

Abstract
Abstract. Reduced cell proliferation and increased levels of cellular glutathione (GSH) are characteristic for cells that overexpress the anti‐apoptotic Bcl‐2 protein. We investigated the influence of various Bcl‐2 domains on both these characteristics. Rat CC531 colorectal cancer cells were stably transfected with the human bcl‐2 gene (CCbcl2 cells) or with bcl‐2 gene constructs missing a coding sequence for a func‐tional domain, BH1 (CCΔBH1 cells), BH3 (CCΔBH3 cells), BH4 (CCΔBH4 cells) or the transmembrane region (CCΔTM cells). We measured GSH levels in exponentially and confluent growing bcl‐2‐transfected cell populations. The fraction of S‐phase cells during exponential growth was significantly reduced in CCbcl2, CCΔBH1, CCΔBH3, and CCΔTM cells compared with parental CC531, neo‐transfected CC531 and CCΔBH4 cells. GSH levels in these bcl‐2 transfectants were significantly higher than in the parental line measured at 50% confluence; at 100% confluence they reached a similar level as found in parental cells. Independently from the presence of BH1, BH3 or TM domains, overexpression of Bcl‐2 reduces cellular proliferation under conditions of increased GSH levels. This apparent link is lost in CCΔBH4 cells; these cells are not reduced in cellular proliferation and harbour significantly higher GSH levels than found in the other transfectants. Studies on the subcellular localization revealed an extremely low expression of the Bcl‐2 protein lacking the N‐terminal BH4 domain in nuclear fractions. Nuclear translocation of Bcl‐2 requires the presence of the BH4 domain and seems prominent in reducing cellular proliferation.
INTRODUCTION
The Bcl‐2 gene family plays a pivotal role in deciding whether cells will live or die (Tsujimoto et al. 1984; Korsmeyer 1999). Its gene products are either anti‐apoptotic molecules (e.g. Bcl‐2, Bcl‐Xl, Bcl‐w and A1) or pro‐apoptotic molecules (e.g. Bax, Bak, Bcl‐Xs, Bad, Bik, Bid and Bim) and the ratio between these two subsets seems an important determinant of sensitivity to cell death signals (Korsmeyer et al. 1993). The ability of many Bcl‐2 family members to form homo‐ as well as heterodimers could be important for activation of specific functions, but also for neutralizing these functions. An additional characteristic is that most members contain a carboxyl‐terminal hydrophobic domain (TM domain) essential for their targeting to membranes and that they can become integral membrane proteins. Many of the anti‐apoptotic members display sequence conservation in four domains (Bcl‐2 homology domains, designated BH1, BH2, BH3 and BH4, respectively), corresponding to alpha‐helical segments. Figure 1 summarizes the positions of the BH domains and TM region within the human Bcl‐2 protein and its multiple properties. Molecules like Bcl‐2 seem to have evolved into multifunctional proteins, acting in part independent from pro‐apoptotic family members (St. Clair et al. 1997). This is illustrated by the finding that Bcl‐2 can interact through its N‐terminal BH4 domain with a variety of different proteins, including Raf‐1 kinase (Wang et al. 1996), the phosphatase calcineurin (Shibasaki et al. 1997) and the transcription factor NF‐kappaB (Grimm et al. 1996).
Figure 1.
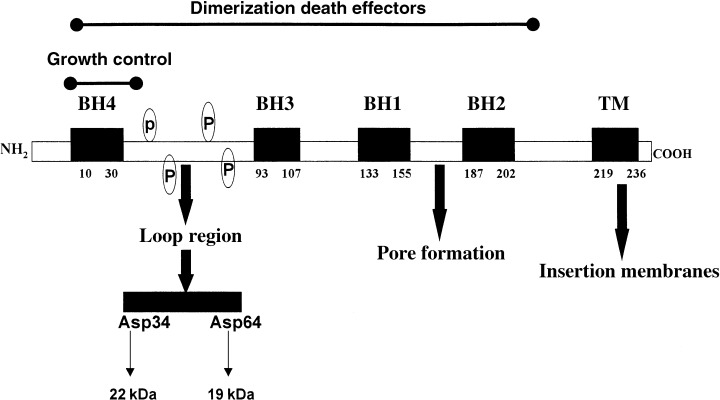
Structure of the human Bcl‐2 protein. The structure of the 26 kDa human Bcl‐2 protein, consisting of 239 amino acids, comprises four Bcl‐2 Homology domains (BH1, BH2, BH3 and BH4) and a carboxyl‐terminal located transmembrane domain (TM). BH domains are denoted in the figure delineated by amino acid numbers. In three‐dimensional context, BH1, BH2, and BH3 domains form an elongated hydrophobic cleft leaving the BH4 domain at the opposite face of the molecule, separated by a flexible loop region. Phosphorylation of serine/threonine residues (p) occurs within the loop region when cells enter the G 2 /M phase of the cell cycle. Cleavage site for caspase‐3 is the Aspartatic (Asp) residue 34 residing in the loop region; also denoted is the putative cleavage site Asp64. Cleavage at Asp64 would yield a 19‐kDa fragment of Bcl‐2 (this report). BH1 and BH2 domains flank two hydrophobic core helices, which are predicted to form ion‐conducting channels in bilayers, requiring insertion in these membranous structures by the transmembrane domain.
Overexpression of Bcl‐2 has repeatedly been associated with a decrease in proliferative activity in vitro and in vivo (Vaux et al. 1988; O’Reilly et al. 1997). Several investigators have provided data suggesting that this effect is due to an inhibition of the cell cycle entry of resting cells (O’Reilly et al. 1996; Linette et al. 1996). The N‐terminal BH4 domain is not only involved in inhibition of cell death, but also seems to be required for this anti‐proliferative effect of Bcl‐2 and possibly of other BH4‐containing anti‐apoptotic family members (Huang et al. 1997). Interestingly, a conserved tyrosine residue located within the BH4 domain of Bcl‐2 appeared to be responsible for an anti‐proliferative effect, while its mutation did not affect the anti‐apoptotic properties; mutation of the corresponding residue in Bcl‐Xl and Bcl‐w had a similar effect (Huang et al. 1997).
It has been speculated that Bcl‐2 may increase the antioxidant capacity of the cell (Hockenbery et al. 1993; 1999, 1993). Overexpression of Bcl‐2 leads to approximately two‐fold higher cellular levels of the ubiquitous tripeptide glutathione (GSH) (Ellerby et al. 1996; Meredith et al. 1998; Rimpler et al. 1999; Voehringer 1999; Schor et al. 2000; Vahrmeijer et al. 2000). GSH, together with certain GSH‐dependent enzymes, plays a critical role in protecting cells from reactive oxygen species generated in mitochondria during normal respiration. In addition, the cellular ratio between reduced and oxidized GSH, the redox balance, has a profound influence upon a myriad of cellular functions, including signal transduction and gene expression. For instance, this balance may profoundly affect the redox‐sensitive transcription factors NF‐AT and NF‐kappaB (Nakamura et al. 1997; Manna et al. 1999; Hutter & Greene 2000) as well as the activation of proteolytic activity of AP24, a serine protease involved in activation of apoptotic DNA‐fragmentation (Wright et al. 1998).
We recently reported that stable overexpression of human Bcl‐2 in rat CC531 colorectal cancer cells led to significantly elevated GSH levels (Vahrmeijer et al. 2000). To investigate the relationship between GSH modulation and the structure of the Bcl‐2 protein, we assayed GSH levels (measured as non‐protein thiols) in CC531 cells stably transfected with full‐length bcl‐2 or bcl‐2 constructs lacking the BH1, BH3, BH4 or TM domain (Hunter et al. 1996). It has been demonstrated by Hunter and co‐workers in transient transfection assays, that these constructs did not have any anti‐apoptotic capacity (Hunter et al. 1996). To establish a relationship between Bcl‐2 domain‐dependent GSH modulation and growth control, measurements of GSH were performed on cultures at 50% confluence (‘exponential’ growth) or 100% confluence (‘plateau’ phase), the latter with strongly reduced proliferative activity. Additionally, we evaluated the efficiency of nuclear translocation of truncated Bcl‐2 proteins as the presence of nuclear residing Bcl‐2 in bcl‐2‐transfected cells might serve as a compartmental control of the cell cycle (Hoetelmans et al. 2000; Hour et al. 2000).
MATERIALS AND METHODS
Plasmids and transfection
The parental and modified CC531 cells were maintained as monolayers in tissue flasks as previously described (Hoetelmans et al. 2000). Using the liposomal transfection reagent Fugene (Roche Diagnostics, Indianapolis, USA), we introduced the full length human bcl‐2 gene, or an empty vector containing the neomycine‐resistance sequence, or bcl‐2 gene constructs lacking the BH1 (aa 138–151), BH3 (aa 90–103), or BH4 (aa 6–31) or TM (aa 203–239) domain (Hunter et al. 1996). All deletion constructs were generously provided by Dr Parslow (University of California, San Francisco, California). Subsequently, neomycin‐resistant clones were selected, designated CCbcl2, CCneo, CCΔBH1, CCΔBH3, CCΔBH4, and CCΔTM, respectively. In addition, we stably transfected CC531 cells with the full‐length human bax gene (CCbax), kindly provided by Dr Wei Zhang (University of Texas M.D. Anderson Cancer Center, Texas).
Western blot analysis, cell fractionation and antibodies
Preparation of whole cell lysates, isolation of nuclear and cytoplasmic fractions, electrophoretic separation, transfer to nitrocellulose membrane and staining of filters was performed as previously described (Hoetelmans et al. 2000). To ensure equal protein loading, we performed the Bradford assay and β‐actin expression was evaluated with the monoclonal antibody (mAb) #C4 (Roche Diagnostics) diluted 1 : 15 000. To compare the efficiency of nuclear translocation of the truncated Bcl‐2 proteins with full‐length Bcl‐2, whole cell and nuclear lysates of each cell line were loaded in a 1 : 3 ratio, ensuring similar intensity of full‐length Bcl‐2 expression in both lysates (Hoetelmans et al. 2000). For detection of the human (truncated) Bcl‐2 protein, we applied the mAb #6C8 (Pharmingen, San Diego, USA), diluted 1 : 500.
Glutathione analysis
Cells were grown in culture flasks for 48 h until c. 50% confluence or 100% confluence by seeding, respectively, 0.25 × 106 or 2.0 × 106 cells. Cell monolayers were washed with phosphate buffered saline (PBS) and harvested by trypsinization. GSH and eventual other non‐protein thiols were assayed by the method of Ellman (1959) with some previously reported modifications (Vahrmeijer et al. 2000). Total protein was determined by the method of Lowry (1951).
Flow cytometry
Cells were fixed for 10 min in 1% paraformaldehyde in PBS and were permeabilized in ice‐cold methanol for 10 min. Cells were washed extensively in PBS and PBS/1% BSA and were, respectively, incubated with RNaseA (1 mg/ml) in PBS (30 min, 37 °C) and propidium iodide (75 µg/ml) in water (30 min, on ice) (both purchased from Sigma Zwyndrecht, The Netherlands). Samples were measured on a FACscan flow cytometer (Becton Dickinson, San Jose, CA, USA). Propidium iodide fluorescence was measured using a 585/42 nm (FL2) bandpass filter and a minimum of 10 000 events was counted. We approached actual sizes of cell cycle fractions by the Modfit2 program, the Synchronization Wizard. The G2/G1‐ratio was set at 1.91 and S‐phase was defined as a one rectangle compartment.
Statistical evaluation
Difference between mean values of GSH content of cell lines under similar growing conditions was tested by one‐way analysis of variance with Dunnett's multiple comparison test with two‐tailed P‐value (P < 0.05). Difference in GSH levels between 50 and 100% populations of the same population was tested with the unpaired t‐test.
RESULTS
CC531 cells were stably transfected with full‐length bcl‐2 and truncated bcl‐2 gene constructs. Expression of the corresponding Bcl‐2 proteins in 50% confluent cell lines was evaluated by western blotting. Using the mAb #6C8, a human Bcl‐2‐specific monoclonal antibody raised against the entire protein, we found a characteristic 26 kDa band for the full‐length Bcl‐2 protein in CCbcl2 cells (Fig. 2). In line with findings of Hunter et al. (1996), we detected various < 26 kDa bands of different intensities in the other cell lines, corresponding to the molecular weights of truncated Bcl‐2 proteins: approximately 25 kDa in CCΔBH1 cells, c. 24 kDa in CCΔBH3 cells, c. 22 kDa in CCΔBH4 cells and c. 25 kDa in CCΔTM cells (Fig. 2). No Bcl‐2 immunoreactivity could be detected with mAb #6C8 in parental CC531 cells or neo‐transfected CC531 cells, or in CC531 cells transfected with the human bax gene (Fig. 2). Equal protein loading in each lane was confirmed by β‐actin staining; variable transfection efficiency resulted in the most abundant expression of Bcl‐2 immunoreactivity in CCbcl2 and CCΔTM cells and somewhat lower in the other cell lines (Fig. 2). The mAb #6C8 also recognized additional Bcl‐2 immunoreactivity of 22 and 19 kDa proteins in CCbcl2 cells. CCΔTM cells displayed the additional 19 kDa immunoreactivity, while no additional immunoreactivity for Bcl‐2 was detected in whole cell lysates of CCΔBH1 and CCΔBH4 cells. In CCΔBH3 cells, the 19 kDa band was more intense than the 24 kDa band representing the Bcl‐2 protein lacking the BH3 domain (Bcl‐2‐ΔBH3).
Figure 2.
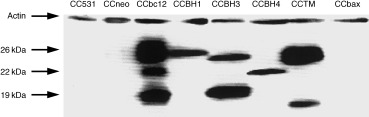
Expression of ( truncated) human Bcl‐2 protein in transfected rat CC531 cells. The expression of human Bcl‐2 on western blot is determined using the mAb #6C8: in parental CC531 cells and CC531 cells transfected with the empty vector (CCneo cells), full length bcl‐2 (CCbcl2 cells) or truncated bcl‐2 gene constructs, missing the coding sequence for the BH1 (CCΔBH1 cells), BH3 (CCΔBH3 cells), BH4 (CCΔBH4 cells), or the TM domain (CCΔTM cells). Additionally, we include CC531 cells transfected with the human bax gene (CCbax cells). The 22 kDa immunoreactivity in CCbcl2 cells could reflect caspase‐(like)‐mediated cleavage of the Bcl‐2 protein at Aspartate residue 34 (Asp34), releasing the BH4 domain. Note that this band appears at similar height on a nitrocellulose filter as the Bcl‐2 protein missing the BH4 domain in CCΔBH4 cells. Immunoreactivity of 19 kDa in CCbcl2, CCΔBH3 (profoundly), and to lesser extent in CCΔTM cells, might result from caspase‐(like) cleavage at Asp64.
Subsequently, parallel cultures of parental CC531 cells and the various bcl‐2 transfectants were grown to approximately 50 or 100% confluence and we assayed both cell cycle distribution by flow cytometry and cellular GSH levels. At 50% confluence, CCbcl2, CCΔBH1, CCΔBH3 and CCΔTM cells were markedly reduced in their cell proliferation compared with parental CC531, neo‐transfected CC531 and CCΔBH4 cells: the G0/G1‐phase was statistically significantly increased and S‐phase fractions were decreased (Table 1). The largest reduction of proliferative activity was observed in CCbcl2 and CCΔTM cells. Proliferative activity was much further reduced when cell cultures had reached total confluence, to approximately similar levels in all cell lines (Table 1).
Table 1.
Cell cycle distribution (%) and GSH levels (nmol GSH/mg total cell protein) of 50 and 100% confluent parental CC531 cell lines , neo‐transfected CC531 cells, and CC531 cells transfected with full‐length bcl‐2 or truncated bcl‐2 gene constructs. a Denotes statistically significant differences in cell cycle distribution at 50 or 100% confluence compared with parental CC531 cells and neo‐transfected cells. Data represent mean ± SD. n = 4–10 for flow cytometric analyses on 50% confluent cell lines, n = 3 for 100% confluent cell populations with all samples in duplicates. a Denotes statistically significant differences in total GSH levels at either 50 or 100% confluence compared with parental CC531 cells and neo‐transfected cells. b Denotes statistically significant differences in GSH levels within one cell line at 50 or 100% confluence. n = 4–6 with samples in duplicates. Data represent mean ± SD
| 50% confluence | 100% confluence | |||||||
|---|---|---|---|---|---|---|---|---|
| Cell cycle | GSH | Cell cycle | GSH | |||||
| G0/G1 (%) | S (%) | G2/M (%) | G0/G1 (%) | S (%) | G2/M (%) | |||
| CC531 | 15.6 ± 3.7 | 63.0 ± 2.7 | 21.4 ± 1.6 | 21.9 ± 2.1 | 54.1 ± 1.0 | 35.1 ± 0.9 | 10.8 ± 0.4 | 30.1 ± 2.3 b |
| CCneo | 17.3 ± 4.2 | 61.7 ± 3.9 | 21.0 ± 2.2 | 22.9 ± 0.8 | 54.3 ± 0.6 | 35.2 ± 0.6 | 10.5 ± 0.3 | 29.6 ± 2.7 b |
| CCbcl2 | 35.5 ± 1.4 a | 46.1 ± 1.3 a | 18.4 ± 0.9 | 30.9 ± 2.0 a | 63.7 ± 1.4 | 23.9 ± 0.3 | 12.4 ± 1.0 | 30.0 ± 2.3 |
| CCΔBH1 | 26.8 ± 2.9 a | 51.1 ± 1.4 a | 22.1 ± 1.5 | 28.4 ± 4.0 a | 51.9 ± 0.5 | 38.0 ± 0.4 | 10.1 ± 0.2 | 31.7 ± 4.0 |
| CCΔBH3 | 25.5 ± 2.4 a | 51.5 ± 3.4 a | 23.0 ± 1.3 | 32.8 ± 2.2 a | 56.4 ± 0.4 | 31.3 ± 5.0 | 12.3 ± 1.4 | 28.2 ± 1.4 b |
| CCΔBH4 | 19.2 ± 2.7 | 60.9 ± 0.9 | 19.9 ± 1.3 | 48.5 ± 7.9 a | 59.8 ± 0.5 | 33.4 ± 2.0 | 6.8 ± 1.2 | 41.9 ± 1.9 a |
| CCΔTM | 29.3 ± 5.7 a | 49.1 ± 3.9 a | 21.6 ± 2.8 | 37.0 ± 4.1 a | 61.2 ± 1.4 | 31.5 ± 0.7 | 7.3 ± 0.3 | 27.8 ± 2.3 b |
Analysis of GSH levels revealed that in 50% confluent cultures, GSH levels were approximately 1.5–2 times higher in cells transfected with full‐length or truncated bcl‐2 than in parental cells, neo‐transfected cells, or CC531 cells transfected with human bax (CCbax cells contained 24.3 ± 3.4 nmol GSH per mg protein). However, in CCΔBH4 cells, GSH levels were even more increased and some 60% higher than the other bcl‐2 transfectants. In 100% confluent cultures, all CC531 cell lines contained very similar GSH levels, except for CCΔBH4 cells in which the GSH level was higher than in other cell lines (Table 1 and Fig. 3).
Figure 3.
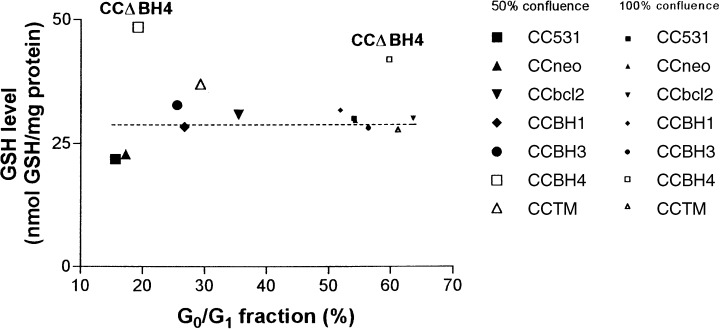
Correlation between growth and cellular GSH levels in parental CC531 cells and bcl‐2 transfectants. GSH levels and cell cycle distribution (based on the G 0 /G 1 ‐phase fraction) of parental CC531 cells and bcl‐ 2 transfected cell lines are plotted. Fifty per cent confluent cell lines are denoted with large icons, 100% confluent cell lines with smaller icons. The dotted line indicates a (presumed) steady‐state level.
We subsequently isolated nuclear fractions of 50% confluent bcl‐2 transfected CC531 cell lines as recently described (Hoetelmans et al. 2000) and evaluated Bcl‐2 immunoreactivity in the nucleus with the mAb #6C8. Nuclear lysates were loaded in such a manner that full‐length Bcl‐2 expression was comparable with expression in whole cell lysate. No immunoreactivity was found in parental, neo‐transfected or bax transfected CC531 cells (data not shown), and no 19 kDa immunoreactivity was detected in any nuclear lysate. At equal protein loading, variable intensities of immunoreactivity of the full‐length or truncated Bcl‐2 protein were found in the nuclear fractions of the bcl‐2 transfectants. Whereas nuclear Bcl‐2‐ΔBH3 was abundantly expressed, the intensity of the Bcl‐2‐ΔBH4 band was extremely low (Fig. 4).
Figure 4.
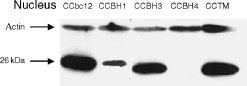
Expression of ( truncated) human Bcl‐2 protein in isolated nuclear fractions of transfected CC531 cells. Western blot analysis is performed on isolated nuclear fractions of CCbcl2, CCΔBH1, CCΔBH3, CCΔBH4 and CCΔTM cells.
DISCUSSION
It has been reported that transfection of cells with the antiapoptotic bcl‐2 gene may lead to an increase of GSH levels (Ellerby et al. 1996; Meredith et al. 1998; Rimpler et al. 1999; Voehringer 1999; Schor et al. 2000; Vahrmeijer et al. 2000). Our data on exponentially growing cells, presented in Table 1, seemed to confirm this. However, the finding that 100% confluent cultures of parental and neo‐transfected cells showed the same GSH levels as cultures of most bcl‐2 transfected cells (both at 50% and 100% confluence) indicates that Bcl‐2 does not up‐regulate GSH synthesis, but rather may prevent the characteristic decrease in GSH levels during the exponential growth phase (Atzori et al. 1990; Hutter et al. 1997). We illustrated this in Fig. 3: the cellular GSH levels in 50% confluent parental and neo‐transfected CC531 cells (G0/G1‐phase fraction of c. 15%) were significantly lower than those in less rapidly dividing cell populations (starting already at G0/G1‐phase fractions of c. 25%), with the exception of CCΔBH4 cells. This raises the question of whether increased GSH levels in some bcl‐2 transfectants reflects reduced mitotic activity in these cells or, alternatively, reduced cellular growth might follow from these relatively high levels of GSH. Regardless of the scenario, these data support a significant role for an overexpression of the Bcl‐2 protein in both cell cycle control and GSH levels.
Reduction of proliferative activity and elevation of cellular GSH levels is comparable among 50% confluent CCbcl2, CCΔBH1, CCΔBH3 and CCΔTM cells. This suggests that both characteristics do not depend on the presence of the BH1, BH3, or TM domain of the Bcl‐2 protein. The finding that CCΔBH4 cells at 50% confluence are not reduced in proliferative activity compared with control CC531 cells, supports earlier findings of Huang et al. (1997), i.e. that the BH4 domain is required for cell growth reduction. Despite the comparable G0/G1‐phase fractions in CCΔBH4, control CC531 and neo‐transfected CC531 cells, control of modulation of GSH levels in CCΔBH4 cells seems to be lost: GSH levels in 50 and 100% confluent CCΔBH4 cells even exceed those of the other bcl‐2 transfectants.
The N‐terminal BH4 domain of Bcl‐2 interacts, among others, with redox‐sensitive transcription factors such as NF‐κB in the cytoplasm (Nakamura et al. 1997). Like Bcl‐2, NF‐κB activation differentially regulates cell cycle progression and apoptosis as was demonstrated in B lymphocytes (Grumont et al. 1998). Hour et al. (2000) and Ivanov et al. (1995) demonstrated competitive binding of Bcl‐2 – through its BH4 domain – with NF‐κB, impairing its activation. Lack of association of Bcl‐2ΔBH4 with NF‐κB in the cytoplasm might result in increased pools of freely moving Bcl‐2ΔBH4, enhancing its potential of increasing GSH levels. Interestingly, Hour et al. (2000) presented data that confirmed the presence of the Bcl‐2‐ NF‐κB complex in nuclear fractions of NIH3T3 cells. The authors speculated that Bcl‐2, when located in the nucleus, may play a role in cell cycle control or apoptosis.
We recently demonstrated that the Bcl‐2 protein resides in interphase nuclei of rat CC531 cells following transfection of the corresponding gene (Hoetelmans et al. 2000). In the present study, we found that Bcl‐2‐ΔBH4 is expressed only at very low levels in the nucleus. Although the cytoplasmic expression of Bcl‐2‐ΔBH4, as detected on a western blot with mAb #6C8, was low compared with full‐length Bcl‐2, it was similar to that of Bcl‐2‐ΔBH1. Nevertheless, CCΔBH1 cells showed a significantly less proliferative activity than CCΔBH4 cells. These data indicate that the nuclear presence of Bcl‐2 requires the presence of the BH4 domain and might explain the lack of reduced proliferation as observed in CCΔBH4 cells. Interestingly, in this context, Voehringer et al. (1998) demonstrated increased nuclear GSH levels in bcl‐2‐transfected HeLa cells. We confirm this observation in preliminary experiments showing that GSH levels in isolated nuclear fractions of 100% confluent parental and neo‐transfected CC531 cells increased from, respectively, 1.9 ± 0.5 and 1.8 ± 0.6 nmol/mg protein to 7.5 ± 0.6 and 7.0 ± 0.2 in 50% confluence. Whereas nuclear GSH levels in CCΔBH4 cells resembled these of control cells, nuclear GSH levels in CCbcl2 cells remained between 5 and 6 nmol/mg protein regardless the proliferative state. These data suggest orchestration of nuclear GSH levels by nuclear residing Bcl‐2 that might relate to control of cell proliferation.
The human Bcl‐2 protein, as overexpressed in CC531 cells, is present in several forms, reflected in western blots as a minor band at 22 kDa and a major band at 19 kDa. In cells undergoing apoptosis, the emergence of a 22‐kDa form has repeatedly been described (Cheng et al. 1997; Fadeel et al. 1999) and it is generally accepted that this cleavage product results from caspase activity (with a pivotal role for caspase‐3). Interestingly, Yamamoto et al. (1998), who applied the same mAb #6C8, reported a Bcl‐2 cleaving activity (directed at the N‐terminal domain), yielding a product of appoximately 19 kDa. This Bcl‐2 fragment was found up‐regulated following apoptosis‐induction and was constitutively present in lymphoid cells (Yamamoto et al. 1998). Cleavage at the Asp64 residue is a likely candidate for yielding this 19 kDa Bcl‐2 fragment (Fig. 1). Although we cannot exclude the possibility that the 22 kDa band detectable in CCbcl2 cells could reflect a subpopulation of apoptotic cells, the abundant presence of 19 kDa Bcl‐2 is unlikely to be related to the process of apoptosis. For instance, in whole cell lysates of CCΔBH3 cells, the majority of Bcl‐2‐ΔBH3 is the 19 kDa form and these cells did not show signs of massive apoptosis. If not related to apoptosis, one could hypothesize that this type of Bcl‐2 cleavage is related to the proliferation‐inhibiting property of Bcl‐2.
In conclusion, our data indicate that proliferating parental and neo‐transfected CC531 cells show lower GSH levels than non‐proliferating cells. Increased GSH levels in cells with an overexpression of the anti‐apoptotic protein Bcl‐2 may reflect (or may lead to) a reduction of cellular proliferation and does not depend on functional presence of the BH1, BH3 or TM domain. Transfection of CC531 cells with a bcl‐2 gene construct missing its N‐terminal BH4 coding sequence does not inhibit cellular proliferation and impairs nuclear translocation of Bcl‐2. Surprisingly, these cells harbour significantly increased GSH levels at both 50 and 100% confluence.
ACKNOWLEDGEMENTS
The authors are indebted to Dr Parslow for providing the bcl‐2 deletion constructs and Dr Wei Zhang for donating the bax gene construct.
REFERENCES
- Atzori L, Dypbukt JM, Sundqvist K, Cotgreave I, Edman CC, Moldeus P, Grafstrom RC (1990) Growth‐associated modifications of low‐molecular‐weight thiols and protein sulfhydryls in human bronchial fibroblasts. J. Cell Physiol. 143, 165. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM (1997) Conversion of Bcl‐2 to a Bax‐like death effector by caspases. Science 278, 1966. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE (1996) Shift of the cellular oxidation‐reduction potential in neural cells expressing Bcl‐2. J. Neurochem. 67, 1259. [DOI] [PubMed] [Google Scholar]
- Ellman GL (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70. [DOI] [PubMed] [Google Scholar]
- Esposti DM, Hatzinisiriou I, McLennan H, Ralph S (1999) Bcl‐2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species‐sensitive probes. J. Biol. Chem. 274, 29831. [DOI] [PubMed] [Google Scholar]
- Fadeel B, Hassan Z, Hellstrom‐Lindberg E, Henter JI, Orrenius S, Zhivotovsky B (1999) Cleavage of Bcl‐2 is an early event in chemotherapy‐induced apoptosis of human myeloid leukemia cells. Leukemia 13, 719. [DOI] [PubMed] [Google Scholar]
- Grimm S, Bauer MK, Baeuerle PA, Schulze‐Osthoff K (1996) Bcl‐2 down‐regulates the activity of transcription factor NF‐kappaB induced upon apoptosis. J. Cell Biol. 134, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, Rourke IJ, O'Reilly LA, Strasser A, Miyake K, Sha W, Gerondakis S (1998) B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF‐kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen‐activated cells. J. Exp. Med. 187, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ (1993) Bcl‐2 functions in an antioxidant pathway to prevent apoptosis. Cell 75, 241. [DOI] [PubMed] [Google Scholar]
- Hoetelmans RWM, Van S looten HJ, Keijzer R, Erkeland S, Van De V elde CJ, Dierendonck JH (2000) Bcl‐2 and Bax proteins are present in interphase nuclei of mammalian cells. Cell Death Differ 7, 384. [DOI] [PubMed] [Google Scholar]
- Hour TC, Chen L, Lin JK (2000) Suppression of transcription factor NF‐kappaB activity by Bcl‐2 protein in NIH3T3 cells: implication of a novel NF‐kappaB p50‐Bcl‐2 complex for the anti‐apoptotic function of Bcl‐2. Eur. J. Cell Biol. 79, 121. [DOI] [PubMed] [Google Scholar]
- Huang DC, O'Reilly LA, Strasser A, Cory S (1997) The anti‐apoptosis function of Bcl‐2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 16, 4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JJ, Bond BL, Parslow TG (1996) Functional dissection of the human Bcl2 protein: sequence requirements for inhibition of apoptosis. Mol. Cell Biol. 16, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter DE, Greene JJ (2000) Influence of the cellular redox state on NF‐kappaB‐regulated gene expression. J. Cell Physiol. 183, 45. [DOI] [PubMed] [Google Scholar]
- Hutter DE, Till BG, Greene JJ (1997) Redox state changes in density‐dependent regulation of proliferation, Exp. Cell Res. 232, 435. [DOI] [PubMed] [Google Scholar]
- Ivanov VN, Deng G, Podack ER, Malek TR (1995) Pleiotropic effects of Bcl‐2 on transcription factors in T cells: potential role of NF‐kappa B p50‐p50 for the anti‐apoptotic function of Bcl‐2. Int. Immunol. 7, 1709. [DOI] [PubMed] [Google Scholar]
- Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE (1993) Bcl‐2 inhibition of neural death: decreased generation of reactive oxygen species. Science 262, 1274. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ (1999) Bcl‐2 gene family and the regulation of programmed cell death. Cancer Res. 59, 1693. [PubMed] [Google Scholar]
- Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN (1993) Bcl‐2/Bax: a rheostat that regulates an anti‐oxidant pathway and cell death. Semin. Cancer Biol. 4, 327. [PubMed] [Google Scholar]
- Linette GP, Li Y, Roth K, Korsmeyer SJ (1996) Cross talk between cell death and cell cycle progression: Bcl‐2 regulates NFAT‐mediated activation. Proc. Natl Acad. Sci. USA 93, 9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH (1951) Protein measurement with the Folin‐Phenol reagent. J. Biol. Chem 193, 265. [PubMed] [Google Scholar]
- Manna SK, Kuo MT, Aggarwal BB (1999) Overexpression of gamma‐glutamylcysteine synthetase suppresses tumor necrosis factor‐induced apoptosis and activation of nuclear transcription factor‐kappa B and activator protein‐1. Oncogene 18, 4371. [DOI] [PubMed] [Google Scholar]
- Meredith MJ, Cusick CL, Soltaninassab S, Sekhar KS, Lu S, Freeman ML (1998) Expression of Bcl‐2 increases intracellular glutathione by inhibiting methionine‐dependent GSH efflux. Biochem. Biophys. Res. Commun 248, 458. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Nakamura K, Yodoi J (1997) Redox regulation of cellular activation, Annu. Rev. Immunol. 15, 351. [DOI] [PubMed] [Google Scholar]
- O'Reilly LA, Harris AW, Tarlinton DM, Corcoran LM, Strasser A (1997) Expression of a bcl‐2 transgene reduces proliferation and slows turnover of developing B lymphocytes in vivo . J. Immunol. 159, 2301. [PubMed] [Google Scholar]
- O'Reilly LA, Huang DC, Strasser A (1996) The cell death inhibitor Bcl‐2 and its homologues influence control of cell cycle entry. EMBO J. 15, 6979. [PMC free article] [PubMed] [Google Scholar]
- Rimpler MM, Rauen U, Schmidt T, Moroy T, De G root H (1999) Protection against hydrogen peroxide cytotoxicity in rat‐1 fibroblasts provided by the oncoprotein Bcl‐2: maintenance of calcium homeostasis is secondary to the effect of Bcl‐2 on cellular glutathione. Biochem. J. 340, 291. [PMC free article] [PubMed] [Google Scholar]
- Schor NF, Rudin CM, Hartman AR, Thompson CB, Tyurina YY, Kagan VE (2000) Cell line dependence of Bcl‐2‐induced alteration of glutathione handling. Oncogene 19, 472. [DOI] [PubMed] [Google Scholar]
- Shibasaki F, Kondo E, Akagi T, McKeon F (1997) Suppression of signalling through transcription factor NF–AT by interactions between calcineurin and Bcl‐2. Nature 386, 728. [DOI] [PubMed] [Google Scholar]
- St. Clair EG, Anderson SJ, Oltvai ZN (1997) Bcl‐2 counters apoptosis by Bax heterodimerization‐dependent and ‐independent mechanisms in the T‐cell lineage. J. Biol. Chem 272, 29347. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM (1984) Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097. [DOI] [PubMed] [Google Scholar]
- Vahrmeijer AL, Hoetelmans RW, Mulder GJ, Schutrups J, Van V lierberghe RL, Van De V elde CJ, Van D ierendonck JH (2000) Development of resistance to glutathione depletion‐induced cell death in CC531 colon carcinoma cells: association with increased expression of bcl‐2. Biochem. Pharmacol. 59, 1557. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM (1988) Bcl‐2 gene promotes haemopoietic cell survival and cooperates with c‐myc to immortalize pre‐B cells. Nature 335, 440. [DOI] [PubMed] [Google Scholar]
- Voehringer DW (1999) Bcl‐2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic. Biol. Med. 27, 945. [DOI] [PubMed] [Google Scholar]
- Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE (1998) Bcl‐2 expression causes redistribution of glutathione to the nucleus. Proc. Natl Acad. Sci. USA 95, 2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, Rapp UR, Reed JC (1996) Bcl‐2 targets the protein kinase Raf‐1 to mitochondria. Cell 87, 629. [DOI] [PubMed] [Google Scholar]
- Wright SC, Wang H, Wei QS, Kinder DH, Larrick JW (1998) Bcl‐2‐mediated resistance to apoptosis is associated with glutathione‐induced inhibition of AP24 activation of nuclear DNA fragmentation. Cancer Res. 58, 5570. [PubMed] [Google Scholar]
- Yamamoto AM, Eloy L, Bach JF, Garchon HJ (1998) N‐terminus cleavage of bcl‐2 by a novel cellular non‐ICE cysteine proteinase. Leukemia 12, 1467. [DOI] [PubMed] [Google Scholar]