

Abstract
Objectives: This study aims to identify new anti‐cancer agents from Cordyceps‐colonizing fungi, using an ecology‐based approach. It also aims to explore their anti‐cell proliferative mechanisms, and to evaluate their anti‐tumour effects in vivo.
Materials and methods: Extracts from Cordyceps‐colonizing fungi were tested on HeLa cells, and active extracts were separated to obtain anti‐tumour metabolites; their structures were elucidated by mass and nuclear magnetic resonance spectroscopy. Cell cycle analysis was evaluated using flow cytometry. Tumour formation assays were performed using C57BL/6J mice.
Results: Based on ecological considerations, the selected extracts were subjected to initial anti‐tumour screening. Bioassay‐guided fractionation of the active extract afforded two new epipolythiodioxopiperazines, named gliocladicillins A (1) and B (2). (A) 1 and B (2) inhibited growth of HeLa, HepG2 and MCF‐7 tumour cells. Further study demonstrated that both preparations arrested the cell cycle at G2/M phase in a dose‐dependent manner, and induced apoptosis through up‐regulation of expression of p53, p21, and cyclin B, and activation of caspases‐8, ‐9 and ‐3. These data imply that gliocladicillins A (1) and B (2) induce tumour cell apoptosis through both extrinsic and intrinsic pathways. In addition, in vivo studies showed that they displayed significant inhibitory effects on cell population growth of melanoma B16 cells imlanted into immunodeficient mice.
Conclusions: Gliocladicillins A (1) and B (2) are effective anti‐tumour agents in vitro and in vivo and should be further evaluated for their potential in clinical use.
Introduction
Cancer is a class of disease characterized by division of cells in a seemingly uncontrolled manner. According to the American Cancer Society, 7.6 million people died from cancer globally in 2007, and cancer is currently responsible for 25% of all deaths in the developed countries (1). Anti‐tumour agents continue to be the foundation of cancer therapies, although development of new anti‐cancer drugs remains a great challenge (2). Natural products have played a major role in cancer treatment. Of all available anti‐cancer agents from the 1940s to 2006, over 70% of them are either natural products themselves or are derivatives of natural products (3). For example, paclitaxel (Taxol), a well‐known drug approved for use in the treatment of ovarian and breast cancers, was originally isolated from the bark of the plant Taxus brevifolia (4, 5). Micro‐organisms are well‐known producers of bioactive natural products with diverse structures, from which many anti‐cancer drugs have been discovered (6); examples include anthracycline, bleomycin, actinomycin, mitomycin and aureolic acids (7, 8, 9). Apoptosis is a key biological pathway of cell death in multicellular organisms; lack of cell death in neoplasms is one of cancer’s major problems. Agents with apoptosis‐inducing effects are of high medical significance in cancer therapy (10). Some natural products have been found to regulate apoptotic pathways; for example, apoptolidin, isolated from Nocardiopsis sp. can selectively induce apoptosis in E1A‐transformed cells (11), and its remarkable selectivity makes it a lead compound in treatment of malignancy.
Cancer cells evolve in part by over‐riding normal cell cycle regulation, resulting in lack of control of cell proliferation, and its inhibition is a successful strategy for development of anti‐cancer drugs. Use of new chemotherapeutic agents derived from natural products with anti‐proliferative effects will be welcome in clinical situations (12). Examples are the fungal metabolite wortmannin (which can inhibit PI3 kinase‐mediated signal transduction pathways) (13), geldanamycin (a natural ansamycin, is a direct protein tyrosine kinase inhibitor) (14) and rapamycins (isolated from Streptomyces hygroscopicus) which can block cell cycle progression in T cells and B cells, as well as in osteosarcoma and rhabdomyosarcoma cell lines (15). Epothilones, from the myxobacterium Sorangium cellulosum are anti‐tumour agents that have a similar mode of action to Taxol, but offer advantages of greater water solubility and sample availability by successful use of fermentation technology. Using combinatorial methodology, more epothilones have been produced and evaluated for their anti‐tumour activity, leading to the discovery of 12,13‐desoxy‐ and 15‐aza‐epothilone B as most promising anti‐tumour agents (16). The search for natural products that can inhibit cell proliferation and induce tumour cell apoptosis continues to be an important approach to discovery of new anti‐cancer drugs.
Chemical ecology of natural products is at the very heart of such drug discovery (17) and application of fungal ecology for new bioactive natural products has proven to be an effective approach (18). Based on ecological considerations, we initiated chemical studies of certain fungi associated with Cordyceps sinensis (Berk.) Sacc. (19). These fungi have been called ‘Cordyceps‐colonizing fungi’ although their exact relationships with C. sinensis still remains ill‐defined. C. sinensis‐ colonizing fungi’ are known as Chinese caterpillar fungus or ‘Dong Chong Xia Cao’. They are famous fungi used in traditional Chinese medicine, and have been widely used as a tonic and/or medicine for hundreds of years in the Asian countries. It is found primarily at high altitude (above 3500 m), on the Qinghai‐Tibetan plateau and endophytically parasitizes dead caterpillars of the moth Hepilus spp. Ongoing exploration of C. sinensis has shown that this species can produce many bioactive compounds, including anti‐cancer agents (20, 21). However, due to its growing popularity, the natural fungus has been over‐harvested to the extent that it is now an endangered species (22). Despite claimed medical benefits, including that of anti‐cancer activity (whether these effects originate from itself or from metabolites produced by the colonizing fungi) remain to be answered. Our long‐term goal is to understand the relationships between the fungi and C. sinensis from mycological and chemical aspects. During an ongoing ecology‐based search for anti‐tumour agents, a library of 200 extracts prepared from solid‐substrate fermentation of 200 selected strains of Cordyceps‐colonizing fungi were screened. The strain producing the most active extract was large scale fermented, and bioassay‐guided fractionation of the resultant extract afforded two new metabolites, gliocladicillins A (1) and B (2), along with 11,11′‐dideoxyverticillin (23) which was known already. Compounds A (1) and B (2) displayed significant anti‐proliferative effects on HeLa cells, and mechanism study revealed that they both induced G2/M cell cycle arrest, and apoptosis, in a dose‐dependent manner. In addition, data from tumour formation assays in mice indicated that A (1) and B (2) showed significant in vivo anti‐tumour effects. These results suggest that gliocladicillins A (1) and B (2) should be further evaluated as candidates for anti‐cancer drugs.
Materials and Methods
Chemicals and reagents
Dulbecco’s modified Eagle’s medium (DMEM) and RPMI 1640 medium were purchased from Life Technologies (Grand Island, NY, USA), foetal bovine serum (FBS) was from YHSM (Beijing, China), and trypsin was obtained from Roche (Indianapolis, IN, USA). Dimethyl sulphoxide (DMSO), 3‐(4,5‐dimethyl thizol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT; CAS# 298‐93‐1), propidium iodide (PI; CAS# 25535‐16‐4), RNase A, and ONPG (ortho‐nitrophenyl‐b‐D‐galactopyranoside; CAS# 369‐07‐3) were purchased from Sigma (St Louis, MO, USA). Polyethylenimine (CAS# 9002‐98‐6) was purchased from Polyscience Inc. (Warrington, PA, USA). PARP1 (sc‐56196), p53 (sc‐6243), p21 (sc‐6246), Bcl‐Xs/l (sc‐1041), cyclin B1 (sc‐7393), and Chk1 (sc8408) were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Anti‐mouse (W4021) and anti‐rabbit (W4011) horseradish peroxidase‐conjugated immunoglobulin G, and Luciferase Assay System (E4530) were purchased from Promega (San Luis Obispo, CA, USA). Western Luminescent Kit and BCA Protein Assay Kit were from Vigorous (Beijing, China).
Instrumentation for structure elucidation
Proton (1H) and carbon‐13 (13C) nuclear magnetic resonance (NMR) data were acquired using Bruker Avance‐400 and ‐600 spectrometers using solvent signals (CDCl3; δH 7.26/δC 77.0) as references. Heteronuclear multiple quantum coherence and heteronuclear multiple bond coherence (HMBC) experiments were optimized for 145.0 and 8.0 Hz, respectively. Electrospray ionization mass spectrometry (ESI‐MS) data were recorded on a Bruker Esquire 3000plus spectrometer, and high resolution electrospray ionization mass spectrometry (HRESI‐MS) data were recorded on a Bruker APEX III 7.0 T spectrometer.
Cell lines and culture
MCF‐7 (breast cancer), HepG2 (hepatocellular carcinoma) and HeLa (cervical cancer) human solid tumour cell lines, and B16 mouse melanoma tumour cell line were obtained from American Type Culture Collection (Rockville, MD, USA). MCF‐7, HepG2 and HeLa cells were maintained in DMEM, whereas B16 cells were maintained in RPMI 1640 medium, all supplemented with 10% heat‐inactivated FBS at 37 °C, in a humidified atmosphere containing 5% CO2.
Fungal material and extract preparation
Strains of Cordyceps‐colonizing fungi were isolated by one of the authors (X.L.) from samples of C. sinensis (Berk.) Sacc. collected in Linzhi, Tibet, in March 2004. The isolate that produced the most active extract was identified as Gliocladium sp. and assigned the accession no. XZC04‐CC‐302 in X.L’s culture collection at the Institute of Microbiology, Chinese Academy of Sciences, Beijing. Extracts were prepared by solid‐substrate fermentation on rice, as described previously (24).
Preparation of the active extract and isolation of compounds 1–3
Solid‐substrate fermentation was carried out in one hundred 500‐ml Erlenmeyer flasks, each containing 150 g of rice. Fermented rice substrate was extracted repeatedly with ethyl acetate (6 × 1 l), and the organic solvent was evaporated to dryness in vacuo to afford crude extract (40 g). Crude extract was fractionated by silica gel column chromatography (CC) (6 × 30 cm) using CH2Cl2–MeOH gradient elution, and the active fraction (1.0 g) eluted with 99 : 1 CH2Cl2–MeOH was subsequently separated by silica gel CC (2.8 × 14 cm) using 99.5 : 0.5 CH2Cl2–MeOH as eluents. One active fraction (150 mg) was further purified by semipreparative reversed‐phase high‐performance liquid chromatography (RP HPLC; Agilent Zorbax SB‐C18 column (Angilent Technologies, USA); 5 μm; 9.4 × 250 mm, 2 ml/min) to obtained gliocladicillin A (1; 30.0 mg, t R 14.3 min; 50–55% acetonitrile in H2O for 5 min, and followed by 55% acetonitrile for 25 min). Purification of a further active fraction (120 mg) by RP HPLC afforded gliocladicillin B (2; 25.0 mg, t R 19.5 min; 60–85% MeOH in H2O for 40 min). A last active fraction (100 mg) was also separated by RP HPLC to afford 11,11′‐dideoxyverticillin (3; 18.0 mg, t R 18.0 min; 60% acetonitrile in H2O for 5 min, and followed by 60–70% for 25 min).
Cytotoxicity and cell viability analyses
Viabilities of control and treated cells were evaluated using MTT assay, in triplicate. Cells (1 × 104/well) were seeded in 96‐well microtitre plates containing 100 μl culture medium, and were permitted to adhere for 16–18 h, washed with PBS, and then treated with the test compounds. After 4‐h treatment, cells were allowed to grow for a further 48 h after medium was replaced by fresh, and then incubated at 37 °C in 50 μl MTT solution (5 mg/ml) for 3 h. After removal of medium and MTT, 200 μl DMSO was added to each well, and the assay plate was read at 570 nm using a microplate reader (SUNRISE‐basic TECAN, TECAN Austria GmbH, Groedig, Austria). Absorbance of untreated cells was considered as 100%.
Cell cycle analysis
HeLa cells were treated with test compounds at indicated concentrations for 24 or 48 h. Cells were fixed in 70% cold ethanol, and then stained with a solution containing 45 μg/ml of PI and 50 μg/ml of RNase A. Flow cytometric analysis was performed using FACScan instrumentation (BD Biosciences, San Jose, CA, USA). Data were analysed using the ModFit LT program.
Western blot analysis
HeLa cells were grown to 70% confluence in 6‐cm dishes and incubated in DMEM with gradient concentrations of test compounds at 37 °C for 24 h. After rinsing in ice‐cold PBS, cells were lysed in 0.1 ml of lysis buffer (50 mm Tris‐HCl, pH 7.4, 1% NP‐40, 0.25% Na‐deoxycholate, 150 mm NaCl, 1.0 mm EDTA, 1.0 mm PMSF, and 1× protease inhibitor cocktail from Roche). Cell lysates were analysed on 8% or 15% sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred to PVDF membranes (GE Healthcare Bio‐Sciences Corp., Piscataway, NJ, USA). Immunoblotting was then performed with indicated antibodies, followed by anti‐mouse or anti‐rabbit horseradish peroxidase‐conjugated immunoglobulin G. Immunoreactive proteins were detected using a luminescence kit reagent (Vigorous).
Detection of apoptosis
Apoptosis was analysed flow cytometrically, apoptotic cells appearing as a sub‐G1 population in PI‐stained preparations. Percentage of cells undergoing apoptosis was determined by three independent experiments; PARP‐1 cleavage and caspase activation were detected by Western blotting with indicated antibodies. For DNA fragmentation analysis, 1 × 106 cells were pelleted and washed in ice‐cold PBS, resuspended in 200 μl PBS (with 0.2 mg/ml protease K), then 200 μl PBS with 2% NP‐40 was added, vortex‐mixed thoroughly, and treated cells were allowed to stay on ice for 30 min. After centrifuging at 13 000 g for 5 min, supernatant was collected and DNA was ethanol‐precipitated and resuspended in 30 μl distilled water. RNaseA (10 mg/ml) was then added to achieve a final concentration of 100 μg/ml. The mixture was first incubated at 37 °C for 30 min, then 6 μl of 6× loading buffer (0.25% bromophenol blue, 0.05% xylene cyanol, and 50% glycerol) was added and DNA was subjected to 2% agarose gel electrophoresis and visualized using ethidium bromide staining.
Transfection and luciferase reporter assay
HeLa cells were seeded into 12‐well plates 24 h before transfection, and media were removed and replaced with 1 ml fresh DMEM 2 h prior to transfection. 1 μg DNA was diluted with 150 mm NaCl solution, in an Eppendorf tube, to achieve a final volume of 50 μl. The solution was mixed thoroughly, 12 μg polyethylenimine (in 50 μl of 150 mm NaCl) was added, and then incubated at room temperature for 15 min. The DNA/polyethylenimine mixture was then added to cells, and medium was changed 8 h after the transfection.
For p53‐ and p21‐luc reporter assays, cells were first transfected with 0.5 μg p53‐luc or p21‐luc and 0.5 μg pCMV‐β‐gal for 24 h, treated with test compounds for 12 h, and then lysed with 1× reporter lysis buffer (Promega). Luciferase activity was measured using Promega SLR luciferase reporter system with a 20/20n luminometer (Turner Biosystems, Sunnyvale, CA, USA). As internal control of transfection, β‐galactosidase activity was measured through ONPG hydrolysis. Absorbance at 420 nm was measured using a UV‐2100 spectrophotometer (Unic, Shanghai, China). Relative luciferase activity was calculated as RLU = luciferase activity/β‐galactosidase activity.
In vivo anti‐tumour activity
Female C57BL/6J mice were purchased from Vital River Laboratories (Beijing, China), and all experiments were carried out using 8‐week‐old mice weighing 18–22 g. B16 cells (5 × 104) were injected subcutaneously into the right axilla region of the mice, and at 24 h after implantation, non‐control mice were intraperitoneally injected with vehicle or test compounds, then once daily for 21 days; all animals were weighed daily. At the end of the experiment, every mouse was killed and tumours were excised and weighed. Inhibition of the tumour growth in vivo was calculated using the following formula: growth inhibition = [(average tumour weight of the control group –average tumour weight of the test group)/average tumour weight of the control group] × 100%.
Results
Screening of extract library and bioassay‐guided fractionation
The 200 crude extracts prepared from Cordyceps‐colonizing fungi were subjected to MTT assay using HeLa cells, and their anti‐proliferative effects were evaluated by GI50 values. Gliocladium sp. produced the most active extract, and its scale‐up fermentation extract reproduced the activity with GI50 value of 15 μg/ml. Bioassay‐directed separation of this extract enriched the activity to a fraction with a GI50 value of 2.0 μg/ml, and further purification of this active fraction led to isolation of three active compounds, with GI50 values of 0.50, 0.10 and 0.25 μg/ml, respectively (Fig. 1a).
Figure 1.
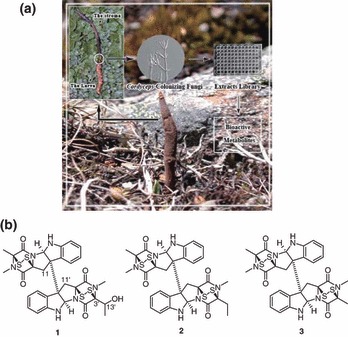
(a) Cordyceps‐colonizing fungi from C. sinensis samples collected in Tibet. (b) Chemical structures of gliocladicillins A (1), B (2), and 11,11′‐dideoxyverticillin (3).
Identification of gliocladicillins A (1), B (2), and 11,11′‐dideoxyverticillin ( 3 )
Structures of extracts 1–3 were elucidated by interpretation of MS and NMR data. Compound 3 was readily identified as 11,11′‐dideoxyverticillin, a cytotoxic metabolite recently discovered as a receptor tyrosine kinase inhibitor, by comparison of its NMR and MS data with those previously reported (19). The molecular formula of A (1) was assigned as C31H30N6O5S4 by HRESI‐MS. Analysis of its 1H and 13C NMR data revealed structural features similar to those found in extract 3, except that the methyl group at C‐3′ was replaced by signals for a 2‐hydroxyethyl unit; this observation was supported by relevant 1H–1H COSY and HMBC correlations. Thus, the structure of gliocladicillin A (1) was elucidated as shown (Fig. 1b). The molecular formula of B (2) was discovered to be C31H30N6O4S4 by HRESI‐MS. Interpretation of its NMR data revealed that the oxymethine (C‐13′; δH/δC 4.46/68.17) was replaced by resonances for a methylene unit (δH/δC 2.17, 2.37/24.81), leading to the recognition of B (2) as the 3′‐deoxy analogue of A (1) (Fig. 1b).
Anti‐proliferative effects of A (1) and B (2) on human cancer cell lines
Anti‐tumour effects of A (1) and B (2) were evaluated against three human cancer cell lines – HeLa, HepG2, and MCF‐7 – using the MTT assay. The GI50 values of A (1) and B (2) ranged from 0.10 to 0.50 μg/ml (Fig. 2). In this assay, MCF‐7 cells showed relatively higher susceptibility (GI50 = 0.20 μg/ml) to A (1) compared to HeLa and HepG2 cells (GI50 = 0.50 μg/ml).
Figure 2.

Anti‐proliferative effects of gliocladicillins A (1) and B (2) on three human tumour cell lines. Cells seeded in 96‐well plates were treated with various concentrations of 1 and 2 for 4 h, and their viabilities were determined using MTT assay. GI50 values are shown as mean ± standard deviation of three independent experiments.
G2/M arrest and apoptosis induced by A (1) and B (2) in HeLa cells
To determine the mechanisms by which A (1) and B (2) inhibit proliferation of tumour cells, cell cycles were analysed in HeLa cells treated with A (1) and B (2), at indicated concentrations, for 24 and 48 h, respectively. The percentage of cells in each phase of the cell cycle was determined by flow cytometry (Fig. 3a,b, respectively; the sub‐G1 population indicated the apoptotic cells). In non‐apoptotic populations, HeLa cells accumulated in G2/M phase when treated with A (1) and B (2). For example, HeLa cells treated with 2.0 μg/ml of A (1) for 24 h resulted in an increase in the percentage of cells from 22.7% to 41.8% in the G2/M phase, suggesting that both A (1) and B (2) inhibited cell proliferation by G2/M phase arrest. The percentage of apoptotic cells increased significantly in treated cells, and the proportion was higher both at 24 and 48 h, with increase in concentration of either A (1) or B (2). These results suggested that A (1) and B (2) both triggered tumour cell apoptosis in a time‐ and dose‐dependent manner. Apoptosis induced by A (1) and B (2) was also confirmed by Annexin V staining (Fig. 3c,d) and DNA fragmentation analysis of treated HeLa cells (Fig. 4a).
Figure 3.

Effects of gliocladicillins A (1) and B (2) on cell cycle distribution and apoptosis. (a,b) Flow cytometry analysis. HeLa cells were incubated in the absence or presence of 1 (a) or 2 (b) for 24 and 48 h, respectively. Cells were harvested and fixed in 70% ethanol for flow cytometry. Data shown are representatives of three independent experiments. (c,d) HeLa cells were incubated in the absence or presence of 0.50 μg/ml of 1 (c) or 0.10 to 0.30 μg/ml of 2 (d) for 24 h. Cells were harvested and stained with Annexin V and propidium iodide for flow cytometry.
Figure 4.

Gliocladicillins A (1) and B (2) both induce apoptosis in HeLa cells. (a) DNA fragmentation analysis. HeLa cells were incubated in the absence or presence of 0.50 to 2.0 μg/ml of A (1) for 24 h, and 25 μg/ml cycloheximide was used as the positive control. Cells were harvested and the genomic DNA was extracted and analysed using agarose gel. (b) Immunoblotting. HeLa cells were incubated in the absence or presence of 0.125 to 2.0 μg/ml of A (1) (left panel) or 0.125 to 1.0 μg/ml of 2 (right panel) for 24 h, and cells were harvested and analysed by immunoblotting for PARP‐1, caspase‐3, caspase‐8, caspase‐9, Bax, Bcl‐XL; β‐actin was used as the loading control.
Activation of the caspase pathway in HeLa cells
HeLa cells were first treated with different concentrations of either A (1) or B (2), and cell lyastes were then harvested for immunoblotting initially with caspase‐8, caspase‐9, effector caspase‐3, and its substrate PARP‐1. Results indicated that caspase‐8 was cleaved into its active form (43/41 kD), while caspase‐9 cleaved to an active large fragment of 35 kD (Fig. 4b). Activation of effector caspase‐3 and its substrate PARP was also examined, with caspase‐3 cleaving into its active forms (17/21 kD) and PARP into an 85‐kD active fragment. All the cleavages of these were shown to act in a dose‐dependent manner.
Protein levels of Bax and Bcl‐XL were examined (Fig. 4b) as increase in the ratio of Bax/Bcl‐XL expression may have modulated subsequent activity of caspase‐3, which functions as the executioner of apoptosis. Together, these results indicated that both A (1) and B (2), activate the caspase pathway and induce HeLa cells to undergo apoptosis through both extrinsic and mitochondrial pathways.
Effects of A (1) and B (2) on proteins involved in cell cycle progression
Flow cytometry data showed that A (1) and B (2) both induced HeLa cells to arrest in the G2/M phase. It is known that induction of p21 and p53 are implicated in G2/M arrest (25, 26); thus, expression of p21 and p53 proteins was examined by Western blot analysis to determine whether A (1) and B (2) could regulate cell cycle‐related protein expression. The result showed that levels of both p21 and p53 increased significantly Fig. 5a). As for transcription of p21 and p53, both p21 and p53 luciferase reporters were activated in A (1)‐ and B (2)‐ treated cells (Fig. 5b), suggesting that A (1) and B (2) may cause G2/M arrest by regulating transcription of p21 and p53.
Figure 5.

Gliocladicillins A (1) and B (2) regulate expressions of cell cycle regulators and transcription of p53 and p21. (a) Immunoblotting results. HeLa cells were incubated in the absence or presence of A (1) or B (2) for 24 h, and cell lysates were harvested and analysed by immunoblotting with indicated antibodies, β‐actin being the loading control. (b) Luciferase assay results. HeLa cells seeded into 12‐well plate were transfected with p21‐ or p53‐luc reporter plasmids and pCMV‐β‐gal was used as the transfection control. Transfectants were incubated in the absence or presence of A (1) (upper 2 panels) or B (2) (lower 2 panels) for 12 h, and cell lysates were harvested and subjected to luciferase assay. Relative luciferase activity, normalized by β‐galactosidase activity shown.
Degradation of the cyclin B subunit of protein kinase CDK2/cyclin B dimer is required for inactivation of CDK2 and ensures mitotic exit, while its inappropriate accumulation causes G2/M phase arrest. Thus, expression of cyclin B and CDK2 were examined; results showed that expression of cyclin B was elevated, whereas that of CDK2 remained unchanged in the treated HeLa cells compared to controls (Fig. 5a), suggesting that A (1) and B (2) might block degradation of cyclin B and therefore cause G2/M arrest.
In vivo anti‐tumour effects of compounds A (1) and B (2)
Due to their potent in vitro anti‐proliferative activities, compounds A (1) and B (2) were further evaluated for their in vivo anti‐tumour effects using a xenograft mouse melanoma tumour model. B16 melanoma cells (5 × 104) were injected into C57BL/6J mice, and after 24 h, test mice were individually injected with A (1) (0.25 and 0.50 mg/kg body weight, respectively) or B (2) (0.10 and 0.40 mg/kg body weight, respectively) once a day for 21 days. All animals were then killed at day 21 with their body weights having been recorded each day before their death. tumours were removed from the bodies and weighed. Average tumour weight from the mice treated with A (1) were 0.54 and 0.24 g, whereas those from mice treated with B (2) were 0.80 and 0.34 g, respectively of indicated dosages (average weight of tumours from the control group was 1.84 g; Table 1). In vivo results indicated that A (1) and B (2) treatment both showed significant anti‐tumour efficacy (P < 0.005) for B16 melanomas in a dose‐dependent manner, with inhibition levels of 69.8–87.2% for A (1) and 56.7–82.5% for B (2).
Table 1.
Anti‐tumour effects of 1 and 2 against B16 in C57BL/6J mice
| Treatment | Dosage (mg/kg/day) ×day | Mice (n) (initial/end) | Body weight (g) (initial/end) | Tumour weight (g) | Inhibition rate (%) |
|---|---|---|---|---|---|
| Normal saline | 8/8 | 18.26/21.07 | 1.84 ± 0.70 | ||
| 1 | 0.25 × 21 | 8/8 | 18.79/19.68 | 0.56 ± 0.22* | 69.8 |
| 1 | 0.50 × 21 | 8/8 | 18.41/18.32 | 0.24 ± 0.12* | 87.2 |
| 2 | 0.10 × 21 | 8/8 | 18.32/18.87 | 0.80 ± 0.36** | 56.7 |
| 2 | 0.40 × 21 | 8/8 | 18.53/18.40 | 0.32 ± 0.17* | 82.5 |
B16 cell suspensions were injected subcutaneously into the right axilla region of C57BL/6J female mice (18 ± 2 g), and daily intraperitoneal injections of normal saline or test samples commenced 1 day after injection of tumour cells. All mice were killed on day 21 and the tumours were dissected out and weighed. Data are expressed as mean ± standard deviation.
*P < 0.001 versus normal saline group. **P < 0.005 versus normal saline group.
Discussion
Natural products have been demonstrated to play a major role in discoveries of novel anti‐cancer drugs (3, 8). High throughput screening techniques have been widely used to screen appropriate libraries of large numbers of natural extracts and synthetic compounds against diseases to identify quickly active agents that may serve as initiation points for desperately needed drugs. Here, in this work, an ecology‐based approach has been applied to select unique fungi and to prepare their extract details; initial screening of 200 extracts obtained from selected Cordyceps‐colonizing fungi identified a fungus, Gliocladium sp., that produced the most active derivatives. Subsequent bioassay‐guided fractionation of the scaled‐up sample led to isolation and identification of new metabolites, gliocladicillins A (1) and B (2), and 11,11′‐dideoxyverticillin (3) (23). These metabolites were responsible for all the anti‐tumour effects of the crude extract.
Compounds 1–3 are epipolythiodioxopiperazines (ETP), produced only by fungi, and cell toxicity of ETPs has made them attractive as potential therapeutic agents against cancer. As a member of the ETPs, gliotoxin has been reported to be a dual inhibitor of farnesyltransferase and geranylgeranyltransferase I, and has shown pronounced activity against rat mammary carcinomas without detectable general toxicity in vivo (27). Sch52900, a verticillin type of ETP, has been reported to induce differentiation of leukaemia cells (28), while chaetocin, another ETP closely related to the verticillins, has been recently identified as an anti‐myeloma agent with in vitro and in vivo activity (29). The known ETP recognised here, 11,11′‐dideoxyverticillin ( 3 ), has been shown to be a dual inhibitor of EGFR/ErbB‐2 and VEGFR‐1 tyrosine kinase, and has also displayed in vivo anti‐tumour efficacy (23, 30). Based on evidence from the literature, gliocladicillins A (1) and B (2) are two new ETPs isolated in this study, thus were also evaluated to throw light on their anti‐tumour mechanisms. They were found to cause G2/M cell cycle arrest and to induce apoptosis in HeLa cells, by activating multiple signalling pathways. Anti‐tumour activity of A (1) and B (2) were closely associated with their ability to induce G2/M phase arrest, as demonstrated by flow cytometry results (Fig. 3), and their interference with proteins of G2/M transition and mitotic exit (that is, p21, p53, CDK2, and cyclin B) could have contributed to this effect. Our results suggest that A (1) and B (2) both activated expression of p21 and p53, and caused cyclin B accumulation, in a dose‐dependent manner, which themselves may induce G2/M arrest and interfere with mitotic exit. Even though activation of p21 and p53 expression and accumulation of cyclin B play a major role in the molecular basis of the A (1)‐ and B (2)‐induced cell cycle arrest, further studies are needed to elucidate the detailed mechanisms in which A (1) and B (2) regulate expression of p53 and block degradation of cyclin B. Previous evaluation of known ETPs for their anti‐tumour effects, have suggested that they may be involved in multiple pathways in cell proliferation and apoptosis (23, 28, 29); our results obtained from this study suggest that A (1) and B (2) are both involved in regulating multiple pathways controlling the cell cycle.
Tumour cells often evade apoptosis by expressing anti‐apoptotic proteins, down‐regulating pro‐apoptotic genes, and altering signalling pathways, which provide a survival advantage for them (31). Induction of apoptosis in tumour cells is therefore, one of the strategies explored for anti‐cancer drug development (32, 33), and activation of caspases is important for induction of death signals by apoptosis. Caspases can be divided into initiator (caspase‐8 and caspase‐9) and effector caspases (caspase‐3). The extrinsic pathway of caspase activation begins with activation of caspase‐8, and active caspase‐8 can activate the effector caspases, such as caspase‐3, which then act downstream to cleave substrates such as PARP, resulting in programmed cell death. In this study, A (1) and B (2) were found to activate caspase‐8 and eventually activate caspase‐3, implying that they are involved in activating the extrinsic pathway. The intrinsic pathway of apoptosis starts with the release of cytochrome c from mitochondria into cell cytoplasm, where it forms a complex with Apaf‐1 to recruit and activate caspase‐9, and then activated caspase‐9 triggers the effector caspases ‐ promoting apoptosis. The Bcl‐2 family of anti‐apoptotic proteins, including Bcl‐2 and Bcl‐XL, can inhibit this caspase activation pathway, in part by forming pores that stabilize mitochondrial membranes. These proteins also block apoptosis by forming inactivating heterodimers with pro‐apoptotic proteins such as Bak and Bax. Tumour cells can acquire resistance to apoptosis by expressing anti‐apoptotic proteins such as Bcl‐2 or by down‐regulating pro‐apoptotic proteins such as Bax, and expression of both Bcl‐2 and Bax are regulated by the p53 tumour suppressor gene (34, 35). Here, compounds A (1) and B (2) were found to up‐regulate expression of p53, to change the ratio of Bax/Bcl‐XL, and to activate caspase‐9, indicating that A (1)‐ and B (2)‐induced apoptosis was also mediated through the intrinsic pathway.
In vivo anti‐tumour activities of A (1) and B (2) were evaluated in C57BL/6J mice, a xenograft model. Our results showed that both reagents displayed promising inhibitory efficacies for population growth of melanoma B16 cells, without causing any obvious decrease in body weights of the mice (which would have indicated damage to normal tissues). The remaining question to answer is whether in vivo success was caused solely by the anti‐proliferative effects of A (1) and B (2). Because tumour growth and metastasis are dependent on angiogenesis, tumours must continuously stimulate growth of new capillary blood vessels to sustain tumour cell replication; the known ETP 11,11′‐dideoxyverticillin ( 3 ) has been reported to display anti‐angiogenic activity (30). These new gliocladicillins should be further investigated for their role in signalling pathways involved in angiogenesis too, in order to understand these mechanisms of their in vivo anti‐tumour effects also.
In this study, the ecology‐based approach for discovery of new bioactive natural products has again been demonstrated to be valuable. This approach may help to explore relationships between these fungi and their host C. sinensis, and to further study the ecology of fungi involved, from the mycological perspective. For chemists studying natural products, it would be appealing to work on a small number of extracts if such an effort resulted in discovery of new bioactive natural products. Isolation of new bioactive ETPs, gliocladicillins A (1) and B (2), from the Cordyceps‐colonizing fungus Gliocladium sp., provided further evidence for usefulness of such an approach.
In conclusion, new gliocladicillins A (1) and B (2) were found to inhibit proliferation of HeLa, HepG2 and MCF‐7 tumour cells and in treated HeLa cells, to induce cell cycle arrest in the G2/M phase, through activation of the p53 pathway and causing cyclin B accumulation. The new derivatives also induced tumour cell apoptosis by activating both intrinsic and extrinsic pathways and they also exhibited significant in vivo anti‐tumour activity in mice. These results suggest that gliocladicillins A (1) and B (2) are effective both in vitro and in vivo as anti‐proliferative and pro‐apoptotic agents and should be further evaluated for their potential in clinical usage.
Acknowledgements
This work was supported by the State Key Basic Research Project Grant 2004CB719601 (Y. Che), the Key Project of National Hi‐Tech Research and Development Grant 2007AA021506 (Y. Che), the Key Project of Chinese Academy of Sciences Grant KSCX2‐YW‐G‐013 (X. Ye), and the National Natural Science Foundation of China Grants 30771098 (X. Ye) and 30870057 (Y. Che).
Y. Chen and H. Guo contributed equally to this work.
References
- 1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J. Clin. 58, 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Ismae GFV, Rosa DD, Mano MS, Awada A (2008) Novel cytotoxic drugs: Old challenges, new solutions. Cancer Treat. Rev. 34, 81–91. [DOI] [PubMed] [Google Scholar]
- 3. Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 70, 461–477. [DOI] [PubMed] [Google Scholar]
- 4. Ojima I, Chakravarty S, Inoue T, Lin S, He L, Horwitz SB, Kuduk SD, Danishefsky SJ (1999) A common pharmacophore for cytotoxic natural products that stabilize microtubules. Proc. Natl. Acad. Sci. USA 96, 4256–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiménez‐Barbero J, Amat‐Guerri F, Snyder JP (2002) The solid state, solution and tubulin‐bound conformations of agents that promote microtubule stabilization. Curr. Med. Chem. Anticancer Agents 2, 91–122. [DOI] [PubMed] [Google Scholar]
- 6. Tobert JA (2003) Lovastatin and beyond: the history of the HMG‐CoA reductase inhibitors. Nat. Rev. Drug Discov. 2, 517–526. [DOI] [PubMed] [Google Scholar]
- 7. Binaschi M, Farinosi R, Borgnetto ME, Capranico G (2000) In vivo site specificity and human isoenzyme selectivity of two topoisomerase II‐poisoning anthracyclines. Cancer Res. 60, 3770–3776. [PubMed] [Google Scholar]
- 8. Chen J, Ghorai MK, Kenney G, Stubbe J (2008) Mechanistic studies on bleomycin‐mediated DNA damage: multiple binding modes can result in double‐stranded DNA cleavage. Nucleic Acids Res. 36, 3781–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Da Rocha AB, Lopes RM, Schwartsmann G (2001) Natural products in anticancer therapy. Curr. Opin. Pharmacol. 1, 364–369. [DOI] [PubMed] [Google Scholar]
- 10. Reed JC (2006) Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat. Clin. Pract. Oncol. 3, 388–398. [DOI] [PubMed] [Google Scholar]
- 11. Salomon AR, Voehringer DW, Herzenberg LA, Khosla C (2001) Apoptolidin, a selective cytotoxic agent, is an inhibitor of F0F1‐ATPase. Chem. Biol. 8, 71–80. [DOI] [PubMed] [Google Scholar]
- 12. Bailly C (2008) Ready for a comeback of natural products in oncology. Biochem. Pharmacol 77, 1447–1457. [DOI] [PubMed] [Google Scholar]
- 13. Cardenas ME, Sanfridson A, Cutler NS, Heitman J (1998) Signal‐transduction cascades as targets for therapeutic intervention by natural products. Trends Biotechnol. 16, 427–433. [DOI] [PubMed] [Google Scholar]
- 14. Germano S, Barberis D, Santoro MM, Penengo L, Citri A, Yarden Y, Gaudino G (2006) Geldanamycins trigger a novel Ron degradative pathway, hampering oncogenic signaling. J. Biol. Chem. 281, 21710–21719. [DOI] [PubMed] [Google Scholar]
- 15. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL (2008) Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN‐deficient glioblastoma. PLoS Med. 5, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou TC, O’Connor OA, Tong WP, Guan Y, Zhang ZG, Stachel SJ, Lee C, Danishefsky SJ (2001) The synthesis, discovery, and development of a highly promising class of microtubule stabilization agents: curative effects of desoxyepothilones B and F against human tumor xenografts in nude mice. Proc. Natl. Acad. Sci. USA 98, 8113–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meinwald J, Eisner T (2008) Chemical ecology in retrospect and prospect. Proc. Natl. Acad. Sci. USA 105, 4539–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gloer JB (1997) Applications of fungal ecology in the search of new bioactive natural products In: Wicklow DT, Soderstrom B, eds. The Mycota IV: Environmental and Microbial Relationships, pp. 249–268. Berlin, Germany: Springer‐Verlag. [Google Scholar]
- 19. Guo H, Hu H, Liu S, Liu X‐Z, Zhou Y, Che Y (2007) Bioactive p‐terphenyl derivatives from a Cordyceps‐colonizing isolate of Gliocladium sp. J. Nat. Prod. 70, 1519–1521. [DOI] [PubMed] [Google Scholar]
- 20. Zhu JS, Halpern GM, Jones K (1998) The scientific rediscovery of an ancient Chinese herbal medicine: Cordyceps sinensis: part I. J. Altern. Complement. Med. 4, 289–303. [DOI] [PubMed] [Google Scholar]
- 21. Yoshikawa N, Nakamura K, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M (2004) Antitumour activity of cordycepin in mice. Clin. Exp. Pharmacol. Physiol. 31(Suppl. 2), S51–S53. [DOI] [PubMed] [Google Scholar]
- 22. Paterson RR (2008) Cordyceps: a traditional Chinese medicine and another fungal therapeutic biofactory? Phytochemistry 69, 1469–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang YX, Chen Y, Guo XN, Zhang XW, Zhao WM, Zhong L, Zhou J, Xi Y, Lin LP, Ding J (2005) 11,11′‐dideoxy‐verticillin: a natural compound possessing growth factor receptor tyrosine kinase‐inhibitory effect with anti‐tumor activity. Anticancer Drugs 16, 515–524. [DOI] [PubMed] [Google Scholar]
- 24. Liu L, Liu S, Jiang L, Chen X, Guo L, Che Y (2008) Chloropupukeananin, the first chlorinated pupukeanane derivative, and its precursors from Pestalotiopsis fici . Org. Lett. 10, 1397–1400. [DOI] [PubMed] [Google Scholar]
- 25. Dulic V, Stein GH, Far DF, Reed SI (1998) Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M‐phase transition. Mol. Cell. Biol. 18, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taylor WR, Stark GR (2001) Regulation of the G2/M transition by p53. Oncogene 20, 1803–1815. [DOI] [PubMed] [Google Scholar]
- 27. Vigushin DM, Mirsaidi N, Brooke G, Sun C, Pace P, Inman L, Moody CJ, Coombes RC (2004) Gliotoxin is a dual inhibitor of farnesyltransferase and geranylgeranyltransferase I with antitumor activity against breast cancer in vivo . Med. Oncol. 21, 21–30. [DOI] [PubMed] [Google Scholar]
- 28. Erkel G, Gehrt A, Anke T, Sterner O (2002) Induction of differentiation in acute promyelocytic leukemia cells (HL‐60) by the verticillin derivative Sch 52900. Z. Naturforsch. 57C, 759–767. [DOI] [PubMed] [Google Scholar]
- 29. Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC (2007) Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood 109, 2579–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y, Zhang YX, Li MH, Zhao WM, Shi YH, Miao ZH, Zhang XW, Lin LP, Ding J (2005) Antiangiogenic activity of 11,11′‐dideoxyverticillin, a natural product isolated from the fungus Shiraia bambusicola . Biochem. Biophys. Res. Commun. 329, 1334–1342. [DOI] [PubMed] [Google Scholar]
- 31. Letai AG (2008) Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 8, 121–132. [DOI] [PubMed] [Google Scholar]
- 32. Ehrhardt H, Häcker S, Wittmann S, Maurer M, Borkhardt A, Toloczko A, Debatin KM, Fulda S, Jeremias I (2008) Cytotoxic drug‐induced, p53‐mediated upregulation of caspase‐8 in tumor cells. Oncogene 27, 783–793. [DOI] [PubMed] [Google Scholar]
- 33. Melet A, Song K, Bucur O, Jagani Z, Grassian AR, Khosravi‐Far R (2008) Apoptotic pathways in tumor progression and therapy. Adv. Exp. Med. Biol. 615, 47–79. [DOI] [PubMed] [Google Scholar]
- 34. Chipuk JE, Kuwana T, Bouchier‐Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014. [DOI] [PubMed] [Google Scholar]
- 35. Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293–299. [DOI] [PubMed] [Google Scholar]