

Abstract
Objective: CD44 is a transmembrane glycoprotein and can facilitate signal transduction by serving as a platform for molecular recruitment and assembly. A number of studies have suggested that CD44 can either positively or negatively regulate cell proliferation. The purpose of this study was to investigate how CD44 can inhibit cell proliferation.
Materials and methods: We engineered E6.1 Jurkat cells to express CD44. Importantly, these cells lack endogenous CD44 expression. Molecular pathways involved with cell proliferation were studied using RT2‐PCR array, siRNA, Western blotting and by employing pharmacological inhibitors of ERK1/2, p38 and the PI3K/Akt pathways.
Results: We found that CD44 expression significantly inhibited cell proliferation and down‐regulated EGR‐1 expression and EGR‐1 targets cyclin D1 and cyclin D2. Transfection of control E6.1 Jurkat cells with EGR‐1 siRNA also inhibited cell proliferation, confirming its role. Disruption of the PI3K/Akt pathway with pharmacological inhibitors reduced both EGR‐1 expression and cell proliferation, recapitulating the properties of CD44 expressing cells. Akt was hypophosphorylated in cells expressing CD44 showing its potential role in negatively regulating Akt activation. Strikingly, constitutively active Akt rescued the proliferation defect showing requirement for active Akt, in our system.
Conclusion: Our results suggest a novel pathway by which CD44 inactivates Akt, down‐regulates EGR‐1 expression and inhibits cell proliferation.
Introduction
CD44, a type I transmembrane glycoprotein, has diverse roles in a number of cell functions including: (i) leucocyte trafficking (1), (ii) angiogenesis (2) and (iii) cell proliferation (3, 4, 5, 6). Although CD44 does not contain any signalling domain, it can act as a platform for recruitment and assembly of molecular machinery for signal transduction (7). For example, CD44 clustering in T cells can recruit tyrosine phosphatase CD45 to the CD44 cluster. CD45 then dephosphorylates the negative regulatory tyrosine of Src family kinase, Lck. In turn, this signalling event results in F‐actin ring formation and round cell spreading (8).
A number of reports have shown that CD44 can induce or augment cell proliferative responses. For example, CD44 can trigger mobilization of Ca2+ in aortic endothelial cells which in turn leads to their proliferation (9). Other mechanisms whereby CD44 induces cell proliferation have also been reported, including activation of MAP kinases (10). By contrast, negative regulation of cell proliferation by CD44 has been less frequently described with only one report showing that CD44 can inhibit proliferation of the NB4 cell line (3). Thus, it is tempting to speculate that CD44 has the potential to regulate cell proliferation in both positive and negative fashions. The purpose of this study was to evaluate the molecular basis for CD44‐mediated anti‐proliferative effects, using the E6.1 Jurkat cell system (11). Importantly, E6.1 Jurkat cells do not express endogenous CD44. Therefore, the molecular and biochemical mechanisms whereby CD44 regulates cellular processes can be directly investigated in E6.1 Jurkat cells expressing CD44, by comparison with their open vector control counterparts (11).
Here, we show that CD44 expression in E6.1 Jurkat cells inhibited their proliferation compared to cells transfected with the open vector control. Moreover, CD44 reduced expression of early growth response‐1 (EGR‐1) gene. EGR‐1 protein is a zinc finger transcription factor that can bind and activate promoters of many genes, whose products can influence cell proliferation (12, 13, 14, 15, 16). Indeed, transfection of E6.1 Jurkat cells with EGR‐1 siRNA, but not siRNA control, inhibited cell proliferation, confirming its role in proliferation of these cells. In terms of a mechanism for how CD44 could regulate EGR‐1, we found that EGR‐1 expression was regulated via the PI3K/Akt pathway in E6.1 Jurkat cells. Moreover, we found that CD44 disrupted Akt activation as assessed by Western blotting and that constitutively active Akt rescued the proliferation defect. Thus, our results suggest a novel pathway in which CD44 can negatively regulate cell proliferation via Akt inactivation and down‐regulated EGR‐1 expression.
Materials and methods
Cell lines
E6.1 Jurkat cells were purchased from the American Type Culture Collection and were maintained as suggested by the supplier. The human lymphoma cell line HuT78 was kindly provided by Dr Elisa Fleming (U.T. Southwestern Medical Center, Dallas, TX) and cultured in RPMI supplemented with 10% heat‐inactivated FBS, 1% sodium pyruvate, 25 mm HEPES and 1% penicillin/streptomycin/glutamine.
Antibodies and reagents
Pan anti‐CD44 antibody, clone IM7 was from BD Biosciences, San Jose, CA, USA. Goat anti‐human EGR‐1 and rat IgG isotype control were from R&D Systems, Tustin, CA, USA. FITC‐conjugated anti‐rat secondary antibody was from Caltag Laboratories Inc. (Burlingame, CA, USA) and secondary antibodies labelled with alkaline phosphatise, for Western blotting, were purchased from Invitrogen, Carlsbad, CA, USA. Antibodies against β‐actin, Akt and phosphorylated Akt were all from Cell Signaling (Danvers, MA, USA). Pharmacological inhibitors wortmannin, LY294002, SB239063 and U0126 and bovine testis hyaluronidase (EC 3.2.1.35, type 1‐S) were from Sigma‐Aldrich (St. Louis, MO, USA). Sodium hyaluronan was from Acros Organics (Geel, Belgium). All SYBR‐labelled primers used for RT2‐PCR were from SABiosciences (Frederick, MD, USA).
Cloning of CD44
Total RNA was extracted from HuT78 cells using TRIzol Reagent (Invitrogen). Standard form CD44 transcripts were amplified using SuperScript III one‐step RT‐PCR system (Invitrogen), and forward primer 5′‐CCCAAGCTTGGATCCTCCAGCTCCTTTCG‐3′ containing an engineered HindIII site (underlined) and reverse primer 5′‐GCTCTAGATCCCAGCTCCCTGTAATGGT‐3′ containing engineered XbaI site (underlined). Cycling conditions were as follows: 50 °C for 20 min, 94 °C for 2 min, 35 cycles of 94 °C for 30 s, 59 °C for 30 s and 72 °C for 90 s, and finally 72 °C for 7 min. Gel‐purified cDNA was then digested with HindIII and XbaI and ligated into pcDNA3.1 expression vector (Invitrogen) double digested with HindIII/XbaI. Purified CD44‐pcDNA3.1 was transfected into E6.1 Jurkat cells using FuGENE 6 transfection reagent (Roche, Basel, Schweiz). Cells were then collected for CD44 expression by FACS. Integrity of the CD44 sequence was confirmed by DNA sequencing in both forward and reverse directions.
Cell surface expression of CD44
Cells were incubated with anti‐CD44 monoclonal antibody IM7 (1:500 dilution) or IgG isotype control (1:500 dilution) in HBSS, containing 1% FCS, for 30 min on ice. After washing, cells were incubated with FITC‐conjugated anti‐rat antibody (1:500 dilution) in HBSS containing 1% FCS for 30 min, on ice. Cells were washed, stained with propidium iodide (1 μg/ml) and then subjected to FACS. Dead cells were gated out of the analysis, based on propidium iodide staining.
Cell adhesion assay
To assess CD44 binding to hyaluronan, we performed a cell adhesion assay with 35S‐labelled cells, essentially as described previously (17). Briefly, 100 μl of hyaluronan solution (0.1 mg/ml) was added to wells of an Amine CovaLink plate (NALGENE, NUNC, Rochester, NY, USA), followed by addition of 50 μl of 0.1 N HCl and 50 μl of 0.2 mm 1‐ethyl‐3‐(3‐dimethylaminopropyl)‐carbodiimide. After overnight incubation at room temperature, wells were washed three times in PBS containing 2 m NaCl and 0.04 mm MgSO4, followed by two additional washes with PBS alone. The hyaluronan‐coated plates were then counter‐coated with 3% BSA in PBS for 3 h at 37 °C. E6.1 Jurkat cells (vector control and those expressing CD44) were metabolically labelled in the presence of 30 μCi/ml 35S‐methionine/cysteine (Trans 35S from MP Biomedical, Solon, CA, USA) overnight in methionine/cysteine‐free DMEM medium. After washing, radiolabelled cells were added to the hyaluronan‐coated wells (104/well) and incubated at room temperature for 30 min. After removal of non‐adherent cells, wells were washed three times with PBS, and remaining adherent cells were solubilized in 1% SDS and counted for radioactivity. Percentages of adherent cells were calculated by dividing recovered counts per minute by total counts per minute added to each well.
Doubling time assay
E6.1 Jurkat cells were seeded in a 12‐well plate and cultured for 70.5 h. They were then stained with trypan blue to exclude dead cells and counted using a haemocytometer. Doubling time was calculated using the formula: T d = (t 2−t 1) × log2/log(q 2−q 1), where t 1 is starting time of culture, t 2 is ending culture time, q 1 is cell number at time t 1 and q 2 is cell number at time t 2.
Cell proliferation assays
The cells were cultured in flat‐bottomed 96‐well plates (200, 1000, 5000 or 25 000 cells/well) and were pulsed with [3H]dT (tritiated thymidine) (0.5 μCi/well) for the last 24 h of the culture period. Cells were then harvested on to glass fibre filter paper and read in a β‐counter (Beckman Coulter Inc., Brea, CA, USA).
Measurement of endogenous hyaluronan
Production of endogenously synthesized hyaluronan was measured using the competitive inhibition plate assay similar to a method described previously (18). Briefly, 100 μl of hyaluronan solution (20 μg/ml) was added to wells of an Amine CovaLink plate (Nalge Nunc International), followed by addition of 50 μl of 0.1 N HCl and 50 μl of 0.2 mm 1‐ethyl‐3‐(3‐dimethylaminopropyl)‐carbodiimide. After overnight incubation at room temperature, wells were washed three times in PBS containing 2 m NaCl and 0.04 mm MgSO4, followed by two additional washes with PBS alone. Hyaluronan‐coated plates were then counter‐coated with 3% BSA in PBS for 3 h at 37 °C. Hyaluronan‐coated plates were then counter‐coated with 5% non‐fat milk in TBS‐0.1% Tween 20 for 3 h at 37 °C. HA solution (20, 10, 5, 2.5, 1.25, 0.625, 0.313 μg/ml) or supernates from Jurkat cells expressing CD44, or transfected with open vector control, each mixed with 2 μg/ml biotin‐labelled HABP (Northstar BioProducts, East Falmouth, MA, USA) and equilibrated overnight, were then added to the blocked wells and incubated for 1.5 h at room temperature. After washing three times with TBS‐0.1% Tween 20, steptavidin–alkaline phosphatase‐labelled reagent (Sigma, St Louis, MO, USA) was added to wells and incubated for 1.5 h at room temperature. Again, after washing, substrate (Sigma) was added to the wells. Absorbance was measured at 405 nm after colour development. Concentration of hyaluronan in samples was calculated based on a standard curve.
Impact of hyaluronan on cell proliferation
CD44 or open vector control E6.1 Jurkat cells (5000 cells/well) were treated with 0.01, 0.05 or 0.1 mg/ml hyaluronan, or with 0.1, 1 or 10 U/ml bovine testis hyaluronidase. Impact of hyaluronan or hyaluronidase on cell proliferation was evaluated 48 h later. Cultures were pulsed with [3H]dT (0.5 μCi/well) for the last 24 h of the culture period. Cells were then harvested on to glass fibre filter paper and read in a β‐counter (Beckman Coulter Inc.).
RT2‐PCR array
Total RNA was isolated from the E6.1 Jurkat cells using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and was quantified using NanoDrop 1000 (Thermo Scientific, Wilmington, DE, USA). RNA quality was assessed using an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Human Signal Transduction Pathway Finder PCR array was performed as described by the manufacturer (SABiosciences). RT2‐PCR was conducted using an ABI‐7300 Real‐time PCR System (Applied Biosystems, Foster City, CA, USA) with the following programme: one cycle of 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Quality control of PCR data for consequent analysis was based on Genomic DNA Control, Reverse Transcription Control and Positive PCR Control as described by the manufacturer. Data analysis was conducted using Excel spreadsheets in Windows, provided by SABiosciences.
Western blot assay
Proteins were prepared from E6.1 Jurkat cells using the nuclear extract kit (Active Motif North America, Carlsbad, CA, USA) and protein concentrations were determined using Bio‐Rad assay (Bio‐Rad, Hercules, CA, USA). Samples were boiled for 10 min and then equal concentrations of them were loaded into wells of a 4–20% SDS–PAGE gel. Transfer of proteins to nitrocellulose membranes was performed by transblotting overnight at 4 °C. Membranes were then blocked in TBS containing 5% non‐fat milk and 0.1% Tween‐20 (blocking buffer). Then membranes were incubated with the primary antibodies (1:1000 dilution in blocking buffer), washed in TBS containing 0.1% Tween‐20 followed by incubation with secondary antibodies labelled with alkaline phosphatase (1:1000 dilution in blocking buffer). After washing in TBS containing 0.1% Tween‐20, membranes were developed using the Lumi‐Phos WB chemiluminescence substrate system (Thermo Scientific).
Silencing EGR‐1 mRNA
Open vector control E6.1 Jurkat cells were transfected with 10 μm human EGR‐1 siRNA or with 10 μm of control siRNA as described by the manufacturer (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA). Impact of siRNA on cell proliferation was evaluated 72 h after transfection.
Impact of myristoylated Akt on the proliferation of CD44 expressing E6.1 Jurkat cells
E6.1 Jurkat cells expressing CD44 were infected with 180 MOI of adenovirus engineered with Ad‐CMV‐Akt1 (Myr) expression vector, as described by the manufacturer (Vector Biolabs, Philadelphia, PA, USA). This construct encodes Akt with a myristoylation site for constitutive activity (19). Fresh medium was added to wells 6 h after infection. Impact of activated Akt protein on cell proliferation was evaluated 48 h later using [3H]dT uptake experiments as described earlier.
Statistical analyses
Comparisons between two groups were assessed by two‐tailed Student’s t‐test. Differences between groups were considered significant with P < 0.05. All experiments were performed at least twice to confirm reproducibility.
Results
Expression of CD44
We stably transfected E6.1 Jurkat cells with open vector (pcDNA3.1) or CD44‐pcDNA3.1 expression vector using standard techniques. As shown in Fig. 1a, cells transfected with CD44‐pcDNA3.1, but not cells transfected with the open vector control, expressed mRNA for CD44 as assessed by RT‐PCR. We did not detect CD44 expression in open vector control cells, as assessed by FACS (Fig. 1b, panel 1). In contrast, cells transfected with CD44‐pcDNA3.1 expression vector showed surface expression of CD44 protein (Fig. 1b, panel 2). Next, we tested whether expressed CD44 protein was able to bind its major ligand, hyaluronan, using a cell adhesion assay. As shown in Fig. 1c, the cells stably transfected with CD44‐pcDNA3.1 expression vector, but not vector control, had the ability to adhere to hyaluronan‐coated wells. These results showed that CD44 expressed on cell surfaces maintained ligand‐binding potential.
Figure 1.
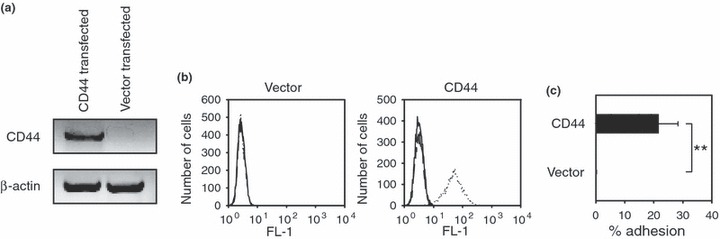
CD44 expression in E6.1 Jurkat cells. (a) The E6.1 Jurkat cell line was transfected with the CD44‐pcDNA3.1 expression vector or with the open vector control (i.e. pcDNA3.1 alone). The expression of CD44 mRNA was then assessed by RT‐PCR. Data shown are ethidium bromide‐stained products separated on a 1% agarose gel. (b) Expression of CD44 protein on the surface of Jurkat cells was evaluated by FACS. The histograms show cells alone (solid lines), the isotype IgG control (dashed lines) or the anti‐CD44 antibody (dotted lines). Results from FACS show that Jurkat cells transfected with the CD44‐pcDNA construct, but not the open vector control, expressed CD44 on their surfaces. (c) The ability of E6.1 Jurkat cells to bind hyaluronan was evaluated using an adhesion assay. E6.1 Jurkat cells expressing CD44, but not the open vector control, exhibited binding to hyaluronan‐coated plates. Results are presented as mean ± SD from triplicate samples. Asterisks indicate statistically significant differences (**P < 0.01).
Impact of CD44 on cell size and doubling time
We next investigated morphology of the cells under bright field microscopy. As shown in Fig. 2a, open vector control cells and cells expressing CD44 showed similar rounded morphology under light microscopy. On the other hand, cells expressing CD44 had a modest but significant reduction in their sizes as compared to open vector control cells (Fig. 2b). These results mirrored previous reports on leukaemic cells, which showed that expression of CD44 reduced cell size compared to counterparts lacking CD44 expression (20).
Figure 2.
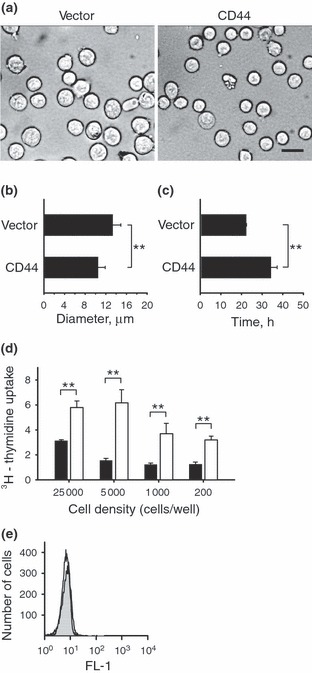
Impact of CD44 on cell size, doubling time, proliferation and apoptosis. (a–b) The sizes of E6.1 Jurkat cells expressing CD44 or transfected with the open vector control were evaluated under bright field microscopy. The E6.1 Jurkat cells expressing CD44 showed a modest but significant reduction in their diameters as compared with the open vector control cells (scale bar, 20 μm). (c) The doubling time of E6.1 Jurkat cells expressing CD44 was significantly increased as compared with the doubling time of open vector control E6.1 Jurkat cells. (d) The E6.1 Jurkat cells expressing CD44 (black bars) or the open vector control (white bars) were cultured at the indicated densities and pulsed with [3H]dT during the last 24 h of the culture period. Results show that reducing the number of cells did not restore the proliferation of CD44 expressing E6.1 Jurkat cells to the same level as the open vector control cells. These results suggest that CD44 did not impair proliferation due to contact inhibition. (e) Apoptosis of open vector control E6.1 Jurkat cells (open histogram) and CD44 expressing E6.1 Jurkat cells (closed histogram) were ascertained using Annexin V staining and FACS. We failed to detect CD44‐induced apoptosis as compared with the open vector control cells. Results show representative FACS histograms from triplicate experiments with similar results. Values in this figure are expressed as mean ± SD from 280 independently measured cells (b) or triplicate samples (c and d). Asterisks indicate statistically significant differences (**P < 0.01).
Next, doubling times of open vector control cells were compared to those of cells expressing CD44. Open vector control cells had significantly shorter doubling time than those expressing CD44 (Fig. 2c). Thus, CD44 expression reduced doubling rate of the cells.
Effect of cell density on proliferation
Morrison et al. have shown that CD44 can inhibit proliferation of schwannoma cells through contact inhibition (21). To test whether CD44 inhibited cell proliferation by a similar mechanism, we cultured the cells at different densities and measured their proliferation based on uptake of [3H]dT. As shown in Fig. 2d, reduction in cell density did not restore proliferation of CD44 expressing cells compared to open vector control cells. Based on these results, we concluded that cell proliferation was not impaired by CD44 due to contact inhibition.
Impact of CD44 on apoptosis
CD44 has been shown to induce apoptosis in some cell types (11). To determine if CD44 expression induced apoptosis in E6.1 Jurkat cells, we compared annexin V staining using FACS. As shown in Fig. 2e, the open vector control (open histogram) and CD44 (closed histogram) expressing cells exhibited similar, if not identical, annexin V staining profiles. These results suggested that CD44 expression did not induce apoptosis in this cell type.
Functional role of hyaluronan in cell proliferation
Hyaluronan is a glycosaminoglycan and serves as a major ligand of CD44; exogenous hyaluronan can facilitate cell proliferation via CD44 (22). Importantly, recent studies using splenic murine T cells have shown that endogenously synthesized hyaluronan can promote their proliferation by an autocrine type mechanism (23). Therefore, we first tested whether CD44 expressing E6.1 Jurkat cells or vector control E6.1 Jurkat cells synthesized hyaluronan. We failed to detect hyaluronan in cell culture supernates (data not shown) suggesting that these cells synthesized little or no hyaluronan. As hyaluronan co‐polymers retained on surfaces of cells could also impact their proliferation characteristics (23), we next cultured CD44 cells and open vector control cells with hyaluronidase, to digest surface‐associated hyaluronan and hence, disrupt the potential impact of this co‐polymer on cell proliferation. As shown in Fig. 3a, we failed to detect differences in proliferation for either CD44 expressing or open vector control cells that were cultured with graded doses of hyaluronidase. Moreover, addition of exogenous hyaluronan to cell cultures failed to have any impact on their proliferation (Fig. 3b). As surface‐expressed CD44 was able to bind hyaluronan (Fig. 1c), our results were not simply because of failure of CD44 to bind the exogenous hyaluronan molecule. Thus, in total, our findings strongly suggested that hyaluronan played little or no role in the reduced proliferation that we observed for the CD44 expressing E6.1 Jurkat cells.
Figure 3.
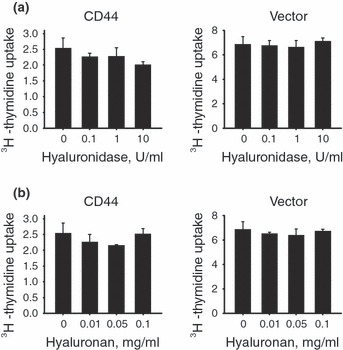
Hyaluronan does not have an effect on E6.1 Jurkat cell proliferation. (a) E6.1 Jurkat cells expressing CD44 or the open vector control were cultured in medium containing graded doses of hyaluronidase and pulsed with [3H]dT as described in the text. Hyaluronidase did not significantly alter cell proliferation as compared with 0 U/ml. (b) CD44 expressing E6.1 Jurkat cells or open vector control E6.1 Jurkat cells were cultured in medium containing graded activities of hyaluronan. Exogenous hyaluronan did not have a significant impact on cell proliferation as compared with 0 mg/ml. Values in this figure are expressed as mean ± SD from triplicate samples.
EGR‐1 expression in CD44 E6.1 Jurkat cells
To identify differences in gene expression between the open vector control and CD44 expressing E6.1 Jurkat cells, we employed the Human Signal Transduction Pathway Finder PCR array available from SABiosciences. This array contains primers for analysing profiles of 84 different genes. As shown in Table 1, CD44 expression modulated a number of genes, based on their down‐ or up‐regulation. Among down‐regulated genes, EGR‐1 was markedly reduced in the CD44 expressing cells. Based on the large number of studies showing that EGR‐1 can play a role in cell proliferation (12, 13, 14, 15, 16), we selected this molecule for further studies.
Table 1.
Differential gene expression induced by CD44 in E6.1 Jurkat cells
| Accession No. | Symbol | Fold up‐ or down‐regulation | Gene name | Description |
|---|---|---|---|---|
| Down‐regulation | ||||
| NM_001964 | EGR1 | −6.81 | AT225/G0S30 | Early growth response 1 |
| NM_021784 | FOXA2 | −4.17 | HNF3B/TCF3B | Forkhead box A2 |
| NM_002982 | CCL2 | −3.40 | GDCF‐2/GDCF‐2 HC11 | Chemokine (C‐C motif) ligand 2 |
| NM_020182 | TMEPAI | −2.85 | PMEPA1/STAG1 | Transmembrane prostate androgen‐induced protein |
| NM_002228 | JUN | −2.61 | AP1/c‐Jun | Jun oncogene |
| NM_000639 | FASLG | −2.41 | APT1LG1/CD178 | Fas ligand (TNF superfamily, member 6) |
| NM_003202 | TCF7 | −2.21 | TCF‐1 | Transcription factor 7 (T‐cell specific, HMG‐box) |
| NM_000589 | IL4 | −2.15 | BSF1/IL‐4 | Interleukin 4 |
| Up‐regulation | ||||
| NM_000584 | IL8 | 15.98 | 3‐10C/AMCF‐I | Interleukin 8 |
| NM_004591 | CCL20 | 10.94 | CKb4/LARC | Chemokine (C‐C motif) ligand 20 |
| NM_000450 | SELE | 5.75 | CD62E/ELAM | Selectin E (endothelial adhesion molecule 1) |
| NM_000598 | IGFBP3 | 5.14 | BP‐53/IBP3 | Insulin‐like growth factor‐binding protein 3 |
| NM_015869 | PPARG | 5.07 | NR1C3/PPARG1 | Peroxisome proliferator‐activated receptor gamma |
| NM_000230 | LEP | 3.68 | OB/OBS | Leptin |
| NM_000418 | IL4R | 3.45 | CD124/IL4RA | Interleukin 4 receptor |
| NM_005551 | KLK2 | 3.21 | KLK2A2/hK2 | Kallikrein‐related peptidase 2 |
| NM_002416 | CXCL9 | z2.62 | CMK/Humig | Chemokine (C‐X‐C motif) ligand 9 |
The expression profiles of 84 different genes were evaluated using a commercially available RT2‐PCR array. A 2‐fold cut‐off was used to identify down‐ or up‐regulated genes in E6.1 Jurkat cells expressing CD44.
As shown in Fig. 4a, expression of the EGR‐1 gene was significantly reduced in CD44 expressing cells relative to open vector control cells, confirming results obtained from the RT2‐PCR array. Importantly, EGR‐1 protein was also significantly reduced in the CD44 cells as assessed by Western blotting (Fig. 4b,c). Thus, our results showed that EGR‐1 was down‐regulated in E6.1 Jurkat cells expressing CD44.
Figure 4.
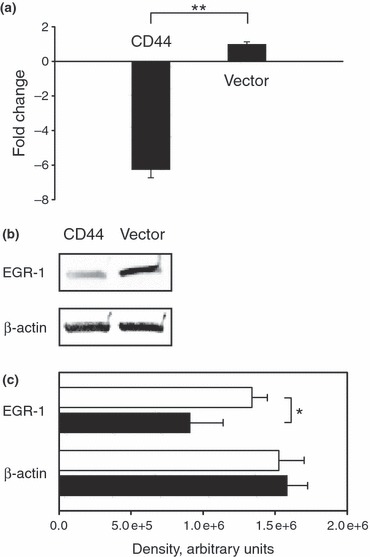
EGR‐1 expression is reduced in E6.1 Jurkat cells expressing CD44. (a) The expression levels of EGR‐1 mRNA were assessed using real‐time PCR. The ΔΔC t‐based fold change was calculated from the raw threshold cycle. The EGR‐1 mRNA expression levels were significantly reduced in E6.1 Jurkat cells expressing CD44 as compared with the open vector control E6.1 Jurkat cells. These results confirm the findings obtained from the RT2‐PCR array (i.e. reduced expression of the EGR‐1 mRNA). (b) The E6.1 Jurkat cells expressing CD44 showed decreased EGR‐1 protein expression as compared with the open vector control E6.1 Jurkat cells as evaluated by Western blotting. (c) The band intensities in Western blots were quantitatively evaluated for EGR‐1 and β‐actin protein expressions. E6.1 Jurkat cells expressing CD44 (closed bars) show reduced concentrations of EGR‐1 as compared with the open vector control E6.1 Jurkat cells (open bars). The concentrations of β‐actin were the same for both cell types. These results show that reduced transcription of the egr‐1 gene is correlated with reduced EGR‐1 protein expression. Results are presented as mean ± SD from triplicate samples. Asterisks indicate statistically significant differences (*P < 0.05; **P < 0.01).
Regulation and function of EGR‐1
To investigate regulation and function of EGR‐1 in E6.1 Jurkat cells, we treated open vector control cells with inhibitors of the PI3K/Akt pathway (wortmannin and LY294002), p38 (SB239063) and ERK 1/2 (U0126). As shown in Fig. 5a, wortmannin and LY294002, but not other inhibitors, significantly reduced proliferation of the cells as compared to their non‐treated counterparts. These results suggested that the PI3K/Akt pathway played a central role in their proliferation. Moreover, wortmannin and LY294002 markedly reduced EGR‐1 expression. In contrast, SB239063 and U0126 did not affect EGR‐1 expression (Fig. 5b). These findings showed that EGR‐1 expression was regulated by the PI3K/Akt pathway.
Figure 5.
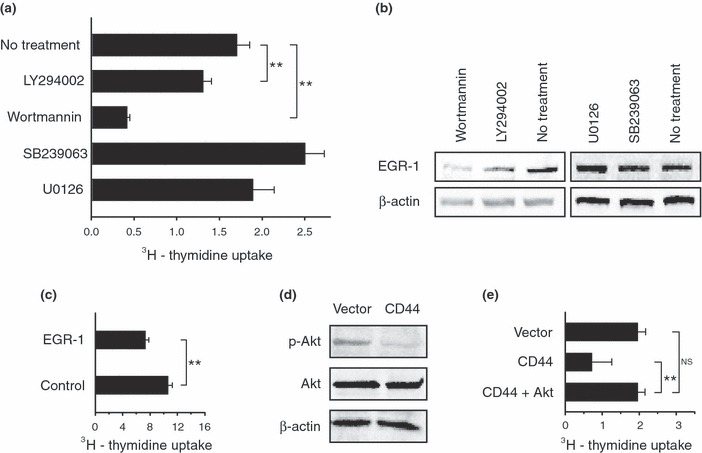
Akt activation is correlated with E6.1 Jurkat cell proliferation and EGR‐1 expression. (a) Open vector control E6.1 Jurkat cells were treated with the indicated inhibitors and the cell proliferation was assessed by the uptake of [3H]dT. Inhibitors of the PI3K/Akt pathway (LY294002 and wortmannin), but not other inhibitors, significantly reduced cell proliferation as compared with the non‐treatment group. These results show that the PI3K/Akt pathway plays a role in the proliferation of E6.1 Jurkat cells. (b) The concentrations of EGR‐1 in E6.1 Jurkat cells treated with the inhibitors in A were evaluated using Western blotting. Inhibitors of the PI3K/Akt pathway reduced EGR‐1 protein concentration showing that EGR‐1 expression is correlated with this pathway. (c) Transfection of open vector control E6.1 Jurkat cells with human EGR‐1 siRNA, but not control siRNA, significantly reduced cell proliferation. These results show that EGR‐1 plays a role in E6.1 Jurkat cell proliferation. (d) The phosphorylation status of Akt in E6.1 Jurkat cells expressing CD44 and the open vector control E6.1 Jurkat cells was evaluated by Western blotting (p‐Akt, phosphorylated Akt; Akt, total protein). The results show that Akt is hypophosphorylated in the CD44 expressing E6.1 Jurkat cells relative to the open vector control E6.1 Jurkat cells. These findings strongly suggest that CD44 expression disrupts Akt activation. (e) Myristoylated Akt was transiently expressed in E6.1 Jurkat cells expressing CD44. The results show that constitutively active Akt rescued the impaired proliferation defect as compared with the open vector control E6.1 Jurkat cells. Results are expressed as mean ± SD from triplicate samples. Asterisks indicate statistically significant differences (**P < 0.01). NS, not significant.
To directly assess whether EGR‐1 plays a role in proliferation of E6.1 Jurkat cells, we transfected them with EGR‐1 siRNA. As shown in Fig. 5c, cells transfected with EGR‐1 siRNA proliferated significantly less than cells transfected with control siRNA. On the other hand, EGR‐1 siRNA did not inhibit proliferation of cells to the same extent as cells expressing CD44 when compared at the same cell densities (5000 cells/well) and pulsed with [3H]dT for the same amount of time (EGR‐1 siRNA‐inhibited proliferation: 30 ± 8%; CD44‐inhibited proliferation: 75 ± 19%). Failure of EGR‐1 siRNA to inhibit cell proliferation to the same extent as CD44 is likely to be due to inability of the siRNA to knock down EGR‐1 expression to the same extent of CD44 down‐regulated EGR‐1 expression (data not shown). It is also possible that CD44 impacts on other factors involved in cell proliferation in addition to EGR‐1. Regardless of source of discrepancy, our results showed that EGR‐1, at least contributes, to proliferation of E6.1 Jurkat cells.
As the above results recapitulated impact of CD44 on EGR‐1 expression and cell proliferation, we hypothesized that CD44 interferes with the PI3K/Akt pathway and consequent PI3K/Akt‐mediated expression of EGR‐1. To test this hypothesis, we evaluated Akt phosphorylation using Western blotting. As shown in Fig. 5d, Akt was hypophosphorylated in E6.1 Jurkat cells expressing CD44 as compared to open vector control cells. As phosphorylation is required to activate Akt fully, we proposed that CD44 inactivated Akt in these E6.1 Jurkat cells. To test if active Akt could complement the defect in cell proliferation, we infected cells expressing CD44 with adenoviruses containing a construct that encoded Akt with a myristoylation sequence. Importantly, myristoylated Akt is constitutively active. As shown in Fig. 5e, activated Akt restored proliferation of CD44 E6.1 Jurkat cells to the same level as open vector control equivalents (P > 0.05). These results strongly suggested that inactivated Akt played a pivotal role in impaired proliferation of CD44 expressing E6.1 Jurkat cells.
Both cyclin D1 and D2 function as regulatory subunits for cyclin‐dependent kinases. Importantly, both cyclin D1 and cyclin D2 are known to be directly regulated by EGR‐1 (24, 25). Therefore, we investigated if levels of cyclin D1 and D2 transrcipts were altered in CD44 expressing E6.1 Jurkat cells as compared to open vector controls, using RT2‐PCR. We found that cyclin D1 was down‐regulated 5.5‐fold and cyclin D2 was down‐regulated 14.5‐fold in E6.1 Jurkat cells expressing CD44 compared to open vector control cells. These results showed that down‐regulation of EGR‐1 expression in CD44 expressing cells resulted in reduced transcription of genes involved in regulation of the cell cycle.
Based on results that EGR‐1 expression is linked to the PI3K/Akt pathway, we proposed that reduced Akt activity ultimately resulted in lowered levels of EGR‐1 expression. In turn, lowered expression levels of EGR‐1 decreased transcription of factors directly regulated by EGR‐1 involved in promoting cell cycle progression (for example, cyclin D1 and cyclin D2) and hence, reduced cell proliferation.
At first glance, results from the PCR array (Table 1) showing increased transcription of IL‐8 would appear to contradict our notion that Akt is inactivated, because the PI3K/Akt pathway is known to play a role in regulating IL‐8. On the other hand, IL‐8 can also be regulated by additional pathways. For example, Tanaka et al. have shown that proteinase‐activated receptor‐2 (PAR2) induced IL‐8 via the MEK/ERK pathway independent from the PI3K/Akt pathway (26). Similarly, Chen et al. have shown that hepatocyte growth factor/scatter factor can induce IL‐8 expression in squamous cell carcinoma cells, by the MEK/ERK pathway in addition to the PI3K/Akt pathway (27). Thus, we propose that IL‐8 transcription is augmented in a PI3K/Akt‐independent fashion in CD44 expressing E6.1 Jurkat cells. It is interesting to note that Huang et al. showed that inhibition of PI3K in T84 intestinal epithelial cells enhanced IL‐8 induced by Salmonella because of altered ERK activation (28).
Discussion
CD44, a cell surface glycoprotein, is expressed in a number of different cell types. Previous investigations have suggested that CD44 may have a role in positively and negatively regulating cell proliferation. To date, the majority of studies have examined how CD44 can induce or enhance cell proliferation. Thus, we decided to explore how CD44 inhibits cell proliferation, using E6.1 Jurkat cells as our model system.
We found that CD44 expression in E6.1 Jurkat cells led to significant reduction in their proliferation, due at least in part, to down‐regulation of EGR‐1 transcription factor. Importantly, EGR‐1 transcription factor regulates a number of genes important for cell growth and cell division. Indeed, transfection of E6.1 Jurkat cells with EGR‐1 siRNA reduced their proliferation, showing a direct role of EGR‐1 in cell proliferation. Moreover, EGR‐1‐regulated cyclin D1 and cyclin D2, factors known to be important for cell cycle progression, were markedly down‐regulated in E6.1 Jurkat cells expressing CD44. Further studies with pharmacological inhibitors showed that EGR‐1 expression is regulated via the PI3K/Akt pathway. Hypophosphorylation of Akt in E6.1 Jurkat cells expressing CD44 showed that CD44 either directly or indirectly impaired Akt activation. The pivotal role of Akt in the proliferation of the CD44 E6.1 Jurkat cells is underscored by our finding that constitutively active Akt completely rescued their proliferation. As EGR‐1 expression was regulated by the PI3K/Akt pathway in E6.1 Jurkat cells, we propose that reduced levels of active Akt ultimately resulted in lower levels of EGR‐1 expression which in turn reduced the transcription of factors such as cyclins D1 and D2 that are required for cell cycle progression. We should point out that others have also shown a linkage between the PI3K/Akt pathway and EGR‐1 expression. For example, low‐density lipoprotein‐induced proliferation of human aortic smooth muscle cells depends on the up‐regulation of EGR‐1, which involves activation of ERK1/2 and the PI3K/Akt pathways (14). Moreover, Abdel‐Malak et al. have shown that angiopoietin‐1‐mediated EGR‐1 expression was involved in the migration, proliferation and capillary‐like tube formation in human umbilical vein endothelial cells. Again, these EGR‐1‐regulated processes were linked to the PI3K/Akt pathway (29).
A previous report has shown that siRNA‐induced knock down of CD44 in colon cancer cells resulted in hyperphosphorylation of Akt (30). Interestingly, we performed the converse experiment (knocked in CD44 expression) and obtained the opposite result (hypophosphorylation of Akt) as compared with experiments in which CD44 was knocked down. Thus, our findings tend to support the notion that the phosphorylation status of Akt can be regulated by CD44. How does CD44 reduce Akt phosphorylation in E6.1 Jurkat cells? The PI3K/Akt pathway is complex and can be regulated at several different checkpoints. Autophosphorylation of receptor tyrosine kinases leads to PI3K activation. The phosphoinositide products of PI3K bind the pleckstrin homology domain of Akt. Binding of phosphoinositides to Akt is followed by its phosphorylation via phosphoinositide‐dependent kinase‐1. In turn, activated Akt binds downstream effectors (e.g. NFκB) that can regulate gene expression (31). A number of proteins fine‐tune Akt activation including inosine‐5′ monophosphate dehydrogenase (32), histone H3 methyltransferase SETDB1 (33) and Src (34). Recently, Hamaïet al. showed that ICAM‐1 decreased PTEN activity with a concomitant increase in Akt activation in human melanoma cells. PTEN is a tumour suppressor that antagonizes PI3K signalling (35). Thus, CD44 could potentially modulate Akt phosphorylation by a number of different mechanisms. Future investigations in our laboratory will seek to address the mechanism(s) by which CD44 regulates Akt phosphorylation.
It is interesting to note that previous studies have suggested a linkage between CD44 expression and EGR‐1 expression. Fitzgerald and O’Neill showed that the CD44 promoter region has an EGR‐1‐binding motif (36). Mutation of this binding site abolished CD44 up‐regulation in the ECV304 endothelial cell line following their stimulation with IL‐1α. These results suggested that EGR‐1 positively up‐regulated CD44 expression. Similarly, Mishra et al. (37) found that lipopolysaccharide‐induced CD44 expression was up‐regulated by EGR‐1 in monocytes. Results presented in this study showed that CD44 expression down‐regulated EGR‐1 expression and in light of the above reports may suggest that there is cross‐talk between CD44 and EGR‐1. Thus, we envisioned that in some cell types (e.g. monocytes), stimulation can induce EGR‐1 expression which then transcribes CD44. The expressed CD44 molecule then down‐regulates EGR‐1 expression potentially via the PI3K/Akt pathway. Thus, CD44 could regulate its own transcription as well as other EGR‐1 transcribed factors, including molecules involved in cell proliferation. Experiments are currently underway in our laboratory to test this EGR‐1/CD44 feedback loop concept.
In conclusion, we have shown that CD44 inhibits cell proliferation in a pathway that involves Akt inactivation and down‐regulation of EGR‐1 expression. Our results and experimental system should provide the framework for further studies aimed at understanding how CD44 negatively regulates cell proliferation.
Acknowledgements
This study was supported by NIH grant RO1 AR48840.
References
- 1. Mummert ME (2005) Immunologic roles of hyaluronan. Immunol. Res. 31, 189–206. [DOI] [PubMed] [Google Scholar]
- 2. Golshani R, Lopez L, Estrella V, Kramer M, Iida N, Lokeshwar VB (2008) Hyaluronic acid synthase‐1 expression regulates bladder cancer growth, invasion, and angiogenesis through CD44. Cancer Res. 68, 483–491. [DOI] [PubMed] [Google Scholar]
- 3. Gadhoum Z, Delaunay J, Maquarre E, Durand L, Lancereaux V, Qi J et al. (2004) The effect of anti‐CD44 monoclonal antibodies on differentiation and proliferation of human acute myeloid leukemia cells. Leuk. Lymphoma 45, 1501–1510. [DOI] [PubMed] [Google Scholar]
- 4. He Y, Wu GD, Sadahiro T, Noh SI, Wang H, Talavera D et al. (2008) Interaction of CD44 and hyaluronic acid enhances biliary epithelial proliferation in cholestatic livers. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G305–G312. [DOI] [PubMed] [Google Scholar]
- 5. Murphy JF, Lennon F, Steele C, Kelleher D, Fitzgerald D, Long AC (2005) Engagement of CD44 modulates cyclooxygenase induction, VEGF generation, and proliferation in human vascular endothelial cells. FASEB J. 19, 446–448. [DOI] [PubMed] [Google Scholar]
- 6. Nasreen N, Mohammed KA, Hardwick J, Van Horn RD, Sanders K, Kathuria H et al. (2002) Low molecular weight hyaluronan induces malignant mesothelioma cell (MMC) proliferation and haptotaxis: role of CD44 receptor in MMC proliferation and haptotaxis. Oncol. Res. 13, 71–78. [PubMed] [Google Scholar]
- 7. Ponta H, Sherman L, Herrlich PA (2003) CD44: from adhesion molecules to signaling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45. [DOI] [PubMed] [Google Scholar]
- 8. Wong NK, Lai JC, Birkenhead D, Shaw AS, Johnson P (2008) CD45 down‐regulates lck‐mediated CD44 signaling and modulates actin rearrangement in T cells. J. Immunol. 181, 7033–7043. [DOI] [PubMed] [Google Scholar]
- 9. Singleton PA, Bourguignon LY (2004) CD44 interaction with ankyrin and IP3 receptor in lipid rafts promotes hyaluronan‐mediated Ca2+ signaling leading to nitric oxide production and endothelial cell adhesion and proliferation. Exp. Cell Res. 295, 102–118. [DOI] [PubMed] [Google Scholar]
- 10. Marhaba R, Bourouba M, Zoller M (2005) CD44v6 promotes proliferation by persisting activation of MAP kinases. Cell. Signal. 17, 961–973. [DOI] [PubMed] [Google Scholar]
- 11. Ruffell B, Johnson P (2008) Hyaluronan induces cell death in activated T cells through CD44. J. Immunol. 181, 7044–7054. [DOI] [PubMed] [Google Scholar]
- 12. Gousseva N, Kugathasan K, Chesterman CN, Khachigian LM (2001) Early growth response factor‐1 mediates insulin‐inducible vascular endothelial cell proliferation and regrowth after injury. J. Cell. Biochem. 81, 523–534. [DOI] [PubMed] [Google Scholar]
- 13. Gururajan M, Simmons A, Dasu T, Spear BT, Calulot C, Robertson DA et al. (2008) Early growth response genes regulate B cell development, proliferation, and immune response. J. Immunol. 181, 4590–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heo KS, Ryoo SW, Kim L, Nam M, Baek ST, Lee H et al. (2008) Cl−‐channel is essential for LDL‐induced cell proliferation via the activation of Erk1/2 and PI3k/Akt and the upregulation of egr‐1 in human aortic smooth muscle cells. Mol. Cells 26, 468–473. [PubMed] [Google Scholar]
- 15. Kaufmann K, Thiel G (2002) Epidermal growth factor and thrombin induced proliferation of immortalized human keratinocytes is coupled to the synthesis of egr‐1, a zinc finger transcriptional regulator. J. Cell. Biochem. 85, 381–391. [DOI] [PubMed] [Google Scholar]
- 16. Mitchell A, Dass CR, Sun LQ, Khachigian LM (2004) Inhibition of human breast carcinoma proliferation, migration, chemoinvasion and solid tumour growth by DNAzymes targeting the zinc finger transcription factor EGR‐1. Nucleic Acids Res. 32, 3065–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mummert ME, Mohamadzadeh M, Mummert DI, Mizumoto N, Takashima A (2000) Development of a peptide inhibitor of hyaluronan‐mediated leukocyte trafficking. J. Exp. Med. 192, 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fosang AJ, Hey NJ, Carney SL, Hardingham TE (1990) An ELISA plate‐based assay for hyaluronan using biotinylated proteoglycan G1 domain (HA‐binding region). Matrix 10, 306–313. [DOI] [PubMed] [Google Scholar]
- 19. Fujio Y, Walsh K (1999) Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage‐dependent manner. J. Biol. Chem. 274, 16349–16354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abecassis I, Maes J, Carrier JL, Hillion J, Goodhardt M, Medjber K et al. (2008) Re‐expression of DNA methylation‐silenced CD44 gene in a resistant NB4 cell line: rescue of CD44‐dependent cell death by cAMP. Leukemia 22, 511–520. [DOI] [PubMed] [Google Scholar]
- 21. Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA et al. (2001) The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 15, 968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galandrini R, Galluzzo E, Albi N, Grossi CE, Velardi A (1994) Hyaluronate is costimulatory for human T cell effector functions and binds to CD44 on activated T cells. J. Immunol. 153, 21–31. [PubMed] [Google Scholar]
- 23. Mahaffey CL, Mummert ME (2007) Hyaluronan synthesis is required for IL‐2‐mediated T cell proliferation. J. Immunol. 179, 8191–8199. [DOI] [PubMed] [Google Scholar]
- 24. Virolle T, Krones‐Herzig A, Baron V, De Gregorio G, Adamson ED, Mercola D (2003) Egr1 promotes growth and survival of prostate cancer cells. Identification of novel Egr1 target genes. J. Biol. Chem. 278, 11802–11810. [DOI] [PubMed] [Google Scholar]
- 25. Xiao D, Chinnappan D, Pestell R, Albanese C, Weber HC (2005) Bombesin regulates cyclin D1 expression through the early growth response protein Egr‐1 in prostate cancer cells. Cancer Res. 65, 9934–9942. [DOI] [PubMed] [Google Scholar]
- 26. Tanaka Y, Sekiguchi F, Hong H, Kawabata A (2008) PAR2 triggers IL‐8 release via MEK/ERK and PI3‐kinase/Akt pathways in GI epithelial cells. Biochem. Biophys. Res. Commun. 377, 622–626. [DOI] [PubMed] [Google Scholar]
- 27. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C (2001) Hepatocyte growth factor/scatter factor‐induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin‐8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 61, 5911–5918. [PubMed] [Google Scholar]
- 28. Huang FC, Li Q, Cherayil BJ (2005) A phosphatidyl‐inositol‐3‐kinase‐dependent anti‐inflammatory pathway activated by Salmonella in epithelial cells. FEMS Microbiol. Lett. 243, 265–270. [DOI] [PubMed] [Google Scholar]
- 29. Abdel‐Malak NA, Mofarrahi M, Mayaki D, Khachigian LM, Hussain SN (2009) Early growth response‐1 regulates angiopoietin‐1‐induced endothelial cell proliferation, migration, and differentiation. Arterioscler. Thromb. Vasc. Biol. 29, 209–216. [DOI] [PubMed] [Google Scholar]
- 30. Subramaniam V, Vincent IR, Gardner H, Chan E, Dhamko H, Jothy S (2007) CD44 regulates cell migration in human colon cancer cells via lyn kinase and AKT phosphorylation. Exp. Mol. Pathol. 83, 207–215. [DOI] [PubMed] [Google Scholar]
- 31. Franke TF (2008) PI3K/Akt: getting it right matters. Oncogene 27, 6473–6488. [DOI] [PubMed] [Google Scholar]
- 32. Ingley E, Hemmings BA (2000) PKB/Akt interacts with inosine‐5′ monophosphate dehydrogenase through its pleckstrin homology domain. FEBS Lett. 478, 253–259. [DOI] [PubMed] [Google Scholar]
- 33. Gao H, Yu Z, Bi D, Jiang L, Cui Y, Sun J et al. (2007) Akt/PKB interacts with the histone H3 methyltransferase SETDB1 and coordinates to silence gene expression. Mol. Cell. Biochem. 305, 35–44. [DOI] [PubMed] [Google Scholar]
- 34. Jiang T, Qiu Y (2003) Interaction between src and a C‐terminal proline‐rich motif of akt is required for akt activation. J. Biol. Chem. 278, 15789–15793. [DOI] [PubMed] [Google Scholar]
- 35. Hamai A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F et al. (2008) ICAM‐1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res. 68, 9854–9864. [DOI] [PubMed] [Google Scholar]
- 36. Fitzgerald KA, O’Neill LA (1999) Characterization of CD44 induction by IL‐1: a critical role for egr‐1. J. Immunol. 162, 4920–4927. [PubMed] [Google Scholar]
- 37. Mishra JP, Mishra S, Gee K, Kumar A (2005) Differential involvement of calmodulin‐dependent protein kinase II‐activated AP‐1 and c‐jun N‐terminal kinase‐activated EGR‐1 signaling pathways in tumor necrosis factor‐alpha and lipopolysaccharide‐induced CD44 expression in human monocytic cells. J. Biol. Chem. 280, 26825–26837. [DOI] [PubMed] [Google Scholar]