

Abstract
Abstract. Dietary oxidants like lipid hydroperoxides (LOOH) can perturb cellular glutathione/glutathione disulphide (GSH/GSSG) status and disrupt mucosal turnover. This study examines the effect of LOOH on GSH/GSSG balance and phase transitions in the human colon cancer CaCo‐2 cell. LOOH at 1 or 5 µm were noncytotoxic, but disrupted cellular GSH/GSSG and stimulated proliferative activity at 6 h that paralleled increases in ornithine decarboxylase activity, thymidine incorporation, expression of cyclin D1/cyclin‐dependent kinase 4, phosphorylation of retinoblastoma protein, and cell progression from G0/G1 to S. At 24 h, LOOH‐induced sustained GSH/GSSG imbalance mediated growth arrest at G0/G1 that correlated with suppression of proliferative activity and enhanced oxidative DNA damage. LOOH‐induced cell transitions were effectively blocked by N‐acetylcysteine. Collectively, the study shows that subtoxic LOOH levels induce CaCo‐2 GSH/GSSG imbalance that elicits time‐dependent cell proliferation followed by growth arrest. These results provide insights into the mechanism of hydroperoxide‐induced disruption of mucosal turnover with implications for understanding oxidant‐mediated genesis of gut pathology.
Introduction
Lipid hydroperoxides (LOOH) represent a class of dietary oxidants that can initiate degenerative processes via a generation of reactive oxygen species (ROS). There is growing recognition that ROS‐mediated degenerative processes can lead to disorders of the digestive system, including intestinal inflammation and cancer (Ames 1983; Parks et al. 1983). Previous studies have shown a causal relationship between the toxicity of dietary polyunsaturated oils with their peroxide contents (Kaneda et al. 1955; Andrews et al. 1960; Kimura et al. 1984), consistent with cytotoxicity associated with consumption of LOOH in vivo. At subtoxic levels, elevated luminal LOOH can induce tissue oxidative stress with potential impact on intestinal integrity. For example, enhancement of tumorigenesis in the colon has been shown to be associated with local administration of oxygenated derivatives of unsaturated fatty acids (Bull et al. 1984; Hara et al. 1966). Intrarectal instillation of hydroperoxy fatty acids was shown to provoke proliferative responses in colonic mucosa in rats (Bull et al. 1984) and rats given oxidized ethyl linoleate developed mucosal hypertrophy of the large intestine (Hara et al. 1966). These studies demonstrate that normal intestinal cell turnover can be disrupted by LOOH, and underscores the tumorigenic potential of oxidized lipids. Despite the implications of LOOH in gut pathology, the mechanism by which hydroperoxides mediate intestinal cell turnover in degenerative pathophysiological processes remains unclear.
Typically, cells are arrested in the quiescent state in terminally differentiated tissues, like the liver, kidney, and brain. While imposition of a severe oxidant stress often results in a cytotoxic biological endpoint, necrotic cell death is not necessarily an obligatory endpoint of all oxidative stress (Flores & McCord 1997). Studies on cell cycle responses have shown that regulatory genetic or environmental barriers govern the entry of cells into death or proliferation (Beach et al. 1988). Shifting these control checkpoints in the direction of reductant or oxidants during oxidative stress could result in a cell that favours quiescence, proliferation or death (Flores & McCord 1997; Cotgreave & Gerdes 1998; Aw 1999). Because the intestinal epithelium has one of the most rapid cell turnover rates of fully differentiated tissues, and organ homeostasis is normally balanced by cell proliferation and cell death (Johnson 1998), induction of oxidative shifts in the cellular redox status by oxidants like LOOH, can enhance cellular mitogenic or apoptotic responses (Aw & Tsunada 1997; Cotgreave & Gerdes 1998; Aw 1999; Wang et al. 2000).
The human colon carcinoma cell line, CaCo‐2, spontaneously exhibits structural and functional characteristics of mature small bowel enterocytes under standard culture conditions (Grasset et al. 1984; Meunier et al. 1995). We previously show that cytotoxic levels of LOOH at 0.1–0.2 mm significantly injure CaCo‐2 cells (Cepinskas et al. 1994) and concentrations at 10–25 µm induce cell apoptosis (Wang et al. 2000). In the current study we have used this CaCo‐2 cell model to test the hypothesis that disruption of redox balance by mild oxidative stress (1–5 µm LOOH) differentially induces exit of cells from quiescence into cell cycle progression, followed by their arrest in G0/G1. The specific objectives of this study were to define the cell cycle effects in CaCo‐2 cells caused by subtoxic concentrations of LOOH and their relationship to LOOH‐induced oxidative shifts in cellular redox (GSH/GSSG) and expression of proliferation markers and cell cycle changes.
Materials and methods
Materials
Menheden oil was a gift from Omega Protein (Reedville, VA). L‐NAC was obtained from Sigma Chemical Company (St. Louis, MO). L‐[1–14C] Ornithine (50 mCi/mmol) was purchased from NEN Products (Boston, MA). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Norcross, GA). DMEM and other cell culture supplies were obtained from Gibco‐BRL (Grand Island, NY). Nitrocellulose membranes and molecular weight markers for western blots were purchased from Bio‐Rad Laboratories (Hercules, CA). Antibodies (Abs) directed against cdk4 and cyclin D1 were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Anti‐PCNA and anti‐Rb were obtained from Oncogene Research Products (Cambridge, MA). The enhanced chemiluminescence (ECL) system for western immunoblot analysis and hyperfilm were purchased from Amersham (Arlington Heights, IL). The kit for DNA extraction was obtained from WAKO Chemicals (Richmond, VA). All other chemicals were of reagent grade and were obtained from local sources.
Culture of CaCo‐2 cells
Human carcinoma cell line (CaCo‐2) was obtained from the American Type Culture Collection (Rockville, MD) and cells were seeded in 25 cm2 culture flasks. The cells were grown in DMEM supplemented with 10% FBS, 1% nonessential amino acids, 100 U/ml penicillin and 10 mg/ml gentamycin. Cell cultures were maintained in a humidified atmosphere with 5% CO2/95% air at 37 °C. The culture medium was changed every 3 days. Cells between 20 and 35 passages were used in the experiments. To test the direct effect of LOOH without the contribution of growth factors, cells were subcultured in serum‐free DMEM prior to all experimentation.
Preparation of lipid hydroperoxide emulsion
LOOH were generated by air oxidation of Menhaden oil for 5 days. HPLC analysis identified the hydroperoxides of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) as the major oxidation products, which typically, were present in equimolar concentrations (Cepinskas et al. 1994). Lipid hydroxide (LOH) constituted less than 1% of the oxidation products. For our studies, the LOOH concentrations of 1 and 5 µm represented the total LOOH content, which was quantified spectrophotometrically by the thiobarbituric acid assay (Buege & Aust 1978). LOOH emulsions were prepared by sonicating 80 µmol oxidized oil and 570 µmol sodium taurocholate in 30 ml phosphate‐buffered saline (PBS), pH 7.4, as previously described (Aw et al. 1992).
Measurement of ODC activity
CaCo‐2 cells (2 × 105) were cultured in 6‐well plates to 70% confluency and were incubated with 1 or 5 µm LOOH for 0–24 h in serum‐free DMEM. In some experiments, cells were used after 7‐day post confluence. At the designated times, cells were collected, washed twice in PBS, resuspended in 100 mm Tris‐HCl, pH 7.4, 1 mm EDTA, 5 mm DTT, 50 µm pyridoxal‐5‐phosphate, and homogenized with a Dura grind Dounce tissue grinder for 20 up and down strokes. The supernatant was incubated in stoppered vials in the presence of 3.5 nmol of L‐[1–14C] ornithine (58 mCi/ml) for 1 h at 37 °C. The reaction was stopped by the addition of 10% trichloroacetic acid (TCA), and the 14CO2 liberated by the decarboxylation of ornithine was trapped on filter paper impregnated with 2 m NaOH. The filter paper was solubilized in scintillation cocktail and the radioactivity of the trapped 14CO2 was counted. ODC activity was standardized for protein content, and the results were expressed as pmoles CO2 release per milligram protein per h. All assays were performed in triplicates.
[3H]‐thymidine incorporation
CaCo‐2 cells were cultured to 70% confluency in 24‐well plates and incubated with 1 or 5 µm LOOH for 6 or 24 h in serum‐free DMEM with or without 1 mm hydroxyurea. One µCi of methyl‐[3H] thymidine was added during the final 4 h incubation. In some experiments, 5 mm NAC was added 30 min before LOOH exposure. At designated times, cells were washed twice with PBS, and precipitated by 5% TCA at 4 °C. The pellets were washed with ice‐cold 95% ethanol and dried at room temperature. The cell pellets were solubilized with 1 m NaOH for 30 min, transferred to 5 ml scintillation cocktail and the cell‐associated radioactivity was counted. The results are expressed as a percentage of total [3H] thymidine incorporated to determine the extent of DNA synthesis vs. DNA repair.
Quantification of GSH and GSSG
Cell GSH and GSSG were determined by the HPLC method of Reed et al. (1980) as we previously described in detail (Noda et al. 2002). Briefly, cells were treated with ice‐cold 5% TCA followed by centrifugation to remove TCA‐insoluble proteins. The acid supernatants were derivatized with 6 mm iodoacetic acid and 1% 2,4‐dinitrofluorobenzene to yield the S‐carboxymethyl and 2,4‐dinitrophenyl derivatives of GSH and GSSG. Separation of GSH and GSSG derivatives was achieved on a 15‐cm × 4.6 mm 10 µm C18‐reversed phase ion‐exchange column.
Determination of 8‐hydroxy 2‐deoxyguanosine (8‐oxoG)
Oxidative damage to DNA was quantified by the formation of 8‐oxoG (Shigenaga et al. 1994). Total DNA was extracted from 3 × 106 cells with a chaotropic iodine protocol (WAKO Chemicals) that minimizes ex vivo DNA oxidation. Briefly, cells were homogenized on ice in 0.1 m NaCl, 30 mm Tris, pH 8.0, 10 mm EDTA, 10 mmβ‐mercaptoethanol, 0.5% Triton‐X‐100 with 6 passes of a Teflon‐glass homogenizer. The nuclei fraction was collected and incubated with RNAse (50 U/ml RNAse A and 100 U/ml RNAse T1) and proteinase K (5 mg/ml) in 10 mm Tris‐HCl, pH 8.0, 5 mm EDTA, and 1% SDS for 1 h at 50 °C to remove contaminating RNA and protein. DNA was extracted from the 10 000 × g supernatant using the iodine protocol consisting of NaI treatment and extraction with 40% 2‐propanol and isopropyl alcohol. The extracted DNA was incubated with 0.1 mg/ml nuclease P1 for 15 min at 65 °C followed by 1 U/µl alkaline phosphatase. The deoxyribonucleosides were purified (10 000 × g for 5 min) on an UltraFree Millipore Eppendorf Filtration system with a 30 000 dalton cutoff, and separated on a 15‐cm × 4.6 mm 3 µm LC‐18 DB Supelco column (Bellefonte, PA) using a 6% linear gradient of methanol in 50 mm KH2PO4 buffer, pH 5.5, and a flow rate of 1 ml/min. 8‐oxoG was detected using an ESA electrochemical detector (0.6 V), and the results are expressed as the number of 8‐oxoG residues per 105 2‐deoxyguanosine (2dG) residues.
Cell cycle analysis by flow cytometry
A 3‐day serum starvation regimen was used to synchronize CaCo‐2 cell growth. Thereafter, cell monolayers were treated with 1 or 5 µm LOOH in serum‐free DMEM for 6 or 24 h. At the designated times, cells were trypsinized, washed with cold PBS, and fixed in 70% ethanol for 30min at −20 °C. Fixed cells were washed with PBS and incubated with DNase‐free RNase for 30 min at 37 °C. Cells (1 × 106) were incubated with propidium iodide (5 µg/ml) at 4 °C for 30 min in the dark. Flow cytometry was performed using a FACS Vantage flow cytometer (Becton Dickinson, San Jose, CA), and the results were analysed with the Cell Quest software.
Western blot analysis
Cell monolayers were washed twice in ice‐cold PBS and lysed at 4 °C in 50 mm Tris‐HCl, pH 7.6, 300 mm NaCl, 0.5% Triton X‐100, 10 µg/ml aprotinin, 10 µg/ml leuptin, 1 mm phenylmethylsulphonyl fluoride, 1.8 mg/ml iodoacetamide, 50 mm NaF, 1 mm DTT. The supernatants were collected and 50 µg of protein was run per lane on an 8% (for Rb), 10% (for cdk4, cyclin D1 and PCNA) sodium dodecyl sulphate polyacrylamide gel (SDS‐PAGE) and electroblotted to nitrocellulose membranes. The membranes were blocked by overnight incubation in 5% dry milk and 0.1% Tween‐20 at 4 °C, and thereafter incubated with the primary antibodies (1 : 40–1 : 200 dilution) for 3 h at room temperature. Each blot was probed with anticdk4, anticyclin D1, anti‐PCNA, or anti‐Rb. The membranes were washed and incubated with horseradish peroxidase‐conjugated goat antimouse or antirabbit immunoglobulin G (1 : 1500 dilution) for 1 h. The immune complexes were visualized by the ECL method according to the manufacture's protocol. Signal intensity was quantified using a Bio‐Rad image analysis system and the results normalized to the signal intensity of β actin for each blot.
Protein assay
Protein was measured using Bio‐Rad Protein Assay kit (Bio‐Rad Laboratories, Hercules, CA) according to the manufacturer's protocol.
Statistical analysis
Results are expressed as mean ± SE. Data were analysed using a one‐way anova with Bonferroni corrections for multiple comparisons. P‐values of < 0.05 were considered statistically significant.
Results
Lipid hydroperoxide dose‐ and time‐dependently promotes cell proliferation or induces growth arrest
The effect of LOOH on cell proliferation was assessed by measurements of cellular activities of ornithine decarboxylase (ODC) and 3H‐thymidine incorporation. Figure 1 summarizes the results on the effect of LOOH on CaCo‐2 cell ODC activity after exposure to 1 or 5 µm LOOH. LOOH dose‐dependently increased ODC activity at 6 h (Fig. 1a), consistent with oxidant‐induced increase in proliferative activity. Notably, by 24 h ODC activity returned to baseline values with 1 µm LOOH, while enzyme activity was inhibited with 5 µm LOOH (Fig. 1b), suggesting that prolonged exposure to LOOH elicited a cessation of proliferative activity consistent with cell growth arrest. In other experiments, we found that neither taurocholate nor lipid hydroxides (LOH), previously reported to evoke proliferative responses (Bull et al. 1984), stimulated ODC activity under these conditions (data not shown), indicating that LOOH was the mitogenic stimulus. Interestingly, the LOOH effect on ODC activity at 6 h was highly dependent on the confluent status of the cells. The results in Fig. 2 show that ODC activity in subconfluent cultures was stimulated by treatment with 1 or 5 µm LOOH to the extents that were similar to that induced by serum (FBS) addition. In contrast, LOOH exposure was without effect in cells that are 7‐day postconfluent. However, unlike LOOH challenge, FBS supplementation in 7‐day postconfluent cells elicited a significant increase in ODC activity that was 4‐fold over the activity induced in subconfluent cells (Fig. 2).
Figure 1.
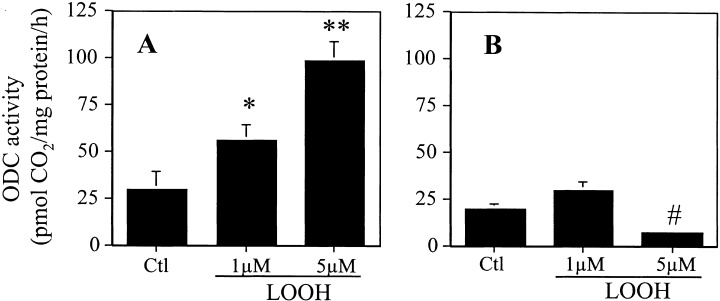
Effect of LOOH on ODC activity. CaCo‐2 cells were grown to 70% confluency and cultured in serum‐free DMEM. Cells were exposed to 1 or 5 µM LOOH for 6 h (a) and 24 h (b) and ODC activity was determined by the liberation of 14CO2. Results are the mean ± SE of five separate experiments performed in triplicates. *P < 0.05 versus untreated control, **P < 0.001 versus control, #P < 0.05 versus control and 1 µM LOOH treatment.
Figure 2.
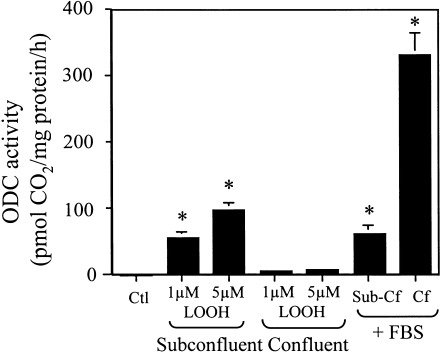
LOOH‐induced CaCo‐2 ODC activity is dependent on cell confluency. Subconfluent (70%) or confluent (7 day) CaCo‐2 cell monolayers were incubated in serum‐free DMEM and exposed to 1 or 5 µM LOOH for 6 h. In parallel experiments, subconfluent or confluent cells grown in serum‐free DMEM were supplemented with 10% fetal bovine serum (FBS) and incubated for 6 h. ODC activity was determined by the liberation of 14CO2. Results are the mean ± SE of four separate experiments performed in triplicate. *P < 0.05 versus untreated control and LOOH‐treated confluent cells.
The results on LOOH effect on [3H]‐thymidine incorporation are summarized in Fig. 3. DNA synthesis and repair activities were determined from [3H]‐thymidine incorporation performed in the absence or presence of hydroxyurea, respectively. At 6 h, 1 or 5 µM LOOH caused significant increases in [3H]‐thymidine incorporation, indicating an increase in DNA synthesis (Fig. 3a) which accounted for > 90% of the incorporated thymidine. In comparison, [3H]‐thymidine incorporation at 24 h was significantly decreased in LOOH‐treated cells (Fig. 3a), of which only 50% of the incorporated thymidine was attributed to DNA synthesis. By contrast, DNA repair activity (thymidine incorporation in the presence of hydroxyurea to inhibit DNA replication) was < 10% at 6 h, but accounted for half the incorporated thymidine at 24 h after LOOH treatment (Fig. 3b). To verify that the increase in DNA repair activity was associated with enhanced oxidative damage to DNA, we quantified the formation of 8 hydroxy 2′‐deoxyguanosine (8‐oxoG). The results in Fig. 4 show that 24 h treatment of cells with 1 or 5 µm LOOH caused a 2‐ and 3‐fold increase in 8‐oxoG levels, respectively. No detectable 8‐oxoG was found after 6 h LOOH treatment (data not shown). Collectively, the results show that subtoxic concentrations of LOOH caused an initial induction of CaCo‐2 cell proliferation at 6 h that was associated with increased ODC activity and DNA synthesis. Prolonged LOOH treatment for 24 h inhibited ODC activity, consistent with cell growth arrest, and promoted DNA repair in association with enhanced oxidative DNA damage.
Figure 3.
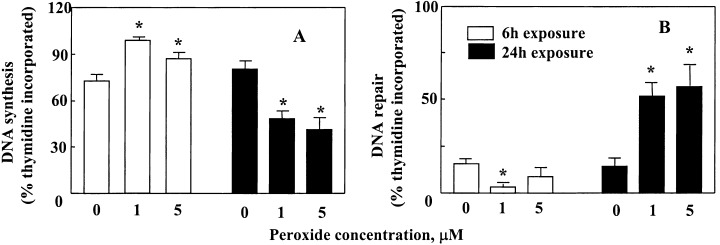
Effect of LOOH on 3H‐thymidine incorporation. Subconfluent (70%) CaCo‐2 cell monolayers in 24‐well plates were incubated with 1 or 5 µM LOOH for 6 or 24 h in serum‐free DMEM in the absence (a) or presence of 1 m m hydroxyurea (b). One µCi of [ 3 H]‐thymidine was added to the cell incubation during the final 4 h incubation. The results are expressed as percentage of total thymidine incorporated. Each value is the mean ± SE of five separate experiments performed in triplicate. * P < 0.05 versus the respective control at 6 or 24 h.
Figure 4.
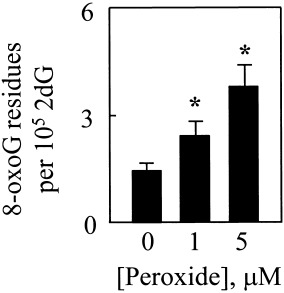
LOOH enhances formation of 8‐oxoG in CaCo‐2 cells. Cells (3 × 10 6 ) were treated with 1 or 5 µ m LOOH for 24 h. Total DNA was extracted and digested with nucleases to yield the deoxyribonucleosides. 8‐oxoG was resolved by HPLC using electrochemical detection, and quantified by comparison to authentic standards. The results are expressed as the number of 8‐oxoG residues per 2dG residues. Each value is the mean ± SE of five separate experiments. * P < 0.05 versus control.
Expression of proliferating markers correlates with lipid hydroperoxide‐induced cell transition
A highly conserved family of serine/threonine protein kinase complex that consists of regulatory cyclins, catalytic cyclin‐dependent kinases (cdks), and the proliferating cell nuclear antigen (PCNA) regulates progression through the cell cycle (Sherr 1995; Prosperi 1997). To further investigate the effect of LOOH on CaCo‐2 proliferation we examined the protein expression of cdk4, cyclin D1, and PCNA. The results in Fig. 5 show that 1 and 5 µm LOOH time‐dependently increased the expression of cdk4 and cyclin D1 after oxidant exposure. Both levels of LOOH significantly increased cdk4 at 3–6 h; thereafter the protein decreased to baseline at 24 h. Cyclin D1 was also strongly up‐regulated at 3–6 h by 1 and 5 µm LOOH. However, cyclin D1 remained elevated at 12 h in cells treated with 1 µm LOOH, but declined to baseline levels in cells treated with the higher LOOH dose. By 24 h, the protein levels of cyclin D1 were similar to control values regardless of LOOH concentration. Notably, the expression of PCNA was relatively constant over the 24 h, suggesting that regulation of PCNA expression is oxidant insensitive. The effect of LOOH on the retinoblastoma protein (Rb) was determined by the expression of its under‐phosphorylated and phosphorylated states (pRb and ppRb, respectively). In the absence of LOOH challenge, the Rb protein was under phosphorylated (Fig. 6), consistent with cell growth arrest after 3 days of serum starvation. Treatment with 1 or 5 µm LOOH increased Rb phosphorylation (ppRb) at 3 and 6 h, consistent with an enhancement of proliferative activity. Interestingly, treatment of cells with 5 µm LOOH also appeared to increase the total Rb protein at 6 h in addition to promoting Rb phosphorylation. Regardless of the LOOH dose, the phosphorylation state of Rb returned to control levels by 24 h post‐treatment that temporally corresponded to a cessation of cell proliferative activity.
Figure 5.
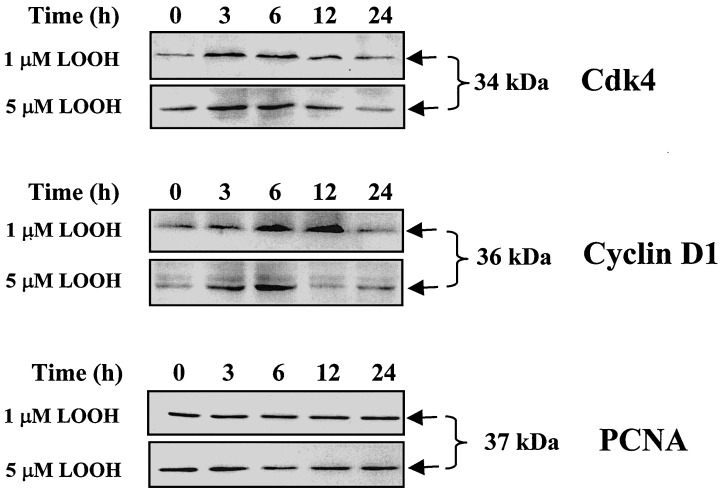
Effect of LOOH on protein expression of cdk4, cyclin D1 and PCNA. CaCo2 cells grown in serum‐free DMEM were treated with 1 or 5 µM LOOH for 0–24 h. At designated times, total proteins were extracted and the protein lysates were subjected to western immunoblot analysis for expression of cdk4 (34 kDa), cyclin D1 (36 kDa) and PCNA (37 kDa). Four separate immunoblots were performed for each proliferation marker and representative immunoblots for each of cdk4 (34 kDa), cyclin D1 (36 kDa) and PCNA (37 kDa) are shown.
Figure 6.
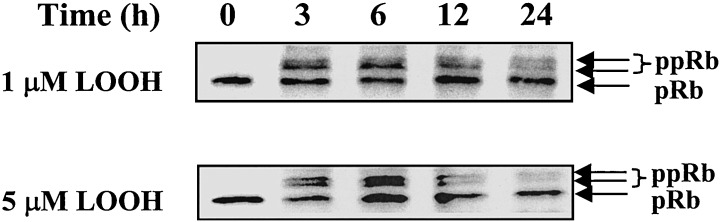
Effect of LOOH on phosphorylation of retinoblastoma protein, Rb. CaCo2 cells grown in serum‐free DMEM were treated with 1 or 5 µM LOOH for 0–24 h. Protein lysates were subjected to western analysis for assessment of the phosphorylation status of Rb (110–114 kDa). The under‐ or hyper‐phosphorylated states of Rb are designated as pRb and ppRb, respectively. A representative immunoblot of four separate experiments is presented.
Cell cycle progression is associated with lipid hydroperoxide‐induced phase transition of CaCo‐2 cells
The effect of LOOH on CaCo‐2 cell transitions was evaluated by cell cycle distribution using flow cytometry. In untreated subconfluent CaCo‐2 cells, serum starvation for 3 days induced growth arrest in 85% of the cells at G0/G1 with diploid DNA content (DNA index, DI = 1.0) (Fig. 7). Interestingly, about 15% of cells were at the S and G2/M phases that exhibited DNA contents between DI of 1.0 and 2.0 (S phase) and DI of 2.0 (G2/M phase), indicating that the transformed CaCo‐2 cells did not completely exit exponential growth with serum deprivation. However, in the presence of serum, 75% of the cells were exponentially growing cells (Fig. 7), showing that serum removal did elicit growth arrest. Treatment of serum starved cells with 1 µM LOOH initiated progression of the G0/G1 cell population into the S phase and a doubling of the DNA content at G2/M phase by 6 h (Fig. 7), that temporally corresponded to the increase in expression of cdk4/cyclin D1, and the phosphorylation of Rb (5, 6, respectively). However, by 24 h post‐LOOH exposure, the cell population was largely growth‐arrested at G0/G1 (Fig. 7), in parallel with decreased cdk4/cyclin D1 expression and de‐phosphorylation of Rb. Exposure to 5 µM LOOH similarly caused entry of the cells into the S and G2/M phases at 6 h and induced growth arrest at G0/G1 at 24 h (Fig. 7) that directly paralleled the temporal changes in proliferating indices (5, 6). Notably, the percent of cells that were growth‐arrested at G0/G1 at 24 h after 1 µm LOOH treatment (80%) was smaller than that caused by 5 µm LOOH (95%, Fig. 7), in accordance with a greater oxidative stress associated with a higher LOOH dose.
Figure 7.
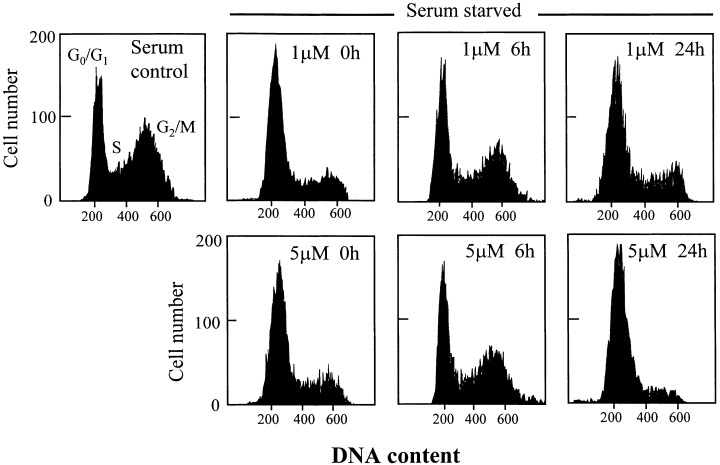
Cell cycle distribution of CaCo‐2 cells exposed to LOOH. CaCo‐2 cells were grown in complete DMEM media (with serum) or cell growth was synchronized by 3‐day serum starvation. Serum starved cells were exposed to 1 or 5 µM LOOH for 0–24 h. At the indicated times, cells were prepared and analysed by flow cytometry as described in Materials and Methods. The cell cycle distribution profile is from one representative of four separate experiments.
Lipid hydroperoxide‐induced disruption of cellular redox balance and linkage to cell transition
Because LOOH can elicit redox imbalance, we sought to determine whether, mechanistically, the differential induction of CaCo‐2 proliferation or growth arrest by LOOH was mediated by a loss of cellular redox balance caused by LOOH. Cellular redox balance is primarily maintained by the distribution of GSH and its oxidized form, GSSG (Kaplowitz et al. 1985), and the ratio of GSH to GSSG is often used as an indicator of the redox status of a cell (Murphy et al. 1992; Wang et al. 2000). Figure 8 shows the effect of LOOH on the kinetics of changes in intracellular GSH and GSSG, and the GSH‐to‐GSSG ratio. Exposure to 1 µM LOOH caused significant decreases in cell GSH at 15 and 30 min (Fig. 8a). From 3 to 24 h, cell GSH recovered to baseline values with an overshoot at 6 h (Fig. 8a). An increase in GSSG paralleled the loss of GSH (30 min to 6 h, Fig. 8b), which resulted in a decrease in the GSH‐to‐GSSG ratio (Fig. 8c). Despite the recovery of cell GSH at 3 to 6 h, the GSH/GSSG ratio only transiently recovered to 50% of control at these times, and remained below the baseline values through 24 h (Fig. 8c). Treatment of cells with 5 µM LOOH caused a similar temporal decrease and recovery of intracellular GSH (Fig. 8d). However, the magnitude of increase in GSSG induced by 5 µm LOOH was 2‐fold higher (Fig. 8e), consistent with a greater oxidation of GSH in accordance with a greater oxidant load. Accordingly, 5 µm LOOH significantly decreased the GSH‐to‐GSSG ratio (50–70% of control) at all times post‐LOOH treatment and remained low throughout 24 h (Fig. 8f), indicating that prolonged oxidant stress elicits a sustained redox imbalance.
Figure 8.
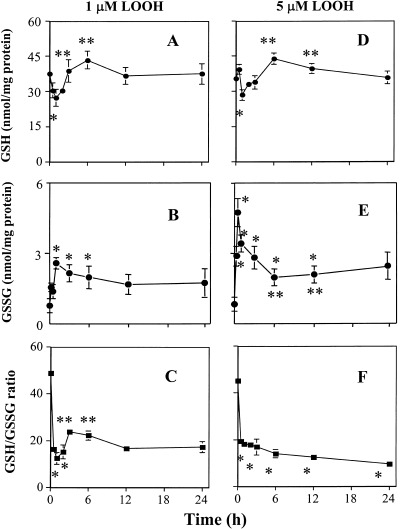
Kinetics of LOOH‐induced changes in cellular GSH, GSSG and GSH‐to‐GSSG ratio in CaCo2 cells. CaCo‐2 cells grown in serum‐free DMEM were treated with 1 or 5 µM LOOH for 0–24 h. At designated times, cells were harvested and processed for HPLC analysis of cellular GSH/GSSG status. Panels (a), (b) and (c) represent GSH, GSSG and GSH‐to‐GSSG ratio in cells treated with 1 µM LOOH, respectively. Panels (d), (e) and (f) represent GSH, GSSG and GSH‐to‐GSSG ratio in cells treated with 5 µM LOOH, respectively. Each value is the mean ± SE of four separate experiments. * P < 0.05 versus 0 min control; ** P < 0.05 versus 30 min.
To verify that the disruption of normal cellular GSH/GSSG status was responsible for the differential effect of LOOH on cell proliferation and growth arrest, we pretreated CaCo‐2 cells with L‐N‐acetyl cysteine (NAC, 5 mm), an important precursor of GSH with thiol reducing activity (Moldeus & Cotgreave 1994). The results in Fig. 9 show that NAC completely abrogated the respective increase or decrease in 3H thymidine incorporation induced by LOOH at 6 h (Fig. 9a) or 24 h (Fig. 9b). Accordingly, pretreatment with NAC attenuated the LOOH‐induced increase in cellular GSSG (Fig. 9c), consistent with a restoration of the cellular GSH/GSSG balance. Collectively, these results support our suggestion that the disruption of cellular GSH/GSSG imbalance by LOOH induces CaCo‐2 cell phase transition. Notably, a shorter duration of LOOH exposure and an acute loss of GSH/GSSG balance (e.g. 6 h) favours cell proliferation while prolonged LOOH stress and extended GSH/GSSG imbalance (e.g. 24 h), favours growth arrest.
Figure 9.
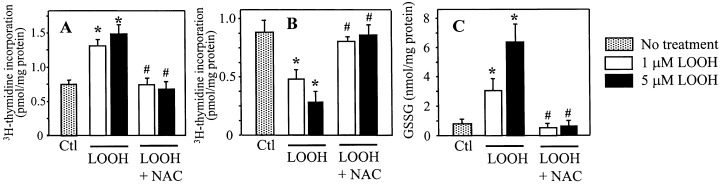
N‐acetylcysteine ( NAC) pretreatment attenuates LOOH‐induced thymidine incorporation and GSSG elevation. CaCo‐2 cells grown in serum‐free DMEM were treated with 1 or 5 µM LOOH for 6 or 24 h in the absence or presence of NAC (5 m m ). Whenever present, NAC was added to cells 30 min before LOOH challenge. At designated times, cells were harvested for determination of 3 [H]‐thymidine incorporation. In parallel experiments, cells were treated with LOOH with or without NAC and cells were harvested at 30 min post‐LOOH treatment. Cell extracts were processed for quantification of GSSG contents by HPLC. Panels (a) and (b) represent thymidine incorporation at 6 and 24 h. Panel (c) represents cellular GSSG contents measured at 30 min. Results are mean ± SE of four separate experiments. * P < 0.05 versus respective untreated controls; # P < 0.05 versus LOOH treatment.
Discussion
It is well recognized that ROS, including LOOH, are agents of cytotoxicity. However, it is increasingly apparent that ROS at low concentrations can play an important role in mediating specific cell responses and expression of genes (Flores & McCord 1997; Aw 1999). As pertains to the intestine, existing evidence implicates a role for ROS in the pathogenesis of degenerative disorders of the gut, such as inflammation and cancer (Ames 1983; Parks et al. 1983). The contribution of redox imbalance, which often results from oxidative stress, to the cellular and molecular events that lead to disruption of intestinal integrity, is poorly understood. The present study provides evidence to support our hypothesis that acute disruption of normal cellular GSH/GSSG status by LOOH promotes an early proliferative response in CaCo‐2 cells in association with enhanced expression of proliferative markers and stimulation of cell cycle progression. At later times, prolonged and sustained LOOH‐induced redox imbalance induces cell growth arrest in association with increased oxidative DNA damage and down regulation of proliferative activity. The doses of 1–5 µm LOOH are physiologically relevant LOOH concentrations that are present in common foods cooked in oils or fats (Aw 1999), and it is significant that this mild oxidative stress can perturb the cellular redox status to the extent that influences cell transition in the absence of cell toxicity. Notably, the disruption of cellular GSH/GSSG status by LOOH was a kinetically rapid process that resulted in marked GSH decreases and GSSG elevation within 15–30 min post‐treatment, indicating that loss of redox homeostasis is an early event that contributes to the cell proliferative responses to oxidant challenge. Significantly, this redox stimulation of proliferative responses can be completely prevented by blockade of the early increases in GSSG by NAC (Fig. 9). Moreover, it is notable that, despite an early recovery of GSH (within 3 h), the GSH/GSSG ratio remained low and compromised for 24 h (Fig. 8), which directly correlated with a cessation of cell proliferative activity and induction of growth arrest. These temporal relationships between LOOH‐induced redox changes and cell transition suggest that the cell responses were mediated predominantly by changes in the GSH‐to‐GSSG ratio rather than by changes in levels of GSH per se.
An early response to the loss of GSH/GSSG balance resulting from acute LOOH exposure was an elevation in ODC activity. Typically, ODC is expressed in growing CaCo‐2 cells and plays a major role in the replication process (D’Agostino et al. 1989). Kinetically, the stimulation of ODC activity directly correlated with increased DNA replicative activity as evidenced by enhanced thymidine incorporation at 6 h after LOOH exposure. Thus, these temporal relationships between an early induction of cellular GSH/GSSG redox imbalance (within minutes) and a later increase in ODC function and DNA replication (within 6 h) are consistent with LOOH‐induced redox change representing an initiating event that governs cell proliferation. In comparison, the sustained loss of GSH/GSSG balance over 24 h caused a suppression of ODC activity and DNA synthesis, suggesting that prolonged LOOH challenge and oxidative shift in the redox status mediates growth arrest of cells in favour of quiescence following one complete progression of the cell cycle. Consistent with these interpretations, our data show that perturbation of the GSH/GSSG status was initially associated with enhanced expression of cyclin D1/cdk4 and increased Rb phosphorylation that was temporally linked to the increase in ODC activity and DNA synthesis at 3–6 h post LOOH exposure. By 12–24 h, the sustained decrease in GSH/GSSG ratio elicited a down‐regulation of these parameters. Thus, our current results provide evidence that LOOH induces differential cell fate in favour of proliferation or growth arrest that is mediated by LOOH‐induced GSH/GSSG shifts. The abrogation of cell transition responses with NAC that directly corresponded to the restoration of cellular redox balance is consistent with this interpretation.
Previous studies have shown that progression through the G1 phase of the mammalian cell division cycle is governed by the function of the interacting complex of PCNA, cyclins, cdks, and p21 (Xlong et al. 1993). Moreover, stimulation of the cdk4/cyclin D1 assembly facilitates exit of cells from G1 by the phosphorylation of Rb (Brugarolas et al. 1995) whereby the release of the proliferative transcription factor, E2F (Weinberg 1995; Bartek et al. 1997) releases the mitotic constraints and stimulates DNA synthesis and S phase gene expression (Bartek et al. 1997; Mihara et al. 1989). That mild LOOH stress and the accompanying GSH/GSSG imbalance is a mitogenic signal in CaCo‐2 cells is supported by cell cycle analysis wherein LOOH treatment increases cell entry into S phase and DNA synthesis (Fig. 7). Taken together, these results are in agreement with previous observations that oxidatively modified lipids can provoke proliferative responses in the gut (Bull et al. 1984). It is notable, however, that the mitogenic effect of LOOH was short‐lived. One possible explanation for the lack of a sustained mitogenic effect may be related to the induction of oxidative DNA damage with extended LOOH exposure time. This interpretation is consistent with the observations of elevated concentrations of 8 oxoG (Fig. 4) and stimulation of DNA repair activity at 24 h after LOOH challenge (Fig. 3b). Indeed, the collective results on measurements of ODC activity and thymidine incorporation, the down regulation of cdk4/cyclin D1, and the under‐phosphorylation of Rb all support a cessation of cell proliferative activity that is consistent with cell entry into quiescence after 24 h LOOH exposure.
The current finding of redox‐mediated cell growth arrest is in agreement with the results of our recent study wherein we treated CaCo‐2 cells with diamide, a cell‐permeant thiol oxidant that directly oxidized GSH to GSSG independent of ROS generation (Noda et al. 2001). Interestingly, however, this direct chemical induction of GSH/GSSG imbalance without the participation of ROS elicited growth arrest at the G1‐to‐S transition and G2/M (Noda et al. 2001), in contrast to growth arrest at G0/G1 exhibited by LOOH‐induced by redox imbalance. This interesting difference in cell cycle response indicates a clear distinction in checkpoint regulation in the two redox models. In the current study, the presence of accumulated damaged DNA resulting directly from ROS damage or from sustained LOOH‐induced redox imbalance may provide one explanation as to why cells were prevented from entry into S‐phase and DNA synthesis so as to allow for repair of the damaged DNA and thereby preventing its propagation in the cell cycle. In the chemical (diamide) model of redox change, we found no evidence of accumulated damaged DNA (data not shown). Thus, despite the similarities in chemical (e.g. diamide) and oxidant (e.g. LOOH) induction of cellular GSH‐to‐GSSG shifts, the differences in their target checkpoints in the cell cycle do underscore an important mechanistic distinction between signalling that are mediated by ROS (which also involves redox changes) and those that are mediated directly through redox changes (which does not involve ROS) (Kokura et al. 2001). Other investigators have similarly demonstrated entry of the human colorectal cancer cell, HCT116, into a sustained G2 phase of cell cycle after exposure to γ‐irradiation or anticancer agents (Waldman et al. 1996; Bunz et al. 1998). Whether these stimuli mediate G2/M arrest via induction cellular redox shift independently of ROS generation is not known.
The observation that LOOH differentially mediates cell proliferation and cell growth arrest at different times after oxidant challenge clearly indicates a fundamental difference in the signalling mechanisms induced by cellular oxidative and redox shifts from that mediated by other mitogenic stimuli such as serum (FBS). In addition, the dependence on the state of cell confluency is another notable difference between the mitogenic effects of LOOH versus FBS. In our study, LOOH strongly stimulated ODC activity in subconfluent, but not in confluent, cell monolayers, while FBS activated ODC activity in both subconfluent and confluent cells (Fig. 2). Moreover, while ODC activity was stimulated to the same degree by LOOH and FBS in subconfluent cells, only FBS activated ODC function, and to a greater degree in day 7 confluent cells (Fig. 2). The reasons for these interesting differences are unclear and warrant further investigation.
In summary, the current study shows that subtoxic LOOH initiates a confluence of cellular events that ultimately lead to early proliferative responses and cell cycle progression and late growth arrest in the human CaCo‐2 cell. Our data is consistent with an early loss of GSH/GSSG redox homeostasis induced by LOOH exposure that mediates these cellular responses. Importantly, our results underscore a pivotal role that redox status may play in colonic tumour cell transition. Indeed, redox modulation of regulatory checkpoints in the cell cycle may suggest a potential mechanism by which oxidants like LOOH influence colonic cell turnover with important implications for understanding oxidant‐mediated development of gut pathology.
Acknowledgements
This study was supported by a grant from the National Institutes of Health DK‐44510.
References
- Ames BN (1983) Dietary carcinogens and anticarcinogens. Science 221, 1256. [DOI] [PubMed] [Google Scholar]
- Andrews JS, Griffith WH, Mead JF, Stein RA (1960) Toxicity of air‐oxidized soybean oil. J. Nutr. 70, 199. [DOI] [PubMed] [Google Scholar]
- Aw TY (1999) Molecular and cellular responses to oxidative stress and changes in oxidation‐reduction imbalance in the intestine. Am. J. Nutr. 70, 557. [DOI] [PubMed] [Google Scholar]
- Aw TY, Tsunada S (1997) Persistent subtoxic lipid peroxide challenge in vivo causes thiol redox imbalance and cytostasis in rat intestine. FASEB J. 11, A219 (Abstract). [Google Scholar]
- Aw TY, Williams MW, Gray L (1992) Absorption and lymphatic transport of peroxidized lipids by rat small intestine in vivo: role of mucosal GSH. Am. J. Physiol. 262, G99. [DOI] [PubMed] [Google Scholar]
- Bartek J, Bartkova J, Lukas J (1997) The retinoblastoma protein pathway in cell cycle control and cancer. Exp. Cell. Res. 237, 1. [DOI] [PubMed] [Google Scholar]
- Beach D, Basilico C, Newport J, eds. (1988) Cell cycle control in eukaryotes In: Current communication in molecular biology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ (1995) Radiation‐induced cell cycle arrest compromised by p21 deficiency. Nature 377, 552. [DOI] [PubMed] [Google Scholar]
- Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Meth. Enzymol. 52, 302. [DOI] [PubMed] [Google Scholar]
- Bull AW, Nigro ND, Golembieski WA, Crissman JD, Marnett LJ (1984) In vivo stimulation of DNA synthesis and induction of ornithine decarboxylase in rat colon by fatty acid hydroperoxides autoxidation products of unsaturated fatty acids. Cancer Res. 44, 4924. [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1487. [DOI] [PubMed] [Google Scholar]
- Cepinskas G, Kvietys PR, Aw TY (1994) Omega 3‐lipid peroxides injure CaCo‐2 cells: relationship to the development of reduced glutathione antioxidant systems. Gastroenterology 107, 80. [DOI] [PubMed] [Google Scholar]
- Cotgreave IA, Gerdes RG (1998) Recent trends in glutathione biochemistry–glutathione–protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 242, 1. [DOI] [PubMed] [Google Scholar]
- D’Agostino L, Daniele B, Pignata S, Gentile R, Tagliaferri P, Contegiacomo A, Silvestro G, Polistina C, Bianco AR, Mazzacca G (1989) Ornithine decarboxylase and diamine oxidase in human colon carcinoma cell line CaCo‐2 in culture. Gastroenterology 97, 888. [DOI] [PubMed] [Google Scholar]
- Flores SC, McCord JM (1997) Redox regulation by the HIV‐1 Tat transcriptional factor In: Oxidative stress and the molecular biology of antioxidant defenses. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, p. 117. [Google Scholar]
- Grasset E, Pinto M, Dussaulx E, Zweibaum A, Desjeux JF (1984) Epithelial properties of human colonic carcinoma cell line CaCo‐2: electrical parameters. Am. J. Physiol. 247, C260. [DOI] [PubMed] [Google Scholar]
- Hara H, Miyashita K, Ito S, Kasai T (1966) Oxidized ethyl linoleate induces mucosal hypertrophy of the large intestine and affects cecal fermentation of dietary fiber in rats. J. Nutr. 126, 800. [DOI] [PubMed] [Google Scholar]
- Johnson LR (1998) Regulation of gastrointestinal mucosal growth. Physiol. Rev. 68, 456. [DOI] [PubMed] [Google Scholar]
- Kaneda T, Sakai H, Ishii S (1955) Nutritive value or toxicity of highly unsaturated fatty acids. J. Biochem. 42, 561. [Google Scholar]
- Kaplowitz N, Aw TY, Ookhtens M (1985) The regulation of hepatic glutathione. Ann. Rev. Pharmacol. Toxicol. 25, 715. [DOI] [PubMed] [Google Scholar]
- Kimura T, Iida K, Takei Y (1984) Mechanisms of adverse effect of air‐oxidized soybean oil‐feeding in rats. J. Nutr. Sci. Vitamin 30, 125. [DOI] [PubMed] [Google Scholar]
- Kokura S, Wolf RE, Yoshikawa T, Granger DN, Aw TY (2001) NFκB signalling is post‐hypoxic endothelial cells: relevance to E‐selectin expression and neutrophil adhesion. J. Vascular Res. 38, 47. [DOI] [PubMed] [Google Scholar]
- Meunier V, Bourrie M, Berger Y, Fabre G (1995) The human intestinal epithelial cell line CaCo‐2; pharmacological and pharmacokinetic applications. Cell Biol. Toxicol. 11, 187. [DOI] [PubMed] [Google Scholar]
- Mihara M, Cao XR, Yen A, Chandler S, Driscoll B, Murphree AL, T’ang A, Fung YT (1989) Cell cycle‐dependent regulation of phosphorylation of the human retinoblastoma gene product. Science 246, 1300. [DOI] [PubMed] [Google Scholar]
- Moldeus P, Cotgreave IA (1994) N‐acetylcysteine. Meth. Enzymol. 234, 482. [DOI] [PubMed] [Google Scholar]
- Murphy ME, Scholich H, Sies H (1992) Protection by glutathione and other thiol compounds against the loss of protein thiols and tocopherol homologs during microsomal lipid peroxidation. Eur. J. Biochem. 210, 139. [DOI] [PubMed] [Google Scholar]
- Noda T, Iwakiri R, Fujimoto K, Aw TY (2001b) Induction of mild intracellular redox imbalance inhibits proliferation CaCo‐2 cells. FASEB J. 15, 2131. [DOI] [PubMed] [Google Scholar]
- Noda T, Iwakiri R, Fujimoto K, Rhoads CA, Aw TY (2002) Exogenous cysteine and cysteine promote cell proliferation in CaCo‐2 cells. Cell Prolif. 35, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DA, Bulkley GB, Granger DN (1983) Role of oxygen‐derived free radicals in digestive tract diseases. Surgery 94, 415. [PubMed] [Google Scholar]
- Prosperi E (1997) Multiple roles of the proliferating cell nuclear antigen: DNA replication, repair and cell cycle control. Prog. Cell. Res. 3, 193. [DOI] [PubMed] [Google Scholar]
- Reed DJ, Babson JR, Beatty PW, Brodie AE, Ellis WW, Potter DW (1980) High‐performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Anal. Biochem. 106, 55. [DOI] [PubMed] [Google Scholar]
- Sherr CJ (1995) D‐type cyclins. Trends Biochem. Sci. 20, 187. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Aboujaoude EN, Chen Q, Ames BN (1994) Assays of oxidative DNA damage biomarkers 8 oxo‐2′‐deoxyguanosine and 8 oxoguanine in nuclear DNA and biological fluids by high‐performance liquid chromatorgraphy with electrochemical detection. Meth. Enzymol. 234, 16. [DOI] [PubMed] [Google Scholar]
- Waldman T, Lengauer C, Kinzler KW, Vogelstein B (1996) Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 381, 713. [DOI] [PubMed] [Google Scholar]
- Wang TG, Gotoh Y, Jennings MH, Rhoads CA, Aw TY (2000) Cellular redox imbalance induced by lipid hydroperoxide promotes apoptosis in human colonic CaCo‐2 cells. FASEB J. 14, 1567. [DOI] [PubMed] [Google Scholar]
- Weinberg RA (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323. [DOI] [PubMed] [Google Scholar]
- Xlong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R., Beach D (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366, 701. [DOI] [PubMed] [Google Scholar]