright NA, Alison MR (1984) The Biology of Epithelial Cell Populations. Oxford, UK: Oxford University Press. [Google Scholar]