

Abstract
In mouse fertilized eggs, correct assembly and distribution of the actin cytoskeleton are intimately related to cleavage in early‐stage embryos. However, in mouse fertilized eggs, mechanisms and involved factors responsible for regulating the actin cytoskeleton are poorly defined. In this study, mTORC2, PKB/Akt and Girdin were found to modulate division of mouse fertilized eggs by regulating distribution of the actin cytoskeleton. RNA interference (RNAi)‐mediated depletion of mTORC2, Akt1 or Girdin disrupted F‐actin rearrangement and strongly inhibited egg development. PKB/Akt has been proven to be a downstream target of the mTORC2 signalling pathway. Girdin, a newly found actin cross‐linker, has been proven to be a downstream target of the Akt signalling pathway. Furthermore, phosphorylation of both Akt1 and girdin was affected by knockdown of mTORC2. Akt1 positively regulated development of the mouse fertilized eggs by girdin‐mediated F‐actin rearrangement. Thus it seems that girdin could be a downstream target of the Akt1‐mediated signalling pathway. Collectively, this study aimed to prove participation of mTORC2/Akt in F‐actin assembly in early‐stage cleavage of mouse fertilized eggs via the function of girdin.
Objectives
In mouse fertilized eggs, the proper assembly and distribution of actin cytoskeleton is intimately related with the cleavage of early‐stage embryo. However, in mammals, especially in mouse fertilized eggs, the mechanisms and involved factors responsible for regulating the actin cytoskeleton are poorly defined. The aim of this study was to determine the role of mTORC2,PKB/Akt and Girdin in early development of fertilized mouse eggs, via regulating the distribution of actin cytoskeleton.
Materials and methods
Changes of F‐actin after treatting with mTORC2 shRNA, Akt siRNA or Girdin siRNA were observed by Immunofluorescence staining and laser‐scanning confocal microscopy. Levels of phosphorylated Girdin at Se1417 were detected by Western immunoblotting. Percentages of cells undergoing division were determined by counting, using a dissecting microscope.
Results
RNA interference (RNAi)‐mediated depletion of mTORC2, Akt1 or Girdin disrupts F‐actin rearrangement, and remarkably inhibited the development of mouse‐fertilized eggs. PKB/Akt has been proved to be a downstream target of the mTORC2 signaling pathway. Girdin, the newly found actin‐cross linker, has been proved to be a downstream target of the Akt signaling pathway. Furthermore phosphorylation of both Akt1 and Girdin were affected by knockdown of mTORC2. Akt1 positively regulates the development of mouse‐fertilized eggs by Girdin mediated F‐actin rearrangement. Girdin could be a downstream target of the Akt1‐mediated signaling pathway.
Conclusions
Collectively, this study aimed to prove the participation of mTORC2/Akt in F‐actin assembling in early‐stage cleavage of mouse fertilized eggs via the function of Girdin.
1. Introduction
Actin filaments (microfilaments) regulate many dynamic events during oocyte meiotic maturation and fertilization, such as sperm incorporation, cortical granule exocytosis, spindle rotation (in the mouse), second polar body emission and contractile ring formation during cleavage.1, 2 However, in mammals, and especially in mouse fertilized eggs, the mechanism regulating the F‐actin cytoskeleton was poorly defined.
mTORC2 is a multimeric kinase composed of the mammalian target of rapamycin kinase (mTOR), mSin1, mLST8 and rictor. It has shown functions in controlling cell growth and actin cytoskeletal assembly and is insensitive to rapamycin. In NIH3T3 fibroblasts, siRNA‐mediated knockdown of mTOR, Rictor or mLST8 prevents actin polymerization and cell spreading.3 In human neutrophils, inhibition of mTORC2 function by Rictor knockdown also leads to cell polarity defects and uniform cortical F‐actin accumulation.4 Together, these studies highlight the possibility that mTORC2 may have effects on the actin cytoskeleton. However, whether mTORC2 impacts microfilament aggregation of mouse fertilized eggs and alters this aggregation of microfilament assembly requires empirical determination.
Although the direct targets of mTORC2 that mediate signalling to the actin cytoskeleton are unknown, mTORC2 signalling to the actin cytoskeleton may involve PKCα and the small GTPases Rho and Rac. In HeLa cells, the morphology of the actin cytoskeleton in PKC‐α knockdown cells is similar to that of Rictor knockdown cells.5
Recent findings show mTORC2 directly phosphorylated Akt/PKB on Ser473.6 mTORC2 is responsible for insulin‐induced Akt Ser‐473 phosphorylation in 3T3‐L1 adipocytes.7
Akt, which was also known as protein kinase B (PKB), was also a serine/threonine kinase. Studies indicated that Akt1 promoted cell motility in mammalian fibroblasts and tumour cells predominantly by reorganizing actin filaments.8, 9, 10Our early studies reported that Akt1 was expressed in mouse fertilized eggs and revealed that Akt1 overexpression in mouse fertilized eggs promoted cell division.11 Further studies performed in our laboratory revealed that mTORC2 was critical for the phosphorylation of Akt1/PKB at Ser473 during embryogenesis, and Akt1 was positively regulated by mTORC2.12 Based on this, we hypothesized that mTORC2 might alter the aggregation of mouse fertilized egg actin cytoskeleton by phosphorylation of Ser473 PKB/Akt1 site.
A search for Akt1‐binding proteins led to the identification of Girdin (girders of actin filament), also known as Akt1 phosphorylation enhancer (APE) or G‐interacting vesicle‐associated protein (GIV). Girdin was a large 220 kDa protein with unique amino and carboxyl‐terminal domains flanking a long coiled‐coil region. Girdin formed oligomers through its amino‐terminal domain and coiled‐coil region. The carboxyl‐terminal domain contained the actin‐binding site and the phosphatidylinositol phosphate‐binding motif located near the Akt1 phosphorylation site (serine 1416). Therefore, Girdin was postulated to cross‐link actin filaments and to anchor them to the plasma membrane in quiescent cells.13, 14, 15 Jiang et al. demonstrated that the knockdown of Girdin in breast cancer cells led to an increase in the number of cells, which demonstrate rugged cortical actin filaments, and disruption of stress fibres. siRNA‐mediated knockdown of Girdin leads to the disruption of stress fibres in Vero fibroblasts.16 In addition, the cells lost their shape and formed rugged boundaries. Enomoto et al. reported that the Girdin CT1 domain is anchored at the plasma membrane via binding to phosphoinositides, where it subsequently cross‐links actin filaments and anchors cortical actin at the plasma membrane. Since the phosphoinositide‐binding site is located near the Akt phosphorylation site of the CT1 domain of Girdin, it is indicated that Akt1 might control the localization of Girdin by regulation of its phosphoinositide‐binding property. Ni et al. reported that Girdin regulates the migration and invasion of glioma cells via the PI3K‐Akt signalling pathway. The results provide a theoretical foundation for the development of anticancer drugs.17 Based on the above results, we speculate that Girdin may play a key role in regulating actin filaments and thus the modulation cell division in mouse fertilized eggs in an Akt1‐dependent manner.
In this report, we showed that RNA interference–mediated dampening of the expression of mTORC2, Akt1 or Girdin disrupts actin filaments and remarkably inhibits the development of mouse fertilized eggs. Furthermore, we found that Akt1 positively regulates the development of mouse fertilized eggs by Girdin‐mediated actin remodelling. Thus, we speculate that Girdin protein may be a downstream target of the Akt1 signalling pathway.
Furthermore, we also found that Akt1‐ and Girdin‐mediated phosphorylation was affected by knockdown of mTORC2 and that mTORC2 may regulate Girdin in the development of mouse fertilized eggs. Our observations collectively indicate that the mTORC2/Akt1/Girdin signalling pathway is crucial for actin remodelling in mouse fertilized eggs.
2. Materials and methods
2.1. Animals
Kunming genealogy‐specific pathogen‐free mice (females at 4–6 weeks and 18–20 g; males at 8 weeks and 25–30 g) were obtained from the Department of Laboratory Animals, the China Medical University. The animal study proposal was approved by the Institutional Animal Care and Use Committee (IACUC) of the Department of Laboratory Animals, China Medical University, with the permit number: IACUC (SCXK) 2008‐0005. All mouse experimental procedures were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People's Republic of China. All experiments were performed at China Medical University in accordance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals.
2.2. Reagents and plasmids
Reagents, unless otherwise specified, were all from Sigma. Anti‐β‐actin monoclonal antibody and anti‐RICTOR monoclonal antibody were obtained from Santa Cruz Biotechnology (San Diego, CA, USA). All other antibodies included anti‐AKT1 polyclonal antibody (Cell Signaling Technology, MA, USA), anti‐phosphorylated AKT1 polyclonal and monoclonal antibodies (Cell Signaling Technology MA, USA), horseradish peroxidase–conjugated anti‐rabbit/mouse IgG (Dako DEN) and Alexa Fluor 488/594‐conjugated goat anti‐rabbit IgG (Invitrogen CA). HRP‐conjugated anti‐mouse and anti‐rabbit IgGs were from Beijing Zhongshan Biotechnology (Bei jing China). The ECL detection system was obtained from Promega (USA); M2 medium, M16 medium and Tyrode's buffer were all obtained from Sigma (IL USA). The plasmid pcDNA3.1/myc‐His B was obtained from Invitrogen Corporation (CA, USA). The mMESSAGE/mMACHINE kit was obtained from Ambion (Austin, TX, USA). The RNA PCR Kit (AMV) Ver 2.1 was obtained from Takara Technology Co. Ltd (Japan). The constructs of Rictor shRNA were provided by Dr. Estella Jacinto (University of Basel, Switzerland). siRNA for mouse Girdin was purchased from Santa Cruz (Girdin siRNA sc‐145407 CA, USA). siRNA for mouse Akt1 was purchased from Santa Cruz (Akt1 siRNA (m): sc‐29196 CA, USA). The constructs encoding the wild‐type AKT (pCIS2‐Akt1‐WT), myristoylated AKT (pCIS2‐myr‐Akt1) and kinase‐deficient AKT (pCIS2‐Akt1‐KD) were gifts from Michael J. Quon (National Institutes of Health).
The coding sequences of wild‐type AKT (Akt1‐WT), kinase‐deficient AKT (Akt1‐KD) and myristoylated AKT (myr‐Akt1) were amplified by PCR using the pCIS2‐Akt1‐WT, pCIS2‐Akt1‐KD and pCIS2‐myr‐Akt1, respectively, as templates and introduced the enzyme‐incision site of HindIII and BamHI into 5 and 3 end, respectively. The products were cloned into the mRNA expression vector pcDNA3.1/myc‐His B (Invitrogen) and named pcDNA3.1‐Akt1‐WT, pcDNA3.1‐Akt1‐KD and pcDNA3.1‐myr‐Akt1 and were constructed by Dr. Chen Feng in our group.11
2.3. Collection and culture of one‐cell stage mouse embryos
We collected and cultured one‐cell stage mouse embryos according to the previously described protocol by Hogan and Constantini (1986). Female mice were first injected with 10 IU of pregnant mare serum gonadotropin (PMSG) and then injected with 10 IU of human chorionic gonadotropin (hCG) 48 hours late. A female mouse was placed with a single male mouse of the same strain. Twenty‐four hours after the hCG injection, we collected one‐cell stage embryos from the oviducts of females possessing a vaginal plug and then placed them immediately in M2 medium. The embryos were cultured in M16 medium under paraffin oil at 37°C in a humidified atmosphere of 5.0% CO2 in air until embryos at different stages were needed.
The development model of mouse fertilized eggs were listed as follows: G1 phase: 12–21 hours after injecting hCG; S phase: 21–27 hours after injecting hCG; G2 phase: 27–30 hours after injecting hCG; M phase: 30–33 hours after injecting hCG (Fig. 1a).
Figure 1.
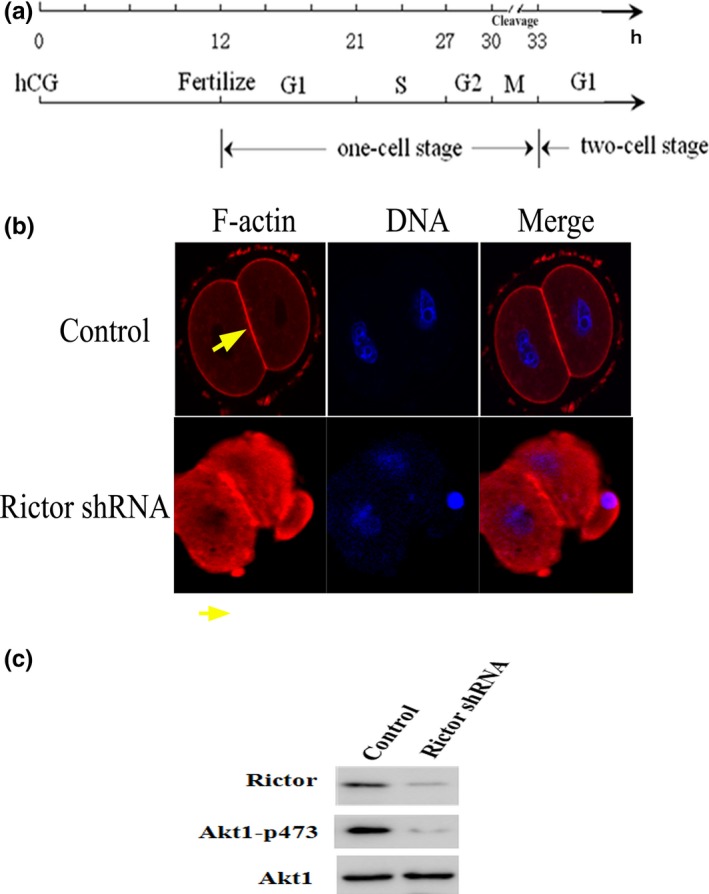
mTORC2 affects rearrangement of the F‐actin cytoskeleton in mouse fertilized eggs. (a) The development model of mouse fertilized eggs. G1 phase: 12–21 hours after injecting hCG; S phase: 21–27 hours after injecting hCG; G2 phase: 27–30 hours after injecting hCG; M phase: 30–33 hours after injecting hCG. (b) Staining for F‐actin (red) and DNA (blue) revealed the organization of the F‐actin cytoskeleton in mouse fertilized eggs transfected with RICTOR shRNA (F‐actin: yellow arrow). Scale bar: 20 μm. (c) Western immunoblotting detection of mTORC2 in 200 mouse fertilized eggs treated with shRNA targeted against mTORC2. WB: Western blotting
According to the development model of mouse fertilized eggs, we mostly injected siRNA or shRNA into one‐cell stage embryos at G1 stage of the cell cycle about 18–20 hours after hCG. Embryos were collected the next day (38–42 hours after the hCG injection), so the siRNA or shRNA may function more than 20 hours after injection.
2.4. In vitro transcription
All the constructs in pcDNA3.1/myc‐His B were cut singly with Age I and transcribed into 5′‐capped mRNA for microinjection using the mMESSAGE/mMACHINE kit. The mRNAs synthesized in vitro were dissolved in nuclease‐free 5 mmol/L Tris and 0.5 mmol/L EDTA (TE) at pH 7.4. The mRNA yield was determined by measuring the absorbance at 260 nm and by running modified non‐denaturing gels loaded with RNA.
2.5. mRNA/shRNA microinjection and observation of the mouse embryos
Various mRNAs/shRNAs were injected into one‐cell stage embryos at the G1 phase of the cell cycle, and the aid of Eppendorf transferman manipulators mounted on an Olympus IX‐70 inverted microscope equipped with DIC optics. Embryos were placed in a drop of M2 medium under paraffin oil on the lid of a 3 cm Falcon culture dish. The injection volume was 5% of the total cell volume or 5–10 pL per egg. The mRNA/shRNA species were diluted to predetermined concentrations in nuclease‐free TE buffer (pH 7.4). Embryos in the control groups were microinjected with/without TE buffer. The percentage of eggs undergoing division was counted 30–35 hours after injection of hCG under a dissecting microscope, and the data were analysed statistically.
2.6. RNA interference
The 21‐nucleotide synthetic duplexes, which caused depletion (knockdown) of Girdin or Akt1, were obtained from Santa Cruz. One‐cell fertilized eggs were microinjected with Girdin siRNA, Akt1 siRNA and a 21‐nucleotide control RNA (Santa Cruz)
2.7. Western immunoblotting
Protein extracts were obtained from the mouse fertilized eggs and prepared by adding approximately 200 eggs in a minimal volume of collection medium to 20 ul of protein extraction buffer (100 mmol/L NaCl, 20 mmol/L Tris‐HCL, pH 7.5, 0.5% Triton X‐100 and 0.5% Nonidet P‐40) containing 1 mmol/L phenylmethylsulfonyl fluoride. Then, Laemmli sample buffer was added to the protein extracts, and the mixture was boiled for 5 min then resolved on a 10% or 6% SDS‐PAGE gel. Afterwards, the fractionated proteins were transferred to a nitrocellulose membrane. The membrane was then blocked with 3% BSA in Tris‐buffered saline containing 0.05% Tween 20 and probed with primary antibodies against Akt1(1:500), Girdin (1:200), Girdin 1417 (1:200) and RICTOR (1:400; Santa Cruz Biotechnology) overnight at 4°C. The membrane was incubated with a horseradish peroxidase‐conjugated anti‐mouse, anti‐rabbit or anti‐goat secondary antibody at 1:3000 dilution (Beijing Zhongshan Biotechnology). The proteins were detected using an enhanced chemiluminescence detection system (Pierce Biotechnology). Phospho‐Girdin 1417 antibody was raised in New Zealand White rabbits against keyhole limpet haemocyanin‐conjugated phosphopeptide.
2.8. Immunofluorescence staining and laser‐scanning confocal microscopy
Eggs were fixed for 10 minutes at room temperature in Dulbecco's phosphate‐buffered saline (DPBS) supplemented with 3% paraformaldehyde and 0.01% glutaraldehyde and then washed in a solution of 3% foetal calf serum (FCS; Biological Industries, Beit‐Haemek, Israel) in DPBS (DPBS/FCS). ZPs were removed post‐fixation by treatment with 0.25% pronase (Sigma) in DPBS/FCS. The ZP‐free eggs were washed in DPBS/FCS. For labelling egg proteins, the plasma membranes of ZP‐free eggs were permeabilized by 10 minutes of incubation in 0.05% NP‐40 in DPBS/FCS solution followed by several rinses in 0.005% NP‐40 in DPBS/FCS. For actin labelling of eggs, specimens were incubated for 2 hours in the presence of rhodamine‐phalloidin (20 μmol/L) or with anti‐Girdin (1:100) followed by an incubation in the presence of a fluorescence‐labelled secondary antibody (1:300). Chromatin labelling and the fertilization stage of the eggs were assessed by incubating the eggs for 10 minutes in the presence of 1 mg/ml Hoechst (Sigma). Actin, Girdin and DNA were visualized and photographed by a Zeiss confocal laser‐scanning microscope (CLSM; Oberkochen, Germany). The Zeiss LSM 410 was equipped with a 25 mW krypton–argon laser, a 10 mW helium/neon laser (488, 543 and 633 maximum lines) and an UV laser (Coherent Inc. Laser Group, Santa Clara, CA, USA). Eggs were scanned using the CLSM through the Z‐axis so that images were taken of sections at the equatorial plane of the egg. The stain intensity was measured by collecting mean density values obtained by the LSM software.
2.9. Statistical analysis
All experiments were performed independently and at least three times. Student's t test and analysis of variance (ANOVA) were used to evaluate the differences between multiple experimental groups using SSPS 13.0 software, and the differences were considered statistically significant at P<0.001.
3. Results
3.1. mTORC2 affects rearrangement of the F‐actin cytoskeleton in mouse fertilized eggs
To explore the function of mTORC2 in mouse fertilized eggs, we used shRNA to silence the gene expression of RICTOR. Mouse one‐cell stage embryos at the G1 phase were cultured in M16 medium after microinjection of Rictor shRNA. After 20 h, the embryos were collected to quantity gene and protein expression of RICTOR. To clarify the roles of mTORC2 in F‐actin rearrangement, we measured the intracellular distribution and rearrangement of F‐actin by immunofluorescence confocal microscopy. In control mouse fertilized eggs, F‐actin localized to the cell cortex explicitly around contractile ring. By contrast, RICTOR knockdown fertilized eggs showed cytoplasmic actin aggregates and less prominent presence of contractile ring (Fig. 1b). Using siRNA to down‐regulate gene expression, we almost completely inhibited RICTOR protein expression in mouse fertilized eggs (Fig. 1c).
3.2. Akt1 regulates cell division and F‐actin rearrangement in mouse fertilized eggs
We investigated whether Akt1 affects cell division and F‐actin rearrangement. This was done by culturing one‐cell stage mouse fertilized eggs in M16 medium after microinjection of the eggs with 0.03 ng mRNA of Akt1‐WT, myr‐Akt1 (constitutively active form of Akt1), KD‐Akt1 or 20 μmol/L Akt1 siRNA. In the myr‐Akt1 construct, the Akt1‐coding region is fused to a myristoylation sequence from pp60 csk. The dominant inhibitory mutant of Akt1 that we used in this study is a kinase‐deficient Akt1 that results from the substitution of alamine for lysine at position 179 in the canonical ATP‐binding domain. This mutant is not only catalytically inactive but has been shown to inhibit the activity and actions of endogenous Akt1. The control for these procedures was microinjection of fertilized eggs with scrambled sequence siRNA or TE buffer. The effect of Akt1 siRNA on Akt1 protein expression was evaluated by Western immunoblot analysis (Fig. 2a). The cleavage of fertilized mouse eggs in each group was calculated after manual counting under a dissecting microscope 35 hours after hCG injection. In the control groups, 61.89% of embryos reached the two‐cell stage 35 hours after the injection of hCG. Only 29.73% and 26.81% of embryos microinjected with KD‐Akt1 mRNA or 20 μmol/L Akt1 siRNA reached the two‐cell stage 35 hours after the hCG injection, respectively. In contrast, the cleavage rate was up to 92.30% 35 hours after the hCG injection with mRNA of myr‐Akt1 (Fig. 2b). These results demonstrated that Akt1 knockdown or KD‐Akt1 clearly interfered with the division of one‐cell stage fertilized eggs. Further, microinjection of myr‐Akt1 prompted the division of mouse fertilized eggs. Next, we measured the intracellular distribution and organization of F‐actin in each experimental group by immunofluorescence confocal microscopy. In the Akt1‐WT and myr‐Akt1 group, there was a distinct increase in the stain of F‐actin around contractile ring after injection; Akt1 mRNAs prompted microfilament aggregation, thus facilitating advanced cell division. When the fertilized eggs overexpressed kinase‐deficient Akt1 or cells were microinjected with Akt1 siRNA, the stain weakened around contractile ring and cells appeared to display an abnormal pattern of cell division. Akt1 expression reduced aggregation and affected the state of the microfilaments and thus affected normal cell division (Fig. 2c). These results suggest that Akt1 can indeed affect the early development of fertilized eggs by affecting the rearrangement of the F‐actin cytoskeleton.
Figure 2.
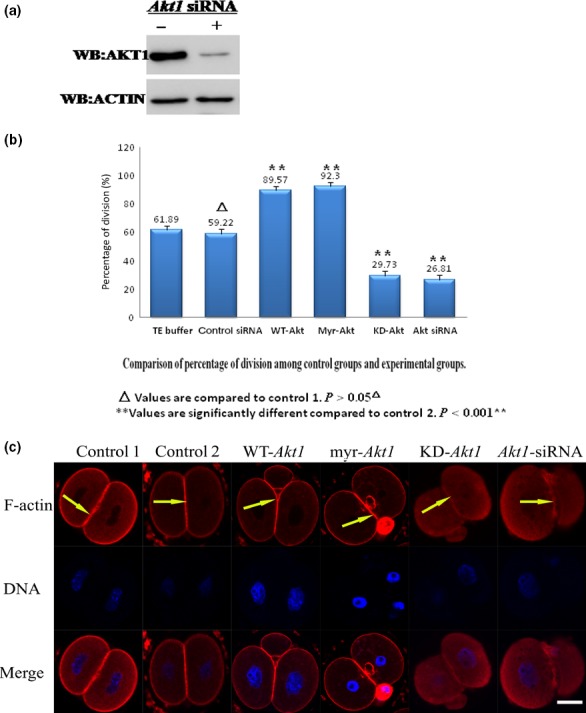
Akt1 regulates cell division and F‐actin rearrangement in mouse fertilized eggs. (a) Western immunoblotting detection of Akt1 in 200 mouse fertilized eggs treated with siRNA targeted against Akt1. WB: Western blotting. (b) The division rate in cultured mouse embryos after 0.03 ng of mRNA‐encoding Akt1‐WT, myr‐Akt1, Akt1‐KD or Akt1 siRNA injection. The percentage of cells undergoing cell division and cell survival was calculated after manual counting under a dissecting microscope 35 hours after injection of human chorionic gonadotropin in female mice. The numbers of eggs undergoing cell division (hatched bars) or survival are given above each bar graph. (c) Cortical remodelling of F‐actin is induced by Akt1 activation. Mouse fertilized eggs were stained with rhodamine‐phalloidin (red fluorescence) to visualize F‐actin cytoskeleton. Various scenarios were studied, which included cells injected with mRNA‐encoding Akt1‐WT, myr‐Akt1, Akt1‐KD or siRNA targeted against Akt1. Fertilized eggs were fixed and labelled with rhodamine‐phalloidin (10 or 20 μmol/L; F‐actin labelling, indicated by white arrows) and with Hoechst 33258 (1 mg/ml; DNA labelling) and imaged by laser‐scanning confocal microscope. Scale bar: 20 μm
3.3. The mTORC2/Akt1 pathway rearranges the F‐actin cytoskeleton of one‐cell stage fertilized eggs
To explore the function of mTORC2 and Akt1 in mouse fertilized eggs, we also monitored Akt1 phosphorylation. We found that Akt1 Ser 473 phosphorylation was abolished by Rictor's knockdown. RNA interference–mediated suppression of rictor inhibited Akt1 Ser473 (Fig. 1c). To further explore the relation of mTORC2 and Akt1, we first microinjected one‐cell stage fertilized eggs with Rictor shRNA and then with the mRNA‐encoding wild‐type Akt1. Next, we observed the rearrangement of the actin cytoskeleton by immunofluorescence confocal microscopy. We found the phenotype seems to be rescued (Fig. 3). Our findings indicated that RICTOR regulates the organization of the actin cytoskeleton and that Akt1 is a mediator of this function.
Figure 3.
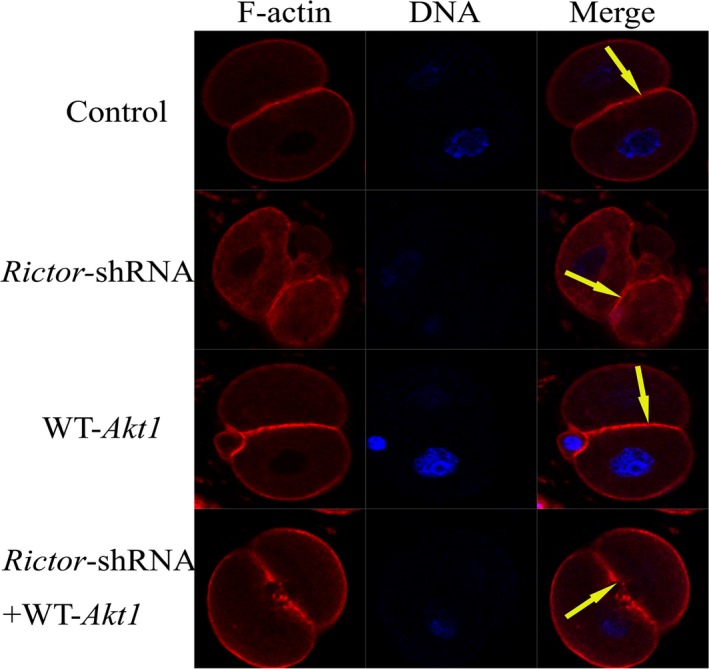
The mTORC2/Akt1 pathway rearranges the F‐actin cytoskeleton of one‐cell stage fertilized eggs. Microinjection of Rictor shRNA then with myr‐Akt1 mRNA into mouse one‐cell stage embryos. Staining for F‐actin (red) revealed the organization of the F‐actin cytoskeleton in mouse fertilized eggs (F‐actin is shown by the yellow arrow). Scale bar: 20 μm
3.4. Girdin is essential for rearrangement of the F‐actin cytoskeleton and the development of mouse fertilized eggs
A search for binding proteins of Akt1 led to the identification of Girdin, an actin‐binding protein. Thus, we examined the functions of Girdin on the rearrangement of the F‐actin cytoskeleton and the development of mouse fertilized eggs. We microinjected several Girdin small (21 nucleotide) interfering RNAs (Girdin siRNA) and 2‐nucleotide, irrelevant or scrambled sequence RNAs (control siRNA) into mouse fertilized eggs at the G1 phase of cell cycle. According to the development model of mouse fertilized eggs, we microinjected Girdin siRNA into one‐cell stage fertilized eggs at G1 stage of the cell cycle about 18–20 hours after hCG. Fertilized eggs were collected the next day (38–42 hours) after the hCG injection. Western immunoblot analyses showed that microinjected with the Girdin siRNAs (20 μmol/L) effectively reduced the expression levels of Girdin (Fig. 4A). The percentage of cell division and death in each group was calculated after manual counting under a dissecting microscope 35 hours after of hCG. In the control groups, about 83% of the embryos reached the two‐cell stage 35 hours after the injection of hCG, and there was no significant difference between the two control groups (P>0.05). Microinjection of embryos with 10 μmol/L Girdin siRNA resulted in 28.59% of the embryos reaching the two‐cell stage at 35 hours following hCG injection. However, only 23.91% reached the two‐cell stage when microinjected with 20 μmol/L Girdin siRNA. Depletion of Girdin inhibited cleavage of mouse fertilized eggs (Fig. 4b)
Figure 4.
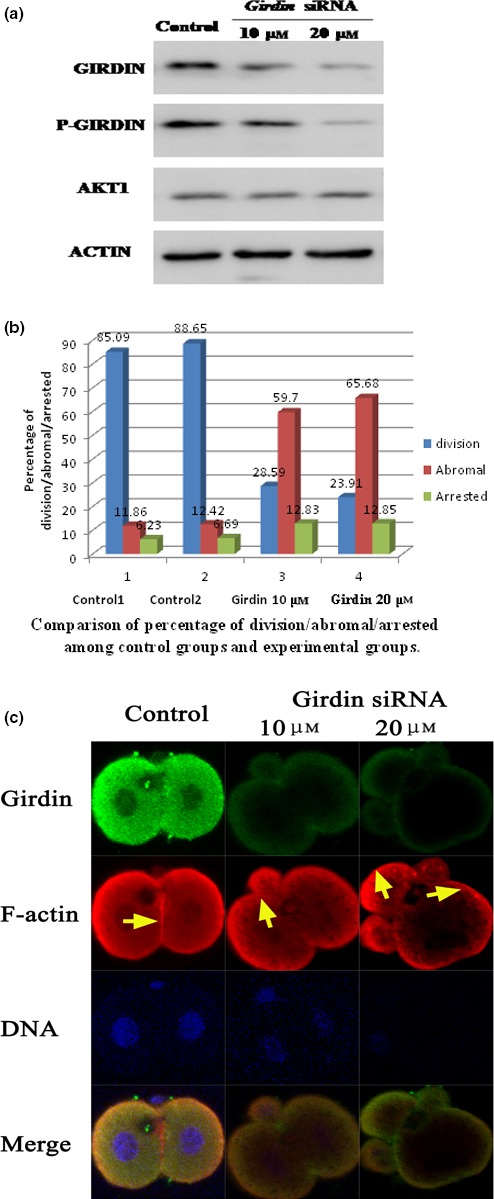
Girdin is essential for rearrangement of the F‐actin cytoskeleton and the development of mouse fertilized eggs. (a) Depletion of Girdin in mouse one‐cell staged fertilized eggs by siRNA. Total cell extracts from control siRNA‐ and Girdin siRNA‐injected one‐cell staged fertilized eggs were subjected to Western blot analyses and immunodetection with anti‐Girdin, anti‐p‐Girdin, anti‐Akt1 and anti‐actin antibodies. (b) The cleavage rate in cultured mouse embryos after Girdin siRNA injections shows that the total number of eggs undergoing cell division is given above each bar graph from three independent experiments. (c) F‐actin cytoskeleton of embryos derived from fertilized eggs treated with Girdin siRNA. Mouse fertilized eggs were treated with control or Girdin siRNAs and fixed 48 hours later, followed by staining with rhodamine‐phalloidin and anti‐Girdin antibody. In situ validation of the interaction between Girdin and polymerized F‐actin by confocal microscopy is shown. One‐cell stage mouse fertilized eggs were treated with 10 or 20 μmol/L Girdin siRNA. Immunolocalization of Girdin is revealed by green staining (antibody), and immunolocalization of actin is revealed by red staining. Control fertilized eggs (21 hours after hCG): one‐cell arrested embryos derived from fertilized eggs that were injected with Girdin siRNA 21 hours after hCG and cultured for 24 hours; control two‐cell embryo: in control fertilized eggs, F‐actin forms a regular ring in the cell cortex. In embryos derived from fertilized eggs treated with girdin siRNA, polymerized F‐actin was observed in the cytoplasm and formed irregular patches that were scattered randomly in the cortex. Scale bar: 20 μm
To test whether Girdin promotes cross‐linking of F‐actin filaments, we examined the effects of Girdin knockdown on the rearrangement of the F‐actin cytoskeleton. Immunofluorescent staining with anti‐Girdin Ab showed that Girdin expression was very low in the Girdin siRNA‐microinjected fertilized eggs. Staining of F‐actin‐rich structures with phalloidin revealed that the correct orientation of the filament is affected and cannot be gathered at the contractile ring in the Girdin siRNA‐microinjected mouse fertilized eggs (Fig. 4c). The depletion of Girdin disrupted the rearrangement of the actin cytoskeleton in mouse eggs, resulting in abnormal cleavage and asymmetrical cytokinesis. These observations suggested that Girdin is essential for rearrangement of F‐actin filaments.
3.5. The Akt1/Girdin pathway regulates rearrangement of the F‐actin cytoskeleton of one‐cell stage fertilized eggs
We used Scansite software (http://scansite.mit.edu) and the Clustal W (1.83) multiple sequence alignment programme (http://www.ebi.ac.uk/clustalw/) to predict relevant target sites of Akt1 on mouse Girdin. Using this approach, we found that mouse Girdin had a serine residue at position 1417 (Ser 1417), which corresponded to Ser 1416 of human Girdin, which was also a residue that was phosphorylated by human Akt1. To gain insight into the role of phosphorylation of Girdin by Akt1, we assessed the location of phosphorylated Girdin in fertilized eggs by staining with anti‐p‐Girdin 1417Ab. One‐cell stage mouse embryos were first microinjected with Akt1‐WT mRNA, myr‐Akt1 mRNA or Akt1 siRNA, and then stained with rhodamine‐phalloidin and anti‐P‐Girdin 1417Ab. We found that the actin filaments and P‐Girdin were clustered in the cell membrane and the cleavage furrow (Fig. 5a). We next examined whether anti‐P‐Girdin Ab could detect endogenously phosphorylated Girdin in response to activation by Akt1. We found that treatment with myr‐Akt1 caused significant increases in phosphorylation of Girdin 1417, whereas only minor phosphorylation was seen with WT‐Akt1. In addition, treatment with KD‐Akt1 mRNA or siRNA‐mediated Akt1 knockdown blocked phosphorylation of Girdin (Fig. 5b). These results strongly suggested that Akt1 could directly phosphorylate Girdin on Ser1417 and promoted its function in mouse fertilized eggs. In additional studies, we first microinjected one‐cell stage eggs with myr‐Akt1 mRNA, as mentioned above, followed by microinjected with Girdin siRNA. Embryos in each experiment were assessed by immunofluorescence confocal microscopy for their F‐actin organization 20 h after microinjection. As anticipated, we detected the disrupted microfilament aggregation after microinjected of Akt1 mRNA and Girdin siRNA. Similar effects were seen in Girdin siRNA‐microinjected groups (Fig. 5c). These findings indicate that Girdin‐ and Akt1‐mediated phosphorylation play major roles in the development of mouse fertilized eggs. Taken together, our observations indicate that the Akt1/Girdin pathway regulates division of one‐cell stage fertilized eggs.
Figure 5.
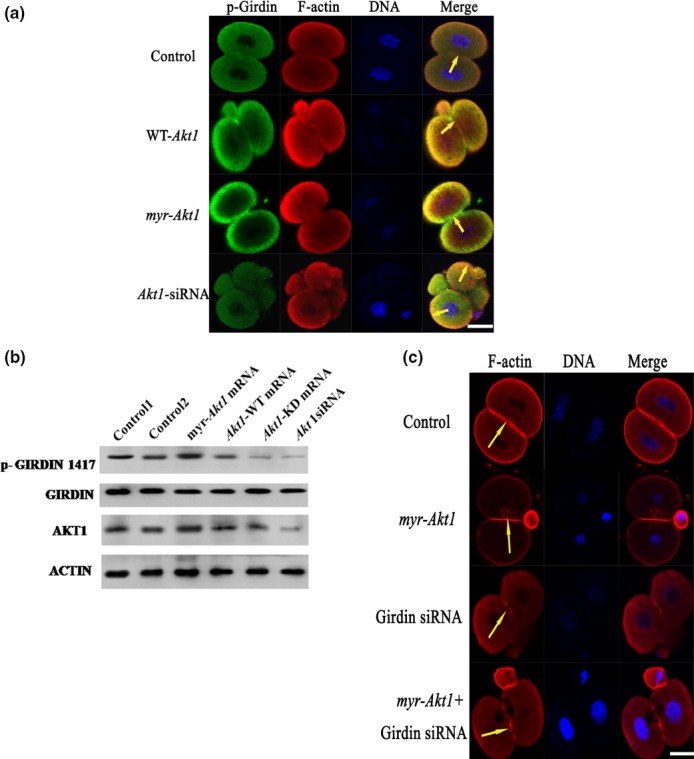
The Akt1/ Girdin pathway regulates rearrangement of the F‐actin cytoskeleton of one‐cell stage fertilized eggs. (a) One‐cell stage mouse embryos were first microinjected with Akt1‐WT mRNA, myr‐Akt1 mRNA or siRNA, then stained with rhodamine‐phalloidin (20 μmol/L; actin labelling as shown in red) and anti‐P‐Girdin (1:50; Girdin labelling; as shown in green). Merged colour detection between P‐Girdin and polymerize F‐actin appears as yellow stained images. The areas of co‐localization are highlighted with yellow arrowheads. Scale bar: 20 μm. (b) 200 fertilized eggs were treated with mRNA coding for Akt1‐WT, myr‐Akt1, Akt1‐KD or siRNA against Akt1. Western immunoblot analyses assessed following immunoreaction with anti‐P‐Girdin (upper panel) and anti‐Girdin (lower panel) antibodies are shown. (c) One‐cell stage mouse embryos were first microinjected with 0.03 ng of myr‐Akt1 mRNA as described in the Materials and Methods, and then 1–2 hours later with Girdin siRNA, following which specimens were stained with rhodamine‐phalloidin (20 μmol/L; actin labelling shown in red). Scale bar: 20 μm
3.6. The mTORC2/Akt1/Girdin pathway rearranges the F‐actin cytoskeleton in one‐cell stage fertilized eggs
To further explore the role for mTOR complexes in the regulation of the Girdin, we examined Girdin phosphorylation in the Rictor knockdown fertilized eggs. In control fertilized eggs, we noted that Girdin was highly phosphorylated at 1417. Mouse fertilized eggs treated with Rictor shRNA exhibited decreased phosphorylation of Girdin 1417. Thus, knockdown of mTORC2 prevented Girdin phosphorylation (Fig. 6a). Furthermore, we examined Girdin phosphorylation in the Rictor shRNA and WT‐Akt1 mRNA injection fertilized eggs. In WT‐Akt1 mRNA‐treated fertilized eggs, we noted that Girdin was highly phosphorylated at 1417. But fertilized eggs treated with Rictor siRNA and WT‐Akt1 exhibited decreased phosphorylation of Girdin 1417 (Fig. 6b). Our data indicate that Akt1‐ and Girdin‐mediated phosphorylation was affected by knockdown of mTORC2 and that mTORC2 may regulate Akt1 and Girdin in the development of mouse fertilized eggs.
Figure 6.
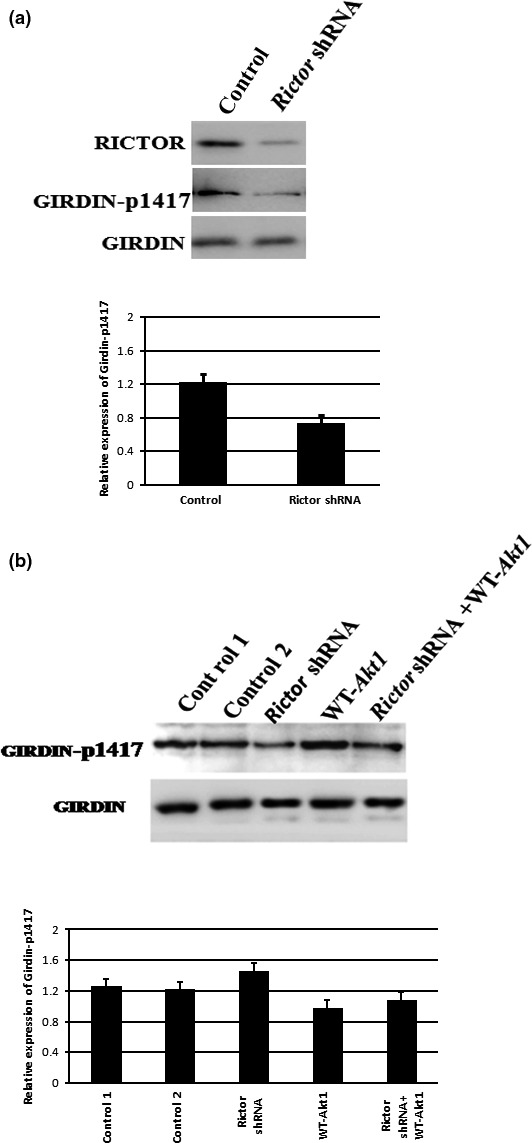
The mTORC2/Akt1/Girdin pathway rearranges the F‐actin cytoskeleton in one‐cell stage fertilized eggs. (a) The effect of gene knockdown on the expression of Rictor and the phosphorylation of Girdin‐1417. Extracts of mouse fertilized eggs were resolved by 6% SDS‐PAGE, transferred to nitrocellulose and probed with phosphor‐Girdin‐Ser1417 antibody and Girdin antibody. (b) We examined Girdin phosphorylation in the fertilized eggs treated with Rictor shRNA, WT‐Akt1 mRNA, Rictor shRNA and WT‐Akt1 coinjection. In WT‐Akt1 mRNA treated cells, we noted that Girdin was highly phosphorylated at 1417. But cells treated with Rictor siRNA and WT‐Akt1 exhibited decreased phosphorylation of Girdin 1417
4. Discussion
Although some reports reveal that polymerization of microfilaments is important for mouse fertilization,18, 19 the signalling pathway is unclear, particularly with regard to the regulation of microfilament rearrangement in mouse fertilized eggs. In this study, we investigated the effects of mTORC2, Akt1 and Girdin on the remodelling of the actin cytoskeleton in mouse fertilized eggs.
Many studies have shown that mTORC2 is associated with the organization of the actin cytoskeleton. Some groups found that mTORC2 may be related to small GTPases (RhoA, Cdc42 and Rac1), which are critical regulatory proteins in cytoskeletal organization and cell migration.20 mTORC2 plays a major role in the phosphorylation and activation of the AGC protein kinase family member Akt/PKB and does so at the stimulatory Ser473 residue. Earlier reports by us had demonstrated that microinjection Rictor shRNA could block cell division of mouse fertilized eggs. It suggested that RICTOR might play an important role in promoting early mitotic division in early mouse fertilized eggs.12 Our further study showed that the actin cytoskeleton could not be proper aggregation around cleavage furrow after rictor shRNA treated in mouse fertilized eggs. They appeared asymmetric and irregular in division. It suggested that mTORC2 was involved in the regulation of actin cytoskeleton distribution in mouse fertilized eggs. The ability of mTORC2 to regulate actin networks suggests that it may be involved in regulating the cleavage of early‐stage embryo.
Although the direct effect of mTORC2 on the aggregation of the actin cytoskeleton is not clear, mTORC2 is critical for the phosphorylation of Akt1/PKB at Ser473 during embryogenesis and that Akt1 is positively regulated by mTORC2.12 We previously reported an important role of Akt1 in the regulation of the first round of mitosis of mouse fertilized eggs.11 In this report, we have found that Akt1 regulates remodelling of the actin cytoskeleton in mouse one‐cell fertilized eggs. We found that the distribution of the actin cytoskeleton has changed after treated with WT‐Akt1 mRNA, myt‐Akt1 mRNA or Akt1 siRNA. Microfilaments concentrated distribution around cleavage furrow after treated with WT‐Akt1 mRNA or myt‐Akt1 mRNA. On the contrary, the distribution of actin filaments was disordered after treated with Akt1 siRNA .The fertilized eggs could not form normal division groove. The results showed that PKB/Akt can influence the correct aggregation and distribution of microfilament cytoskeleton of mouse fertilized eggs. To explore the function of mTORC2 and Akt1 in mouse fertilized eggs, we also monitored Akt1 phosphorylation. We found that Akt1 Ser 473 phosphorylation was abolished by Rictor's knockdown. We also found that the aggregation of actin filaments was destroyed after co‐injection of Akt1 mRNA and Rictor shRNA. Similar observations were made in Rictor shRNA‐injected eggs. Our findings indicated that RICTOR regulates the organization of the actin cytoskeleton and that Akt1 is a mediator of this function.
However, it is still unknown that how Akt1 affects the regulation of microfilament aggregation in mouse fertilized eggs. Further investigation is also needed to understand which factors are involved in this process. A search for binding proteins of Akt1 led to the identification of Girdin, an actin‐binding protein also known as APE (Akt phosphorylation enhancer), GIV (Gα‐interacting vesicle‐associated protein) or HkRP (Hook‐related protein 1). The human and mouse Girdin genes (referred to as CCDC88A and ccdc88a, respectively) encode large proteins of 1870 (1871 in an isoform of human Girdin) and 1845 amino acids, 220–250 kilodaltons (kDa). These proteins bind directly to actin filaments through their carboxyl‐terminal domain. In addition, the Akt1‐binding site maps to the carboxyl‐terminal domain of Girdin, and Akt1 phosphorylates Girdin at Ser 1416 to regulate its subcellular localization and cell migration in fibroblasts.13 Further data suggest a more complex relationship between Girdin and Akt1. For example, exogenously induced expression of both Akt1 and Girdin induced apoptosis and inhibited cell proliferation, survival and cytokinesis in COS7 and HeLa cells.16 The potential targeting of Akt‐Girdin signalling within the tumour microenvironment to develop novel therapies against human cancer.21 Because previous studies have shown that Akt1 promotes cellular proliferation and inhibits apoptosis in many cell types, these complex findings obscure the mechanism by which Girdin regulates the function of Akt1 in various cellular processes. It will be important to address how Girdin‐mediated Akt1 signal affects cell proliferation.22, 23 Thus, we investigated a possible interaction between Girdin and F‐actin. F‐actin was disrupted in the Girdin siRNA‐microinjected mouse fertilized eggs. These observations suggested that Girdin is essential for organization of F‐actin filaments.
Furthermore, we found that the addition of Akt1‐WT and myr‐Akt1 induced significant phosphorylation of Girdin in mouse one‐cell fertilized eggs. In addition, treatment with KD‐Akt1 mRNA or siRNA‐mediated Akt1 knockdown blocked phosphorylation of Girdin. These results strongly suggested that Akt1 could directly phosphorylate Girdin on Ser1417 and promoted its function in mouse fertilized eggs.
To further explore the role for mTOR complexes, Akt1 and Girdin in the regulation of the actin cytoskeleton, w examined Girdin phosphorylation in the Rictor knockdown fertilized eggs. We also found that the level of Girdin 1417 phosphorylation was dramatically decreased in Rictor knockdown fertilized eggs compared with that of control fertilized eggs.
Furthermore, we examined the phosphorylation of Girdin in the Rictor shRNA and WT‐Akt1 mRNA co‐injection fertilized eggs. In WT‐Akt1 mRNA‐treated fertilized egg, we noted that Girdin was highly phosphorylated at 1417. But fertilized eggs treated with Rictor siRNA and WT‐Akt1 exhibited decreased phosphorylation of Girdin 1417. Our data indicated that Akt1 and Girdin‐mediated phosphorylation was affected by knockdown of mTORC2 and that mTORC2 may regulate Girdin in the development of mouse fertilized eggs.
In summary, we showed that siRNA interference and depletion of mTORC2, Akt1 or Girdin disrupted the rearrangement of actin filament and remarkably inhibited the development of mouse fertilized eggs. Also, we found that Akt1 positively regulated the development of mouse fertilized eggs by Girdin‐mediated actin remodelling. Girdin protein could be a downstream target of the Akt1 signalling pathway. We also found that phosphorylation of Akt1 and Girdin were affected by knockdown of mTORC2 and that mTORC2 may regulate the phosphorylation of Girdin during the development of mouse fertilized eggs. Combined with our prior studies that the Rictor‐induced phosphorylation of Akt1 in Ser473 is required for mTORC2 function during the early development of mouse fertilized eggs, our current data suggest that Akt1 is a mTORC2's substrate and mTORC2 regulates the development of mouse fertilized eggs through Akt1 and Girdin. Our study indicates that the mTORC2/Akt1/Girdin signalling pathway is crucial for remodelling actin in mouse fertilized eggs (Fig. 7).
Figure 7.
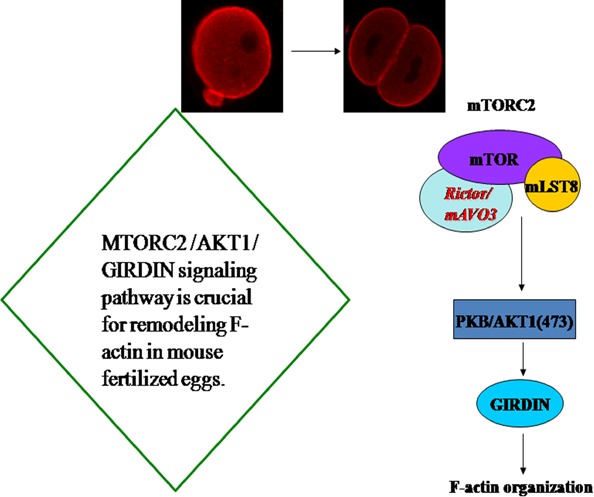
Schematic illustration of the working model for the regulation of F‐actin in mouse fertilized eggs by mTORC2/Akt1/Girdin. Akt1 and Girdin were affected by knockdown of mTORC2. Akt1 positively regulated the development of mouse fertilized eggs by Girdin‐mediated F‐actin remodelling. Girdin protein could be a downstream target of the Akt1 signalling pathway. mTORC2 may regulate Girdin in the development of mouse fertilized eggs. Our current study suggests a critical role of Akt1 as a substrate for mTORC2 and supports an important role for mTORC2 in regulating the development of mouse fertilized eggs through Akt1 and Girdin. Our study indicates that the mTORC2/Akt1/Girdin signalling pathway is crucial for remodelling F‐actin in mouse fertilized eggs
Funding
This work was supported by the National Nature Science Foundation of China (grant no 81270654, 31501162) and Shenyang Bureau of Science and Technology Research Foundation (F11‐264‐1‐69).
Conflict of interest
All authors have contributed significantly and are in agreement with the content of the manuscript. There is no conflict of interest.
Acknowledgement
We thank Dr. Estela Jacinto (University of Basel, Switzerland) for presenting the constructs of Raptor shRNA and Rictor shRNA as a gift.
The copyright line for this article was changed on 26 September 2016 after original online publication.
Contributor Information
Xiuxia Wang, Email: wangxx@sj-hospital.org.
Bingzhi Yu, Email: ybzbiochem@yeah.net.
References
- 1. Pickering SJ, Johnson MH, Braude PR, Houliston E. Cytoskeletal organization in fresh, aged and spontaneously activated human oocytes. Hum Reprod. 1998;3:8–89. [DOI] [PubMed] [Google Scholar]
- 2. Zhu ZY, Chen DY, Li JS, et al. Rotation of meiotic spindle is controlled by microfilaments in mouse oocytes. Biol Reprod. 2003;68:943–946. [DOI] [PubMed] [Google Scholar]
- 3. Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. [DOI] [PubMed] [Google Scholar]
- 4. Liu L, Das S, Losert W, Parent CA. mTORC2 regulates neutrophil chemotaxis in a cAMP‐ and RhoA‐dependent fashion. Dev Cell. 2010;19:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin‐insensitive and raptor‐independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. [DOI] [PubMed] [Google Scholar]
- 6. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science. 2005;307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 7. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for AKT/protein kinase B in3T3‐L1 adipocytes. J Biol Chem. 2005;280:40406–40416. [DOI] [PubMed] [Google Scholar]
- 8. Higuchi M, Masuyama N, Fukui Y, Suzuki A, Gotoh Y. Akt mediates Rac/Cdc42‐regulated cell motility in growth factor‐stimulated cells and invasive PTEN knockout cells. Curr Biol. 2001;11:1958–1962. [DOI] [PubMed] [Google Scholar]
- 9. Kim D, Kim S, Koh H, et al. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953–1962. [DOI] [PubMed] [Google Scholar]
- 10. Morales‐Ruiz M, Fulton D, Sowa G, et al. Vascular endothelial growth factor‐stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res. 2008;6:892–896. [DOI] [PubMed] [Google Scholar]
- 11. Feng C, Yu A, Liu Y, Sun Q‐Y, Yu B. Involvement of protein kinase B/AKT in early development of mouse fertilized eggs. Biol Reprod. 2007;77:560–568. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Z, Zhang G, Xu X, Su W, Yu B. mTOR rictor is the Ser473 kinase for AKT1 in mouse one‐cell stage embryos. Mol Cell Biochem. 2012;361:249–257. [DOI] [PubMed] [Google Scholar]
- 13. Enomoto A, Murakami H, Asai N, et al. Akt/PKB regulates actin organization and cell motility via girdin/APE. Dev Cell. 2005;9:389–402. [DOI] [PubMed] [Google Scholar]
- 14. Anai M, Shojima N, Katagiri H, et al. A novel protein kinase B (PKB)/Akt‐binding protein enhances PKB kinase activity and regulates DNA synthesis. J Biol Chem. 2005;280:18525–18535. [DOI] [PubMed] [Google Scholar]
- 15. Le‐Niculescu H, Niesman I, Fischer T, DeVries L, Farquhar MG. Identification and characterization of GIV, a novel G alpha i/s‐interacting protein found on COPI, endoplasmic reticulum‐Golgi transport vesicles. J Biol Chem. 2005;280:22012–22020. [DOI] [PubMed] [Google Scholar]
- 16. Jiang P, Enomoto A, Jijiwa M, Kato T, Hasegawa T, Ishida M. An actin‐binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 2008;68:1310–1318. [DOI] [PubMed] [Google Scholar]
- 17. Ni W, Fang Y, Tong L, et al. Girdin regulates the migration and invasion of glioma cells via the PI3K‐Akt signaling pathway. Mol Med Rep. 2015;12:5086–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim NH, Chung KS, Day BN. The distribution and requirements of microtubules and microfilaments during fertilization and parthenogenesis in pig oocytes. J Reprod Fertil. 1997;111:143–149. [DOI] [PubMed] [Google Scholar]
- 19. Kim NH, Day BN, Lee HT, Chung KS. Microfilament assembly and cortical granule distribution during maturation, parthenogenetic activation and fertilization in the porcine oocyte. Zygote. 1996;4:145–149. [DOI] [PubMed] [Google Scholar]
- 20. Martin DE, Hall MN. The expanding TOR signaling network. Curr Opin Cell Biol. 2005;17:158–166. [DOI] [PubMed] [Google Scholar]
- 21. Takahashi M, Asai N, Enomoto A. Akt‐Girdin as oncotarget. Oncoscience. 2015;2:811–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia‐Marcos M, Ghosh P, Farquhar MG. GIV is a nonreceptor GEF for G alpha I with a unique motif that regulates Akt signaling. Proc Natl Acad Sci USA. 2009;106:3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghosh P, Garcia‐Marcos M, Bornheimer SJ, Farquhar MG. Activation of galphai3 triggers cell migration via regulation of GIV. J Cell Biol. 2008;182:381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]