

Abstract
Abstract. Haemopoietic stem/progenitor cell (HSPC) development is regulated by extrinsic and intrinsic stimuli. Extrinsic modulators include growth factors and cell adhesion molecules, whereas intrinsic regulation is achieved with many transcription factor families, of which the HOX gene products are known to be important in haemopoiesis. Umbilical cord blood CD133+ HSPC proliferation potential was tested in liquid culture with ‘TPOFLK’ (thrombopoietin, flt‐3 ligand and c‐kit ligand, promoting HSPC survival and self‐renewal), in comparison to ‘K36EG’ (c‐kit‐ligand, interleukins‐3 and ‐6, erythropoietin and granulocyte colony‐stimulating factor, inducing haemopoietic differentiation). TPOFLK induced a higher CD133+ HSPC proliferation (up to 60‐fold more, at week 8) and maintained a higher frequency of the primitive colony‐forming cells than K36EG. Quantitative polymerase chain reaction analysis revealed opposite expression patterns for specific HOX genes in expanding cord blood CD133+ HSPC. After 8 weeks in liquid culture, TPOFLK increased the expression of HOX B3, B4 and A9 (associated with uncommitted HSPC) and reduced the expression of HOX B8 and A10 (expressed in committed myeloid cells) when compared to K36EG. These results suggest that TPOFLK induces CD133+ HSPC proliferation, self‐renewal and maintenance, up‐regulation of HOX B3, B4 and A9 and down‐regulation of HOX B8 and A10 gene expression.
INTRODUCTION
Haemopoiesis is characterized by a high cell turnover requiring the continuous self‐renewal of a primitive stem cell pool as well as controlled differentiation and development of their progenitors into a diverse array of mature blood cells. Such a developmental system calls for complex complementary extrinsic (growth factors and cell adhesion molecules) and intrinsic (signal transduction and transcription factor) stimuli. Dysregulation of one or several of these pathways may lead to impaired haemopoiesis (Tenen et al. 1997).
CD133 is a highly hydrophobic 865 amino acid (120 kDa) glycoprotein. It has five transmembrane domains with eight N‐linked glycosylation sites. CD133 is the human orthologue of the murine prominin 1 and as such was recently described as being a member of the prominin family (Miraglia et al. 1997; Yin et al. 1997; Corbeil et al. 1998; Fargeas et al. 2003). CD133 expression is mainly restricted to a small haemopoietic stem/progenitor cell (HSPC) subset of CD34+ HSPC as well as primitive endothelial cells and several non‐haemopoietic tissues, including the foetal brain, pancreas and kidney (Miraglia et al. 1997; Corbeil et al. 1999; Uchida et al. 2000). Several studies by our group and others have shown that CD133 expression on HSPC precedes that of CD34 on developing HSPC (Bhatia 2001; McGuckin et al. 2003b). CD133 cell function is unknown but the presence of a leucine‐zipper motif (inclined to dimerization) on the second extracellular loop as well as its selective membrane location to plasma membrane protrusions suggests that it could mediate targeted cell–cell communication (McGuckin et al. 1999; Corbeil et al. 2000).
Haemopoietic reconstitution of the blood system relies on the transplantation of sufficient numbers of uncommitted HSPC to recipients to ensure both short‐ and long‐term bone engraftment (Gilmore et al. 2000). One of the main limiting factors of this procedure is often the number of HSPC available from the source, not least when using umbilical cord blood units (Gabutti et al. 1993; Emerson 1996; Gluckman 2000). Our group and others have previously demonstrated the advantage of using cord blood CD133+ HSPC which encompass more uncommitted HSPC than CD34+ counterparts (de Wynter et al. 1998; Forraz et al. 2002a; McGuckin et al. 2003a). Ex vivo expansion technology may circumvent cord blood HSPC limitations and extend their use to adult haemopoietic stem cell transplantation, because it aims to generate in vitro HSPC amplification with growth factors, whilst limiting proliferation‐induced differentiation (Aglietta et al. 1998).
Although the ideal growth factor combination optimized for true stem cell expansion remains to be identified, our group previously reported on the thrombopoietin, flt3‐ligand and c‐kit ligand (TPOFLK) promoting self‐renewal whilst limiting differentiation (2002b, 2004; McGuckin et al. 2004). Further to extrinsic cytokine stimulation, we believe that HSPC ex vivo expansion may benefit from taking into account the intrinsic activation of transcription factors which can modulate HSPC fate both in vivo and in vitro (Zhu & Emerson 2002). For instance, ex vivo expansion of early uncommitted HSPC could be achieved with: activation of P glycoprotein pump genes (Bunting et al. 2000); the exogenous excess addition of all‐trans retinoic acid; soluble Sonic Hedgehog protein (Purton et al. 2000); or the stimulation of Notch family of transcription factors (Varnum‐Finney et al. 2000).
Amongst predominant intrinsic transcriptional pathways, the HOX gene family of transcription factors has been described by several studies as a key group of control genes for regulation (and dysregulation) of the haemopoietic system (Sauvageau et al. 1997; Antonchuk et al. 2002; Krosl et al. 2003). The Hox B4 protein was notably reported as a strong, positive regulator of HSPC self‐renewal (Antonchuk et al. 2002; Kyba et al. 2002). HOX B4 gene over‐expression could specifically enhance the rate of HSPC expansion without impairing normal differentiation or functional repopulation potential and did not induce neoplastic transformation (Sauvageau et al. 1995; Thorsteinsdottir et al. 1999; 2001, 2002). Blocking HOX gene function by the use of anti‐sense oligonucleotides or gene knock‐out, perturbs a number of distinct haemopoietic events. For instance, myeloid, erythroid and lymphoid haemopoiesis are all defective in HOX A9 knock‐out mice (Lawrence et al. 1997), and the anti‐sense ablation of HOX B5, HOX B6, HOX B7 (Takeshita et al. 1993), or HOX A5 (Fuller et al. 1999) blocks erythroid differentiation. Mechanisms modulating the integration between extrinsic growth factor stimulation and intrinsic Hox transcription factors during HSPC ex vivo expansion remain to be further characterized and may help elucidate HSPC ‘proliferation–differentiation’ versus ‘maintenance–self‐renewal’ roles.
A small‐array real‐time HOX (SMART‐HOX) quantitative polymerase chain reaction (Q‐PCR) methodology enabling sensitive analysis of changes in HOX gene expression in human haemopoietic cells has previously been reported and validated (Thompson et al. 2003). This method has been utilized in this study to quantify comparatively the variations in:
-
1
HOX B3, HOX B4 and HOX A9, which are up‐regulated in early, uncommitted HSPC and are down‐regulated during differentiation (1994, 1995; Shimamoto et al. 1998; Dorsam et al. 2003); and
-
2
HOX B8 and HOX A10 gene expression previously linked with committed myeloid cells (Krishnaraju et al. 1997; Bjornsson et al. 2001; Buske et al. 2001; Taghon et al. 2002).
Here, the study investigated a possible correlation between ex vivo expansion kinetics of cord blood CD133+ cells when stimulated by two different growth factor combinations:
-
1
K36EG [c‐kit ligand, interleukin (IL)‐3, IL‐6, erythropoietin and granulocyte colony‐stimulating factor (G‐CSF)] known to recruit cells into proliferation and differentiation (Aglietta et al. 1998; Forraz et al. 2002a; McGuckin et al. 2004); and
-
2
TPOFLK (thrombopoietin, flt‐3 ligand and c‐kit ligand) the synergism of which was reported to amplify early HSPC populations efficiently (Gilmore et al. 2000; McGuckin et al. 2004) and the expression of specific HOX genes (HOX B3, HOX B4, HOX A9, HOX A10 and HOX B8) in expanded cord blood CD133+ cells.
MATERIALS AND METHODS
CD133+ cell immunomagnetic cell separation
Human umbilical cord blood specimens were collected from full‐term, third‐stage labour deliveries after elective Caesarean sections from informed and haematologically normal volunteers in accordance with the South Thames Local Research Ethics Committee. Cord blood mononuclear cells (MNC) were separated through a density gradient method using research grade Ficoll–Paque solution (d: 1.077 g/cm3, Pharmacia Biotech, Uppsala, Sweden). CD133+/– cells were obtained from MNC after immunomagnetic separation using the CD133 mini‐magnetic activated cell sorting (MACS) selection kit (Miltenyi Biotec, Bergish Gladbach, Germany) following the manufacturer's instructions as previously reported (Forraz et al. 2002a; Whiting et al. 2003; McGuckin et al. 2004).
Immunophenotyping and flow cytometry
Cells were incubated in human gammaglobulins (20 min, 4 °C, 2% in phosphate‐buffered saline, Sigma Aldrich, Poole, UK) to block non‐specific Fc receptors. Cells were subsequently labelled (30 min, 4 °C) with monoclonal mouse anti‐human phycoerythrin‐conjugated CD133 (Miltenyi Biotec) and peridinin chlorophyll protein‐conjugated CD34 antibodies (BD‐Pharmingen, San Diego, CA, USA). Following appropriate washing procedures cells were finally fixed in paraformaldehyde (1%, BDH, Poole, UK). Fluorescent events were acquired on a Becton Dickinson FACScan flow cytometer with cellquest software prior to analysis with winmdi software.
Cord blood LinNeg expansion in TPOFLK‐stimulated liquid cultures
Cord blood CD133+ cells were grown in duplicate in 9‐cm2 tissue culture slide flasks (Nunc, Hereford, UK) at a concentration of 2.7 × 104 cells/ml of Iscove's‐modified Dulbecco's medium (IMDM, Life Technologies, Paisley, UK) supplemented with 10% foetal calf serum (PAA Laboratories, Yeoville, UK), gentamicin (50 µg/ml; Life Technologies) and either the TPOFLK cytokines mix: thrombopoietin (10 ng/ml), flt3‐ligand (50 ng/ml) and c‐kit‐ligand (20 ng/ml) or the K36EG cytokines mix: c‐kit‐ligand (20 ng/ml), IL‐3 (50 ng/ml), IL‐6 (20 ng/m), erythropoietin (6 U/ml), G‐CSF (10 ng/ml).
CD133+ cells were cultured at 37 °C, 5% CO2 in a humidified atmosphere and counted weekly. Aliquots were removed for clonogenic assays. All cytokines were purchased from R & D Systems Ltd, Abingdon, UK.
Clonogenic assays in methyl cellulose
Cell aliquots were seeded in 200 µl IMDM, 10% foetal calf serum and 800 µl Methocult solution (H4230, Stem Cell Technologies, London, UK), supplemented with erythropoietin (6 U/mL), c‐kit‐ligand (20 ng/ml), IL‐3 (50 ng/ml), IL‐6 (20 ng/ml), G‐CSF (10 ng/ml), and grown for 14 days at 37 °C, 5% CO2 in a humidified atmosphere, prior to scoring for colony‐forming cells (CFC). All cytokines were purchased from R & D Systems Ltd.
RNA isolation, cDNA preparation and Q‐PCR
Total RNA and cDNA were prepared as previously reported (Thompson et al. 2003) using standard reagents: Trizol, murine Maloney leukaemia virus native reverse transcriptase, random primers (all from Invitrogen, Paisley, UK). As a result of the small numbers of cells, glycogen (Invitrogen) was used as an inert carrier molecule for RNA following the manufacturer's instructions. Real‐time Q‐PCR was carried out using TaqMan probe‐based chemistry (Applied Biosystems, Foster City, USA). The 5′‐reporters for the HOX genes and endogenous controls (18SrRNA) were 6‐carboxyfluorescein (FAM) and VIC™ (Applied Biosystems), respectively. Relative fold expression based on the ΔΔC T method (reviewed by Ginzinger 2002) was corrected for RNA equivalents using 18S rRNA C T (threshold cycle) values.
Statistical analysis
When applicable, results were analysed statistically and expressed as mean ± SEM from experiments performed in at least duplicate. Statistical significance was calculated by the Student's t‐test.
RESULTS
CD133+ cell purification
Cord blood MNC immunolabelled against CD133 or CD34 antigens contained a significantly lower frequency of CD133+ cells [0.39 ± 0.04% (mean ± SEM); range 0.19–0.70] than CD34+ cells (0.59 ± 0.05%; range 0.21–1.36; P = 0.002, n = 31).
However, throughout this study, the mean purity of cord‐blood‐derived CD133+ immunomagnetically selected cells was 94.40 ± 1.02% (range 84.51–97.15, n = 10) and CD133+ cells mean recovery from MNC was 74.80 ± 3.76% (range 60.36–86.64, n = 10). Figure 1 represents CD133 and CD34 antigen percentage distribution within MNC, CD133− and CD133+ harvested cells, respectively.
Figure 1.
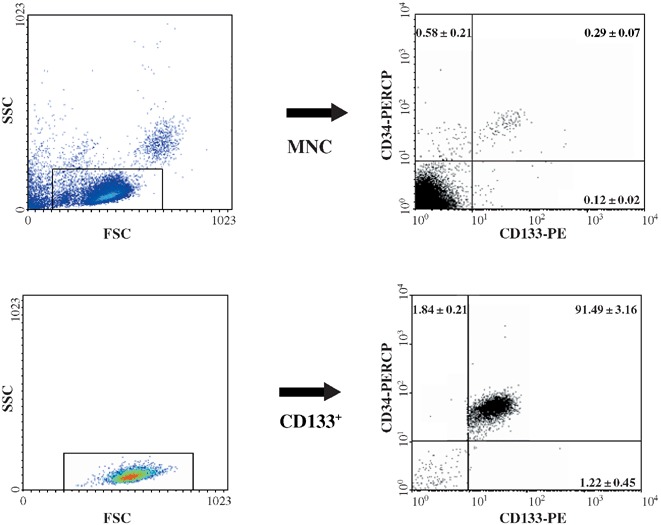
CD133 and CD34 antigen percentage distribution on cord blood MNC and CD133+ cells. The left panels show forward scattered (FSC) versus side scattered (SSC) flow cytometric density plots representing the size and internal complexity/granularity of the three cell populations. The rectangles highlight strategic regions enabling electronic gating of cells with ‘blast’ morphology (medium size, large nucleus, little cytoplasm). This further allowed CD133 and CD34 co‐distribution analysis for each cell population. CD133 and CD34 were labelled with mouse anti‐human specific monoclonal antibodies labelled with phycoerythrin and peridinin chlorophyll protein fluorochromes, respectively. Flow cytometric plots are from a representative example, whereas data are expressed as mean percentage (± SEM) distribution of (clockwise) CD133− CD34+ CD133+ CD34+ and CD133+ CD34− cell subsets of 10 distinct experiments.
TPOFLK induced high proliferation of CB CD133+ cells in 8‐week expansion liquid culture assay
TPOFLK and K36EG cytokine mixes were compared for their ability to expand cord blood CD133+ cells over 8 weeks in vitro. In six distinct experiments, all performed in duplicate, TPOFLK‐stimulated liquid cultures induced a higher cumulative total viable cell fold increase than the K36EG cytokine mix. After 8 weeks, the optimal cumulative cell fold increase was 4.68 × 108 ± 1.51 × 108 in TPOFLK and 7.96 × 106 ± 1.71 × 106 in K36EG‐supplemented cultures (n = 6) (Fig. 2). Interestingly, this difference in expansion was statistically significant from week 3 to week 8 (P < 0.05) of liquid culture. TPOFLK thus appeared to be a very potent stimulus for sustained cord blood CD133+ cell long‐term proliferation.
Figure 2.
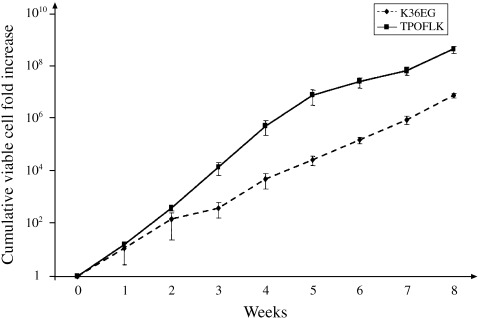
Comparison of cord blood CD133+ cells. Eight‐week expansion in liquid culture stimulated either by TPOFLK or K36EG. TPOFLK‐stimulated liquid cultures induced a higher cumulative total viable haemopoietic cell fold increase from CD133+ cells than K36EG cytokine mix (P < 0.05 from week 3 to week 8). Results are expressed as mean ± SEM of six distinct experiments performed in duplicates.
TPOFLK expansion of cord blood CD133+ cells maintained more primitive CFC than K36EG
After isolation, CD133+ cells contained significantly increased levels of CFC with 547 ± 82 CFC per 104 CD133+ cells compared to the more heterogeneous MNC (5 ± 2 CFC per 104 MNC, n = 6, P < 0.01). Comparative analysis of CFC frequency from expanded CD133+ cells revealed that TPOFLK‐supplemented liquid culture systems maintained more CFC than K36EG supplemented cultures at weeks 4, 6 and 8 (P < 0.05) (Fig. 3). The optimal window for CFC output appeared to be between week 2 and week 6 where TPOFLK‐expanded CD133+ cells produced between 23 ± 5 CFC (weeks 2 and 4) and 25 ± 6 CFC (week 6) per 104 seeded cells. CFC production in TPOFLK‐stimulated liquid cultures significantly decreased at week 8 (6 ± 1 CFC; P < 0.05). TPOFLK cytokine mix therefore induced higher CD133+ cell proliferation whilst expanding more primitive CFC than K36EG.
Figure 3.
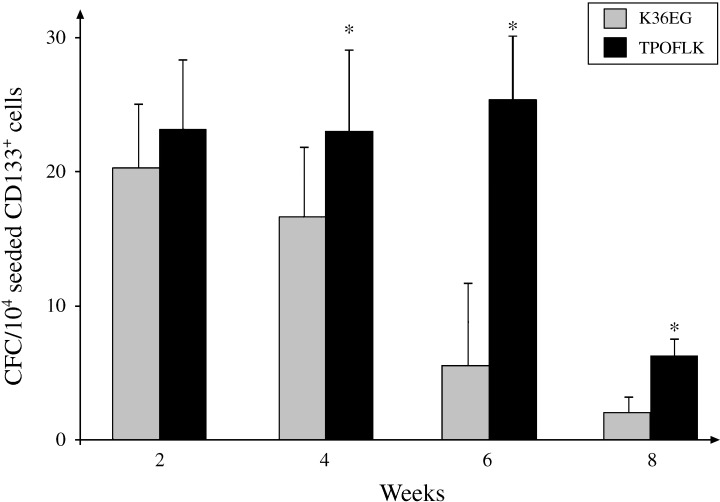
Comparison of 8‐week cord blood CD133+ cell TPOFLK‐ or K36EG‐stimulated expansion in liquid culture clonogenic assay. Methylcellulose‐based clonogenic assays were performed every 2 weeks from aliquots of expanding CD133+ cells under TPOLFK or K36EG stimuli. This permitted frequency calculation of colony‐forming‐cells (CFC) per 104 CD133+ expanding cells. TPOFLK maintained a higher CFC frequency over 8 weeks than K36EG. This was particularly significant at weeks 4, 6 and 8 (*P < 0.01). Results are expressed as mean ± SEM of six distinct experiments performed in duplicates.
TPOFLK and K36EG differentially modulated HOX gene expression in expanding cord blood CD133+ cells
Real‐time Q‐PCR allowed comparative analysis of HOX B3, HOX B4, HOX B8, HOX A9 and HOX A10 gene expression in expanded CD133+ cells in triplicate. Using this method, we compared, from baseline (freshly isolated cord blood CD133+ cells), the fold difference for expression of the above genes in expanded CD133+ cells in either TPOFLK‐ or K36EG‐stimulated liquid cultures.
Such comparison was analysed between week 4 and week 8, during which a significant difference in CD133+ cell proliferation between TPOFLK‐ and K36EG‐treated liquid culture systems was observed (P < 0.05). Furthermore, as a result of the limited number of CD133+ cells available per cord blood unit a compromise had to be reached in the usage of expanding cells for functional assays and gene expression profiling. TPOFLK and K36EG caused different effects on the expression of HOX B3, HOX B4, HOX A9 (associated with proliferating HSPC), and HOX B8 and HOX A10 (associated with committed myeloid progenitors) at week 4 and week 8 of expansion (Fig. 4). In both TPOFLK‐ and K36EG stimulated liquid cultures, expanding CD133+ cells, HOX B3, HOX B4, HOX B8 and HOX A10 gene expression levels were similar after 4 weeks. HOX A9 gene expression at week 4 was slightly higher in K36EG‐expanded CD133+ cells when compared to TPOFLK‐expanded CD133+ cells. From week 4 in TPOFLK‐stimulated CD133+ cells, HOX B3, HOX B4 and HOX A9 gene expression was down‐regulated until week 6 and then progressively up‐regulated to levels between 10 and 100 times higher than in K36EG‐expanded CD133+ cells. Further to this, K36EG induced greater up‐regulation of HOX B8 and HOX A10 gene expression in these genes at week 8 of culture. Taken together, these data suggest a differential effect on HOX gene expression in CD133+ cells expanded with TPOFLK compared to K36EG.
Figure 4.
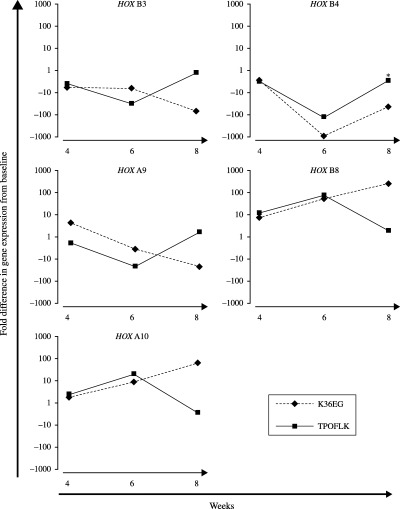
Quantitative RT‐PCR analysis of HOX gene expression in TPOFLK‐ and K36EG‐stimulated cord blood CD133+ cells. The effect of TPOFLK and K36EG stimulations on the expression of several HOX genes in expanding CD133+ cell was analysed by quantitative RT‐PCR by measuring the relative fold difference with day 0 HOX gene expression. At week 8 TPOFLK induced more expression of HOX B3, HOX B4 and HOX A9 genes known to be preferentially expressed in primitive HSPC than K36EG, which favoured expression of HOX B8 and HOX A10 associated with commitment to the myeloid lineage.
DISCUSSION
Activation of transcriptional intrinsic pathways to complement growth factor‐stimulated systems is the focus of growing scientific interest and is important for the development of novel ex vivo HSPC expansion protocols. We and others have shown that CD133 is a good alternative marker for CD34 for HSPC selection when establishing functional assay standards. A majority of CD133+ cells co‐express CD34 but encompass earlier and less committed HSPC than their CD34+ counterparts (de Wynter et al. 1998; Bhatia 2001; McGuckin et al. 2003a).
Previous reports from our group notably demonstrated that CD133+ HSPC were able to produce pan‐lineage haemopoietic differentiation when grown in K36EG cytokine mix (Forraz et al. 2002a; McGuckin et al. 2004). TPOFLK was reported to enhance HSPC self‐renewal and to optimize cell survival by inhibiting apoptosis (Luens et al. 1998; Kohler et al. 1999; McGuckin et al. 2004).
Our 8‐week CD133+ cell expansion assay results are in accordance with these data. Even after 1 week in culture, TPOFLK induced more cord blood CD133+ cell proliferation than K36EG, although such a difference was not initially significant. However, TPOFLK supplementation resulted in a 60‐fold more CD133+ cumulative cell fold increase than the K36EG mix after 8 weeks. TPOFLK did not only produce more HSPC proliferation but cells retained a greater proliferative potential (CFC output) than K36EG, suggesting maintenance at an earlier stage of development. TPOFLK action proved not only to induce high HSPC proliferation whilst maintaining self‐renewal, but TPOFLK extrinsic functional signalling correlated with HOX transcription factors intrinsic transcriptional regulation.
Restricted amounts of RNA available from expanding CD133+ cells limited SMART‐HOX PCR analysis to five carefully selected HOX genes.
In both liquid culture systems (TPOFLK and K36EG), HOX B3, HOX B4 and HOX A9 gene expression levels were below those of baseline after 4 weeks of culture, which may be explained by the impact of artificial expansion methods on the cells’ gene expression profile. However, after 8 weeks, the higher cell proliferation and CFC output from CD133+ cells caused by TPOFLK, was concomitant with a higher gene expression (between 10‐ and 100‐fold difference and back to baseline level) of HOX B3, HOX B4 and HOX A9 than for cells stimulated by K36EG. Interestingly, HOX B3, HOX B4 and HOX A9 gene expression was previously associated with early HSPC (Lawrence et al. 1996; Van Oostveen et al. 1999). Such patterns correlated with down‐regulation of HOX B8 and HOX A10 gene expression (conventionally linked with committed myeloid progenitors; Krishnaraju et al. 1997; Taghon et al. 2002) in cells at week 8 under K36EG stimulation. In this 8‐week CD133+ cell expansion study, TPOFLK maintained a higher transcriptional activity of HOX B3, HOX B4 and HOX A9 genes than K36EG, which favoured HOX B8 and HOX A10 expression. Interestingly, ectopic HOX B8 expression was reported to induce maintenance of myeloid progenitor (Perkins & Cory 1993) and HOX A10 expression is known to be expressed in committed myeloid cells and to be important in regulating haemopoietic lineage determination (Thorsteinsdottir et al. 1997; Taghon et al. 2002).
External HSPC stimulation by cytokines activates a series of signal transduction events mediating intrinsic activation of complex transcription factor pathways (Shivdasani & Orkin 1996). The differential expression of the particular HOX genes investigated here may be linked with different cell cycle dynamics induced by K36EG and TPOFLK, respectively. Quesenberry and colleagues suggested that HSPC could respond differently to external stimuli at diverse points in the cell cycle causing a shift in chromatin and variation in gene expression (Quesenberry et al. 2002). HOX transcription factors have been reported as master regulators of the haemopoietic system (Magli et al. 1997). A study by Sauvageau et al. (1994) established that at least 22 of the 39 known HOX genes were expressed at various levels in five different CD34+ human bone marrow subsets. HOX gene products were later confirmed to play a central role in the proliferation and the differentiation of early HSPC (Giampaolo et al. 1994; Sauvageau et al. 1997). Upon growth factor stimulation, Giampaolo et al. (1994) noticed rapid up‐regulation of HOX B3 expression followed by a delayed up‐regulation of HOX B4. This may be the result of HOX B3 directly activating downstream HOX B4. Expression of HOX A9 followed similar patterns to HOX B3 and HOX B4 gene expression. HOX A9 was previously linked by several studies to influence general haemopoiesis including maintenance of the erythroid, myeloid and lymphoid lineages (Lawrence et al. 1997). Hox A9 transcription factor's mode of action, however, could have more to do with HSPC proliferation modulation than their differentiation per se (Izon et al. 1998; Lawrence et al. 1999).
From week 6 in TPOFLK‐supplemented CD133+ cells, early HSPC‐associated HOX gene (HOX B3, HOX B4 and HOX A9) expression appeared to be rapidly up‐regulated (although those levels were still below those in baseline freshly isolated CD133+ cells) which correlated with the highest CFC output. Interestingly, the reduction in CFC output at week 8 of TPOFLK‐stimulated culture may account for the maintenance of primitive HSPC as it was concomitant with a significant down‐regulation of HOX B8 and HOX A10 (preferably expressed in myeloid progenitors and precursors). Taken together these data thus confirmed TPOFLK's adequacy for HSPC expansion. It may be explained by converging studies demonstrating that thrombopoietin's activation of the p38/MAPK signal transduction pathway resulted in activation of upstream stimulation factor 1 and 2 (USF1 and USF2) transcription factors known to bind to the HOX B4 gene promoter region triggering its transcription (Giannola et al. 2000; Galibert et al. 2001; Kirito et al. 2003).
Work of this nature described here is difficult because of the extreme rarity of the CD133+ HSPC population in cord blood. Furthermore, as a result o f the limited number of colonies that are generated from liquid delta cultures, a compromise must be reached between functional characterization and genetic expression analysis. However, this study is important because, although little is known of the potential of immature cord blood populations beyond haemopoietic use, reports are emerging, including from our laboratory, concerning the potential of cord blood for multiple non‐haemopoietic tissue regeneration (Forraz et al. 2004).
In conclusion, TPOFLK demonstrated potential for cord blood CD133+ HSPC ex vivo expansion inducing slow proliferation, maintenance of early HSPC as well as promoting HSPC survival status. TPOFLK notably induced the maintenance of primitive CFC possibly by modulating transcriptional activation of HOX B3, HOX B4 and HOX A9 genes. Taken together this study suggested that HSPC ex vivo expansion strategy development could further evolve by simultaneously optimizing cytokine synergistic action as well as by better understanding the intrinsic transcriptional pathways (like the HOX family) which balance HSPC self‐renewal and differentiation.
REFERENCES
- Aglietta M, Bertolini F, Carlo‐Stella C, De Vincentiis A, Lanata L, Lemoli RM, Olivieri A, Siena S, Zanon P, Tura S (1998) Ex vivo expansion of hematopoietic cells and their clinical use. Haematologica 83, 824. [PubMed] [Google Scholar]
- Antonchuk J, Sauvageau G, Humphries RK (2001) HOXB4 overexpression mediates very rapid stem cell regeneration and competitive hematopoietic repopulation. Exp. Hematol. 29, 1125. [DOI] [PubMed] [Google Scholar]
- Antonchuk J, Sauvageau G, Humphries RK (2002) HOXB4‐induced expansion of adult hematopoietic stem cells ex vivo. Cell 109, 39. [DOI] [PubMed] [Google Scholar]
- Bhatia M (2001) AC133 expression in human stem cells. Leukemia 15, 1685. [DOI] [PubMed] [Google Scholar]
- Bjornsson JM, Andersson E, Lundstrom P, Larsson N, Xu X, Repetowska E, Humphries RK, Karlsson S (2001) Proliferation of primitive myeloid progenitors can be reversibly induced by HOXA10. Blood 98, 3301. [DOI] [PubMed] [Google Scholar]
- Bunting KD, Zhou S, Lu T, Sorrentino BP (2000) Enforced P‐glycoprotein pump function in murine bone marrow cells results in expansion of side population stem cells in vitro and repopulating cells in vivo. Blood 96, 902. [PubMed] [Google Scholar]
- Buske C, Feuring‐Buske M, Antonchuk J, Rosten P, Hogge DE, Eaves CJ, Humphries RK (2001) Overexpression of HOXA10 perturbs human lymphomyelopoiesis in vitro and in vivo. Blood 97, 2286. [DOI] [PubMed] [Google Scholar]
- Corbeil D, Roper K, Weigmann A, Huttner WB (1998) AC133 hematopoietic stem cell antigen: human homologue of mouse kidney prominin or distinct member of a novel protein family? Blood 91, 2625–2626. [PubMed] [Google Scholar]
- Corbeil D, Roper K, Hannah MJ, Hellwig A, Huttner WB (1999) Selective localization of the polytopic membrane protein prominin in microvilli of epithelial cells – a combination of apical sorting and retention in plasma membrane protrusions. J. Cell Sci. 112, 1023–1033. [DOI] [PubMed] [Google Scholar]
- Corbeil D, Roper K, Hellwig A, Tavian M, Miraglia S, Watt SM, Simmons PJ, Peault B, Buck DW, Huttner WB (2000) The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J. Biol. Chem. 275, 5512–5520. [DOI] [PubMed] [Google Scholar]
- Dorsam ST, Ferrell CM, Dorsam GP, Derynck MK, Vijapurkar U, Khodabakhsh D, Pau B, Bernstein H, Haqq CM, Largman C, Lawrence HJ (2003) The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood in press. [DOI] [PubMed] [Google Scholar]
- Emerson SG (1996) Ex vivo expansion of hematopoietic precursors, progenitors, and stem cells: the next generation of cellular therapeutics. Blood 87, 3082. [PubMed] [Google Scholar]
- Fargeas CA, Florek M, Huttner WB, Corbeil D (2003) Characterization of prominin‐2, a new member of the prominin family of pentaspan membrane glycoproteins. J. Biol. Chem. 278, 8586–8596. [DOI] [PubMed] [Google Scholar]
- Forraz N, Pettengell R, Deglesne PA, Mcguckin CP (2002a) AC133+ umbilical cord blood progenitors demonstrate rapid self‐renewal and low apoptosis. Br. J. Haematol. 119, 516. [DOI] [PubMed] [Google Scholar]
- Forraz N, Pettengell R, Mcguckin CP (2002b) Haemopoietic and neuroglial progenitors are promoted during cord blood ex vivo expansion. Br. J. Haematol. 119, 888. [DOI] [PubMed] [Google Scholar]
- Forraz N, Pettengell R, Mcguckin CP (2004) Characterization of a lineage‐negative stem‐progenitor cell population optimized for ex vivo expansion and enriched for LTC‐IC. Stem Cells 22, 100. [DOI] [PubMed] [Google Scholar]
- Fuller JF, Mcadara J, Yaron Y, Sakaguchi M, Fraser JK, Gasson JC (1999) Characterization of HOX gene expression during myelopoiesis: role of HOX A5 in lineage commitment and maturation. Blood 93, 3391. [PubMed] [Google Scholar]
- Gabutti V, Timeus F, Ramenghi U, Crescenzio N, Marranca D, Miniero R, Cornaglia G, Bagnara GP (1993) Expansion of cord blood progenitors and use for hemopoietic reconstitution. Stem Cells 11(Suppl. 2), 105. [DOI] [PubMed] [Google Scholar]
- Galibert MD, Carreira S, Goding CR (2001) The Usf‐1 transcription factor is a novel target for the stress‐responsive p38 kinase and mediates UV‐induced tyrosinase expression. EMBO J. 20, 5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampaolo A, Sterpetti P, Bulgarini D, Samoggia P, Pelosi E, Valtieri M, Peschle C (1994) Key functional role and lineage‐specific expression of selected HOXB genes in purified hematopoietic progenitor differentiation. Blood 84, 3637. [PubMed] [Google Scholar]
- Giannola DM, Shlomchik WD, Jegathesan M, Liebowitz D, Abrams CS, Kadesch T, Dancis A, Emerson SG (2000) Hematopoietic expression of HOXB4 is regulated in normal and leukemic stem cells through transcriptional activation of the HOXB4 promoter by upstream stimulating factor (USF)‐1 and USF‐2. J. Exp. Med. 192, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore GL, Depasquale DK, Lister J, Shadduck RK (2000) Ex‐vivo expansion of human umbilical cord blood CD34+ hemopoietic stem cells. Exp. Hematol. 28, 1297. [DOI] [PubMed] [Google Scholar]
- Ginzinger DG (2002) Gene quantification using real‐time quantitative PCR: an emerging technology hits the mainstream. Exp. Hematol. 30, 503. [DOI] [PubMed] [Google Scholar]
- Gluckman E (2000) Current status of umbilical cord blood haemopoietic stem cell transplantation. Exp. Hematol. 28, 1197. [DOI] [PubMed] [Google Scholar]
- Izon DJ, Rozenfeld S, Fong ST, Komuves L, Largman C, Lawrence HJ (1998) Loss of function of the homeobox gene Hoxa‐9 perturbs early T‐cell development and induces apoptosis in primitive thymocytes. Blood 92, 383. [PubMed] [Google Scholar]
- Kirito K, Fox N, Kaushansky K (2003) Thrombopoietin stimulates HoxB4 expression: an explanation for the favorable effects of TPO on hematopoietic stem cells. Blood 102, 3172. [DOI] [PubMed] [Google Scholar]
- Kohler T, Plettig R, Wetzstein W, Schaffer B, Ordemann R, Nagels HO, Ehninger G, Bornhauser M (1999) Defining optimum conditions for the ex vivo expansion of human umbilical cord blood cells. Influences of progenitor enrichment, interference with feeder layers, early‐acting cytokines and agitation of culture vessels. Stem Cells 17, 19. [DOI] [PubMed] [Google Scholar]
- Krishnaraju K, Hoffman B, Liebermann DA (1997) Lineage‐specific regulation of hematopoiesis by HOX‐B8 (HOX‐2.4): inhibition of granulocytic differentiation and potentiation of monocytic differentiation. Blood 90, 1840. [PubMed] [Google Scholar]
- Krosl J, Beslu N, Mayotte N, Humphries RK, Sauvageau G (2003) The competitive nature of HOXB4‐transduced HSC is limited by PBX1: the generation of ultra‐competitive stem cells retaining full differentiation potential. Immunity 18, 561. [DOI] [PubMed] [Google Scholar]
- Kyba M, Perlingeiro RC, Daley GQ (2002) HoxB4 confers definitive lymphoid‐myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell 109, 29. [DOI] [PubMed] [Google Scholar]
- Lawrence HJ, Sauvageau G, Humphries RK, Largman C (1996) The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells 14, 281. [DOI] [PubMed] [Google Scholar]
- Lawrence HJ, Helgason CD, Sauvageau G, Fong S, Izon DJ, Humphries RK, Largman C (1997) Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood 89, 1922. [PubMed] [Google Scholar]
- Lawrence HJ, Rozenfeld S, Cruz C, Matsukuma K, Kwong A, Komuves L, Buchberg AM, Largman C (1999) Frequent co‐expression of the HOXA9 and MEIS1 homeobox genes in human myeloid leukemias. Leukemia 13, 1993. [DOI] [PubMed] [Google Scholar]
- Luens KM, Travis MA, Chen BP, Hill BL, Scollay R, Murray LJ (1998) Thrombopoietin, kit ligand, and flk2/flt3 ligand together induce increased numbers of primitive hematopoietic progenitors from human CD34+Thy‐1+Lin‐cells with preserved ability to engraft SCID‐hu bone. Blood 91, 1206. [PubMed] [Google Scholar]
- Magli MC, Largman C, Lawrence HJ (1997) Effects of HOX homeobox genes in blood cell differentiation. J. Cell. Physiol. 173, 168. [DOI] [PubMed] [Google Scholar]
- Mcguckin CP, Pettengell R, Martin F, Jones K, Gordon‐Smith EC, Pearce D (1999) Is AC133 involved in direct cell to cell communication? Exp. Hematol. 28, 160. [Google Scholar]
- Mcguckin CP, Pearce D, Forraz N, Tooze JA, Watt SM, Pettengell R (2003a) Multiparametric analysis of immature cell populations in umbilical cord blood and bone marrow. Eur. J. Haematol. 71, 341. [DOI] [PubMed] [Google Scholar]
- Mcguckin CP, Forraz N, Baradez MO, Lojo‐Rial C, Wertheim D, Whiting K, Watt SM, Pettengell R (2003b) Colocalization analysis of sialomucins CD34 and CD164. Stem Cells 21, 162. [DOI] [PubMed] [Google Scholar]
- Mcguckin CP, Forraz N, Allouard Q, Pettengell R (2004) Umbilical cord blood stem cells can expand hematopoietic and neuroglial progenitors in vitro . Exp. Cell Res. 295, 350–359. [DOI] [PubMed] [Google Scholar]
- Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW (1997) A novel five‐transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 90, 5013–5021. [PubMed] [Google Scholar]
- Van Oostveen JW, Bijl JJ, Raaphorst FM, Walboomers JJM, Meijer CJLM (1999) The role of homeobox genes in normal hematopoiesis and hematological malignancies. Leukemia 13, 1675. [DOI] [PubMed] [Google Scholar]
- Perkins AC, Cory S (1993) Conditional immortalization of mouse myelomonocytic, megakaryocytic and mast cell progenitors by the Hox‐2.4 homeobox gene. EMBO J. 12, 3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton LE, Bernstein ID, Collins SJ (2000) All‐trans retinoic acid enhances the long‐term repopulating activity of cultured hematopoietic stem cells. Blood 95, 470. [PubMed] [Google Scholar]
- Quesenberry PJ, Colvin GA, Lambert JF (2002) The chiaroscuro stem cell: a unified stem cell theory. Blood 100, 4266. [DOI] [PubMed] [Google Scholar]
- Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, Largman C, Lawrence HJ, Humphries RK (1994) Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc. Natl Acad. Sci. USA 91, 12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau G, Thorsteinsdottir U, Eaves CJ, Lawrence HJ, Largman C, Lansdorp PM, Humphries RK (1995) Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 9, 1753. [DOI] [PubMed] [Google Scholar]
- Sauvageau G, Thorsteinsdottir U, Hough MR, Hugo P, Lawrence HJ, Largman C, Humphries RK (1997) Overexpression of HOXB3 in hematopoietic cells causes defective lymphoid development and progressive myeloproliferation. Immunity 6, 13. [DOI] [PubMed] [Google Scholar]
- Shimamoto T, Ohyashiki K, Toyama K, Takeshita K (1998) Homeobox genes in hematopoiesis and leukemogenesis. Int. J. Hematol. 67, 339. [DOI] [PubMed] [Google Scholar]
- Shivdasani RA, Orkin SH (1996) The transcriptional control of hematopoiesis 6. Blood 87, 4025. [PubMed] [Google Scholar]
- Taghon T, Stolz F, De Smedt M, Cnockaert M, Verhasselt B, Plum J, Leclercq G (2002) HOX‐A10 regulates hematopoietic lineage commitment: evidence for a monocyte‐specific transcription factor. Blood 99, 1197. [DOI] [PubMed] [Google Scholar]
- Takeshita K, Bollekens JA, Hijiya N, Ratajczak M, Ruddle FH, Gewirtz AM (1993) A homeobox gene of the Antennapedia class is required for human adult erythropoiesis. Proc. Natl Acad. Sci. USA 90, 3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenen DG, Hromas R, Licht JD, Zhang D‐E (1997) Transcription factors, normal myeloid development and leukaemia. Blood 90, 489. [PubMed] [Google Scholar]
- Thompson A, Quinn MF, Grimwade D, O'Neill CM, Ahmed MR, Grimes S, Mcmullin MF, Cotter F, Lappin TR (2003) Global down‐regulation of HOX gene expression in PML‐RARalpha + acute promyelocytic leukemia identified by small‐array real‐time PCR. Blood 101, 1558. [DOI] [PubMed] [Google Scholar]
- Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Landsorp PM, Lawrence HJ, Largman C, Humphries RK (1997) Overexpression of hoxa10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukaemia. Blood 17, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsteinsdottir U, Sauvageau G, Humphries RK (1999) Enhanced in vivo regenerative potential of HOXB4‐transduced hematopoietic stem cells with regulation of their pool size. Blood 94, 2605. [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL (2000) Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 97, 14720–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum‐Finney B, Xu L, Brashem‐Stein C, Nourigat C, Flowers D, Bakkour S, Pear WS, Bernstein ID (2000) Pluripotent, cytokine‐dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat. Med. 6, 1278. [DOI] [PubMed] [Google Scholar]
- Whiting KA, Mcguckin CP, Wertheim D, Pearce D, Pettengell R (2003) Three‐dimensional analysis of CD34 sialomucin distribution on cord blood and bone marrow. Br. J. Haematol. 122, 771. [DOI] [PubMed] [Google Scholar]
- De Wynter EA, Buck D, Hart C, Heywood R, Coutinho LH, Clayton A, Rafferty JA, Burt D, Guenechea G, Bueren JA, Gagen D, Fairbairn LJ, Lord BI, Testa NG (1998) CD34+AC133+ cells isolated from cord blood are highly enriched in long‐term culture‐initiating cells, NOD/SCID‐repopulating cells and dendritic cell progenitors. Stem Cells. 16, 387. [DOI] [PubMed] [Google Scholar]
- Yin AH, Miraglia S, Zanjani ED, Almeida‐Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW (1997) AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90, 5002–5012. [PubMed] [Google Scholar]
- Zhu J, Emerson SG (2002) Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene 21, 3295. [DOI] [PubMed] [Google Scholar]