

Abstract
Angiogenesis is considered as an essential process for tumour development and invasion. Previously, we demonstrated that cyclin‐dependent kinase inhibition by roscovitine induces a radiosensitization and a synergistic antitumoral effect in human carcinoma but its effect on the microenvironment and tumour angiogenesis remains unknown. Here, we investigated the effect of the combination roscovitine and ionizing radiation (IR) on normal cells in vitro and on tumour angiogenesis in MDA‐MB 231 tumour xenografts. We observed that the combination roscovitine and IR induced a marked reduction of angiogenic hot spot and microvascular density in comparison with IR or roscovitine treatments alone. The Ang‐2/Tie‐2 ratio was increased in presence of reduced vascular endothelial growth factor level suggesting vessel destabilization. In vitro, no radiosensitization effect of roscovitine was found in endothelial, fibroblast, and keratinocyte cells. IR potentiated the antiproliferative effect of roscovitine without inducing apoptosis in endothelial cells. Roscovitine decreased IR‐stimulated vascular endothelial growth factor secretion of MDA‐MB 231 and endothelial cells. A reduction in the endothelial cells invasion and the capillary‐like tube formation in Matrigel were observed following the combination roscovitine and IR. This combined treatment targets angiogenesis resulting in microvessel destabilization without inducing normal cell toxicity.
Introduction
In addition to the genetic and epigenetic changes involved in the acquisition of the cancer cell phenotype, the angiogenesis process was found to have a critical role in tumour progression and invasion (1). In human breast carcinoma, the pro‐angiogenic phenotype was correlated with p53 mutation and up regulation of the vascular endothelial growth factor (VEGF), an important mediator of endothelial cell mitogenesis and survival (2, 3). Several studies have suggested the VEGF level as a predictive as well as a cancer prognostic factor (4, 5).
It is widely accepted that angiogenesis is a dynamic process that implicates vascular remodelling in host tissues surrounding growing tumour. Among the growth factors families, VEGF is a cytokine considered as one of the most potent pro‐angiogenic factor secreted by tumour and surrounding stromal cells through paracrine/autocrine stimulations (3). Both in vitro and in vivo, VEGF was found to mediate the promotion of endothelial cell mitogenesis and survival as a consequence of the activation of mitogen‐activated protein kinase (MAPK) and phosphatidylinositol 3‐kinase (PI3‐K)/Akt signalling pathways, as well as, increasing the expression of proteolytic enzymes involved in stromal degradation leading to neovascularization (6, 7). Similar to VEGF, the angiopoietins Ang‐1 and Ang‐2, and its receptor Tie‐2 are the only known growth factors largely specific for the vascular endothelial cells (8). The activation of Angs/Tie‐2 signal transduction system is considered as an important pathway involved in the vascular maturation, control of vessel quiescence, and vascular integrity (9). Ang‐1 binding to Tie‐2 was found to maintain and stabilize mature vessels by promoting interaction between endothelial cells and surrounding extracellular matrix (8, 9). In contrast, Ang‐2 was identified as a functional antagonist of Ang‐1. It competitively binds to Tie‐2 and plays a crucial role in destabilizing quiescent adult vessels and thus initiates vascular remodelling under VEGF stimulation. However, the binding of Ang‐2/Tie‐2 induced a destabilization of vessels was found to result in regression of vessels in absence of VEGF (9). Therefore, VEGF and the angiopoietin growth factors have a key role in molecular mechanisms underlying the remodelling of host vasculature as well as the angiogenic switch.
Interestingly, studies have shown that the tumour vasculature has a critical role in the tumour response to radiation (10, 11, 12). In particular, Gorski et al. (13) demonstrated that ionizing radiation (IR)‐induced tumour cells promote endothelial cells radioresistance by secreting cytokines such as VEGF that delivers survival signals. The anti‐angiogenic therapies using neutralizing VEGF antibodies showed the enhancement of endothelial cell radiosensitivity resulting in an additive tumour growth delay or a stronger synergistic antitumoral effect than IR alone (13, 14). Recently, Winkler et al. (15) reported the importance of the relationship between the vascular changes after VEGFR2 blockade, tumour hypoxia, and radiation response when anti‐angiogenic therapy and irradiation are combined. The enhancement of radiation therapy was found when VEGFR2 blockade induced a transient vessel normalization leading to an increase of tumour oxygenation.
As a new combined therapy, we previously reported that roscovitine, a cyclin‐dependent kinase (CDK) inhibitor, enhances the antitumoral response when combined with IR, both in vitro and in vivo, in human breast carcinoma (16). However, the effect of the combination roscovitine and IR on the stromal microenvironment, such as neovascularization that contribute to tumour growth remains unknown. To examine this question, we studied the angiogenesis in MDA‐MB 231 xenograft model, the capillary‐like tube formation of endothelial cells, and the response of normal cells to this combination. We analysed in vitro the endothelial cell responses on the expression level of VEGF and its effect on the proliferation, survival, and endothelial cells invasion under paracrine VEGF stimulations.
This study demonstrates that roscovitine decreases the IR‐induced VEGF secretion. This effect is associated with the inhibition of angiogenesis process by a reduction of endothelial cells invasion and the modulation Angs/Tie‐2 pathways resulting in destabilization of tumour vasculature without inducing normal cell radiosensitization.
Materials and methods
Cell culture, reagents, and irradiation conditions
The human breast carcinoma MDA‐MB‐231 cell line was purchased from the American Type Cell Culture and cultured according to its instructions. The primary HMVECs (dermal human microvascular endothelial cells) were from Cambrex (Verviers, Belgium) and cultured at passage 5 in EGM‐2 MV culture medium. The immortalized human microvascular endothelial cell line HMEC‐1 kindly provided by Dr. Randrianarison (CNRS UMR 1582, Institut Gustave Roussy, Villejuif, France) was cultured in MCDB 131 medium (Gibco; Invitrogen, Cergy Pontoise, France) supplemented with 10% foetal calf serum (FCS), 0.1%l‐glutamine, 0.2% penicillin‐streptomycin, 10 ng/mL endothelial growth factor, 1 µg/mL hydrocortisone. The normal keratinocyte HaCat cells kindly provided by Dr. Vozenin–Brotons (IRSN, Fontenay‐aux‐Roses, France) and the immortalized fibroblast MRC5V‐1 were, respectively, cultured in Dulbecco's modified Eagle's medium and RPMI 1640 media supplemented with 10% FCS, 0.1%l‐glutamine, 0.2% penicillin‐streptomycin and maintained in a 5% CO2 humidified atmosphere at 37 °C. Roscovitine (Alexis Coger Co., Paris, France) was dissolved in dimethyl sulfoxide for a stock solution (14 mm) and kept at –20 °C. γ‐Irradiation was delivered by 137Cs source at a dose rate of 1.97 Gy/min.
Clonogenic survival and proliferation assays
Clonogenic survival assays were performed as described elsewhere (16). Following 5 µm roscovitine and/or 4 Gy irradiation treatment cells were cultured up to 12 days at 37 °C in a 5% CO2 humidified atmosphere. Colonies were fixed, stained with crystal violet and counted. Cell proliferation and viability were assessed by Trypan blue exclusion assay. Cells (25 × 103) were plated in triplicate in 6‐well plates. Cells were collected at the indicated time after 4 Gy irradiation and/or 5 µm roscovitine. Total cell number and viable cells (unstained cells) were counted using haemocytometer under a light microscope.
Flow cytometry analysis and apoptosis detection
Cell cycle analysis was performed on exponential growing HMEC‐1 cells. Sham control and treated cells with 4 Gy irradiation and/or 5 µm roscovitine were harvested at the indicated time, washed with ice‐cold phosphate‐buffered saline, fixed with 70% ethanol. DNA content was labelled with propidium iodide in the presence of RNAse (1 mg/mL). Apoptotic cells were determined by counting hyperploids (sub‐G1 peak) and early apoptotic stages were detected using FITC‐labelled annexin V kit (Immunotech, Marseille, France) according to the manufacturer's instruction. Flow cytometry analysis was performed on LeCoulter scan and data were analysed by using multicycle software.
Matrigel invasion assay
HMEC‐1 cell invasion was performed in BIOCOAT MATRIGEL invasion chambers (Becton Dickinson; Invitrogen) that consist of cell culture inserts containing 8 µm pore‐size membranes pre‐coated with Matrigel placed in 24‐well plates. In the lower part of the chamber, 2 × 104 MDA‐MB 231 cells were plated and treated for 48 h with 4 Gy irradiation and/or roscovitine to generate conditioned media. HMEC‐1 cells (5 × 104) (untreated sham and treated with IR or roscovitine or the combination) were added to the upper part of the chamber. The chemoattractant gradient used was the appropriated completed medium with 10% FCS in the upper chambers and 30% FCS in the lower chambers. After 24 h, the HMEC‐1 cells that have invaded through the Matrigel‐coated membrane were fixed with 70% ethanol, stained with crystal violet and counted under a light microscope on the entire membrane surface.
Endothelial cell morphogenesis: tube formation assay
To estimate the formation of endothelial capillary‐like tubes, 12‐well plates were coated with 10 mg/mL Matrigel (BD Bioscience; Invitrogen), allowed to sit at room temperature for 15 min and then incubated at 37 °C for 1 h to the Matrigel to polymerize. HMVEC and HMEC‐1 cells (untreated sham and treated with 4 Gy IR and/or 5 µm roscovitine) were plated, respectively, at the density of 2 × 105 and 105 in 12‐well plate coated with Matrigel and allowed to form capillary‐like tube for 24 h in an incubator at 37 °C in an atmosphere of 5% CO2. Capillary‐like tubes were quantified by counting the anastomoses per field under light microscope with a 5× objective.
Measurement of VEGF levels in conditioned media
MDA‐MB 231 and HMEC‐1 cells were plated in 6‐well plates. Irradiation and/or roscovitine were delivered to exponential growing cells. At the specific time point, VEGF levels in conditioned media were measured by ELISA (human VEGF Quantikine kit, R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions and normalized to cell numbers in each well.
Immunohistochemistry and analyses of microvessels
From tumour growth delay experiments, MDA‐MB 231 xenografts (5 mice/group) were excised on day 30, and divided. To allow comparison, one part of the tumour sample was kept for real‐time quantitative polymerase chain reaction (PCR) analysis (see below). The other part of tumour sample was fixed in ethanol and embedded in paraffin. Tissue sections (4 µm) were subjected to routine deparaffinization and rehydratation before standard avidin–biotin complex peroxidase immunohistochemical staining. Tissue sections were incubated overnight at 4 °C with 1 : 500 dilution of anti‐PECAM‐1 antibody (BD PharMingen, San Diego, CA, USA). Counterstaining was performed with Mayer's haematoxylin before mounting sections in aqueous medium. Image of the whole histological section was recorded using a Nikon SuperCoolscan 8000 ED slide scanner equipped with a FH‐8G1 medical slide holder (Nikon, Champigny‐sur‐Marne, France) and, analysed by an automatic procedure using PixCyt® image analysis software. The resolution was 4000 dots per inch with 1 pixel covering an area of 40 µm2. The microvascular density profiles are expressed as the number of vessels per mm2 of tumour tissue.
RNA isolation and real‐time quantitative PCR
Total RNA was prepared from MDA‐MB 231 xenografts (excised on day 30 of the tumour growth delay experiments) using the total RNA isolation kit (Rneasy, Qiagen, Courtaboeuf, France) according to the manufacturer's instructions. Total RNA quantification and integrity were analysed using Agilent 2100 bioanalyser. First‐strand cDNA was generated from 1 µg of total RNA using SuperScript II (Invitrogen Life Technologies, Carlsbad, CA, USA) and random hexamer primers. cDNA (10 ng) was amplified by PCR using Syber PCR master Mix (Applied Biosystems, Foster City, CA, USA) and 300 nm of primers. The following forward (F) and reverse (R) primers were used for real‐time RT‐PCR: VEGF (F: 5′‐CATCTCTCCTATGTGCTGGCCT‐3′ and R: 5′‐GGGCTGCTGCAATGACGA‐3′), Ang‐1 (F: 5′‐TTAACAGGAGGATGGTGGTTT‐3′ and R: 5′‐GGTTTTGTCCCGCAGTATAGA‐3′), Ang‐2 (F: 5′‐TGGGTCCTGCAGCTACACTTT‐3′ and R: 5′‐TGCACAGCATTGGACACGTA‐3′), PECAM (F: 5′‐GAGCCCAATCACGTTTCAGTTT‐3′ and R: 5′‐TCCTCCCTGCTTCTTGCTAGCT‐3′), and Tie‐2 (F: 5′‐ATGTGGAAGTCGAGAGGCGAT‐3′ and R: 5′‐CGAATAGCCATCCACTATTGTGC‐3′). Thermal cycling conditions consisted of an initial denaturation step at 95 °C, 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min on an ABI PRISM 7700 Sequence Detection System (Applied Biosystems). For each sample, significant PCR fluorescent signals were normalized to a PCR fluorescent signal obtained from the housekeeping gene 18S (Ribosomal RNA control, Applied Biosystems). Relative mRNA quantization was performed by using the comparative ΔΔCT method.
Statistical analysis
Results are expressed as means ± SD. Data were analysed using one‐way anova followed by Student's t‐test. A P‐value of < 0.05 was considered as statistically significant.
Results
Anti‐angiogenic effect of the combination roscovitine and IR in MDA‐MB 231 tumour xenografts
Previously, we reported the antitumoral activity when the roscovitine was combined with IR in MDA‐MB 231 tumours xenografts (16). As shown in Table 1, we measured no tumour growth delaying effect between the roscovitine group and the sham control group. The combined treatment of roscovitine with IR showed a marked growth inhibition of 73% as compared to 54% for the irradiated group (P = 0.02). Importantly, the tumour doubling time for the irradiated group was estimated at 19 days whereas the combination roscovitine and IR groups showed no doubling of tumour volume over 30 days follow‐up. Therefore, we investigated the angiogenesis level in MDA‐MB 231 xenograft samples by using PECAM‐1 immunohistochemistry and real‐time quantitative PCR analysis. In Fig. 1a, PECAM‐1 immunostainning showed in the control group, ‘angiogenic hot spots’ localized heterogeneously within the tumour. Roscovitine or IR alone induced a reduction of 37.5% in microvessel density of tumours. This effect was higher for the combination roscovitine and IR with almost no presence of angiogenic hot spot and a reduction of 63.6% in microvascular density as compared to the control (Fig. 1b). Reliably, we found a significant reduction of mRNA PECAM‐1 expression in the samples treated with the combination as compared to the control (P = 0.01) and treatments alone (P = 0.046). In addition, mRNA VEGF expression was decreased in samples treated with the combination as compared to IR alone (Fig. 1c). To determine if the modulation of mRNA PECAM‐1 and mRNA VEGF levels were associated with an anti‐angiogenic effect of the combination roscovitine and IR, we measured mRNA levels of angiopoietins Ang‐1 and Ang‐2 and their receptor, Tie‐2. Because Ang‐1 and Ang‐2 compete to bind to Tie‐2 and their molar ratio might determine the balance of Angs/Tie‐2 systems. In Fig. 1d, we found that the Ang‐1/Tie‐2 ratio was about 20% lower for the combination roscovitine and IR than the other treatments. However, the Ang‐2/Tie‐2 ratio was about 65% higher (P = 0.02) for the combination than IR or roscovitine alone. The above results collectively show that the combination roscovitine and IR induced a change in the balance of Angs/Tie‐2 system which is characterized by an increase of Ang‐2/Tie‐2 ratio in presence of reduced VEGF level as compared to IR alone or roscovitine alone. These changes are consistent with the decrease of PECAM‐1 and Tie‐2 levels.
Table 1.
Antitumoral activity of the concomitant combination roscovitine plus IR in MDA‐MB 231 xenografts model
| Treatment group | Tumour doubling time (days) | Optimal tumour versus control % (days) | Efficacy scoring a |
|---|---|---|---|
| Control | 7 | ||
| 100 mg/kg | 8 | 100 (11) | – |
| 7.5 Gy | 19 | 50 (28) | + |
| 100 mg/kg + 7.5 Gy | ≥ 30 | 27 (26)* | ++ |
(–) inactive; (+) marginally active; +, + +, + + +, + + + +, active to highly active.
P < 0.05, compared to 7.5 Gy irradiation; Mann–Whitney statistical analysis.
Figure 1.

(a) Localization of microvessels and capillaries in subcutaneous human breast carcinoma MDA‐MB 231 xenografts excised after 30 days of treatments. Paraffin‐tumour embedded sections were stained by immunohistochemistry using PECAM‐1 antibody and counterstained with Mayer's haematoxylin. Black arrows indicate microvessels in tissue section from the irradiated and the combined treatment groups. Images captured at magnification ×200. (b) Quantification of microvessel density and angiogenic hot spot fraction on the whole histological section from images of PECAM‐1 immunostainning by using PixCyt® image analysis software. (c) Real‐time quantitative PCR analysis of relative mRNA levels of PECAM‐1 and VEGF. (d) relative mRNA levels of Ang‐1/Tie‐2 and Ang‐2/Tie‐2 ratios in MDA‐MB 231 xenografts after 30 days of treatments with IR or roscovitine (100 mg/kg) or the concomitant combination. *P < 0.05 (7.5 Gy versus roscovitine + IR); Student's t‐test statistical analysis.
The combination of roscovitine and IR decreases VEGF bioavailability and the invasive potential of HMEC‐1 cells
To investigate in vitro the effect of the combination roscovitine and IR on vascular microenvironment, we first estimated the levels of VEGF released in the conditioned medium of both breast cancer and endothelial cell lines. As shown in Fig. 2a,b, MDA‐MB 231 cells secreted constitutively about 20‐fold more VEGF than HMEC‐1 cells consistent with studies in primary breast carcinomas (2, 5). Roscovitine alone did not significantly affect the level of VEGF released in conditioned media. However, we measured 48 h after IR exposure, a 3‐fold increase of VEGF secretion in MDA‐MB 231 conditioned medium. The level of IR induced VEGF secretion was about 1.8‐fold decrease (P = 0.01) when roscovitine was combined with IR. Similar trends in VEGF bioavailability were measured in roscovitine + IR‐conditioned medium compared to IR‐conditioned medium of HMEC‐1 cells.
Figure 2.

Decrease in radiation‐induced VEGF secretions by roscovitine in (a) MDA‐MB 231 cells and (b) HMEC‐1 cells. The content of VEGF was measured by ELISA on cultured supernatants from exponential growing cells during the indicated incubation time point. (c) Reduction of HMEC‐1 cells invasion by the roscovitine + IR‐conditioned medium of MDA‐MB 231 cells. Invasion of HMEC‐1 endothelial cell was measured by Matrigel invasion chamber assay. Data represent the mean of at least three independent experiments performed in triplicate; error bars, SD. *P < 0.05; Student's t‐test statistical analysis.
To be more closed to the in vivo conditions for the invasion assay, we used a co‐culture system utilizing trans‐well chambers in which MDA‐MB 231 cells were placed in the bottom compartment and endothelial cells were placed in the top compartment, both separated by a Matrigel matrix. In Fig. 2c, we observed that IR‐conditioned medium of MDA‐MB 231 cells induces about a 3‐fold increase in the endothelial cell invasion. This effect was significantly (P = 0.001) reduced by roscovitine + IR‐conditioned medium of MDA‐MB 231 cells leading to about 2.7‐fold inhibition of HMEC‐1 cell invasion. A similar trend of the invasive potential was found between untreated and treated HMEC‐1 cells with the combination, suggesting a major influence of the roscovitine + IR‐conditioned medium of MDA‐MB 231 cells in this effect.
Effect of roscovitine and IR on endothelial tube formation
In addition to proliferation and invasion, the sprouting of endothelial cells and formation of capillary‐like tube structures are important steps in the angiogenic process. In tube formation assay, we observed that both HMEC‐1 and HMVEC endothelial cells were able to form tubes within 24 h of culturing in Matrigel (Fig. 3a). Irradiation alone inhibits the HMEC‐1 and HMVEC tube formation processes, compared to the control and roscovitine treatment. This inhibition was significantly increased when roscovitine and IR was combined simultaneously (Fig. 3). This effect was still observed after 48 h (data not shown). These data suggest that the combination roscovitine and IR could repress capillary‐like tube formation leading to a marked anti‐angiogenic effect in vitro.
Figure 3.

Capillary‐like tube formation in Matrigel. HMEC‐1 and HMVEC cells were treated with roscovitine (5 µm) alone, 4 Gy alone or concomitant combination of roscovitine 5 µm and 4 Gy and allowed to form capillary‐like tubes for 24 h on Matrigel. (a) Microscopic photographs of tube formation are shown (5× objective). (b) Quantitative analysis of capillary‐like tubes per field. Data represent the mean of at least three independent experiments; error bars, SD.
No cytotoxic enhancement of the radiation response of endothelial cells by roscovitine
Next, we investigated whether the decrease in endothelial cell invasion was associated with a cytotoxic effect of the combination roscovitine and IR. The endothelial cell response to the combination roscovitine and IR was estimated by clonogenic survival assay (Fig. 4a). We found no significant decrease in surviving fraction of HMEC‐1 cells when IR and roscovitine were combined in comparison with IR alone. Moreover, we observed no radiosensitization effect of roscovitine in human keratinocyte and fibroblast cell lines (Fig. 4a).
Figure 4.
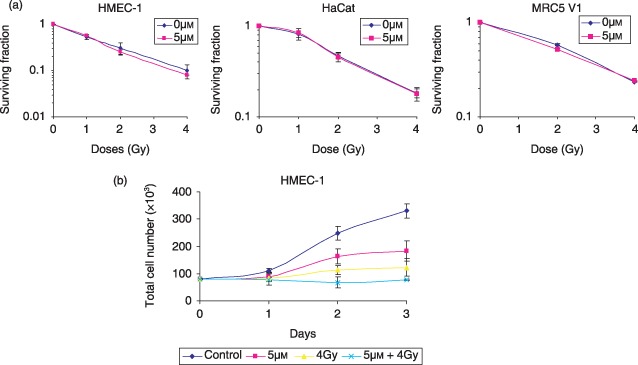
(a) Clonogenic survival assays showing no radiosensitization effect of roscovitine in non‐tumorogenic human cell lines: HMEC‐1 endothelial cells, HaCat keratinocyte cells, and MRC5 V1 fibroblast cells. Data represent the mean of three independent experiments; error bars, SD. (b) Antiproliferative effect of the combination roscovitine and IR in HMEC‐1 cells. Cell number and viability were assessed by Trypan blue exclusion assay. Data represent the mean of three independent experiments; error bars, SD.
The endothelial cell response to the combination was associated with a marked antiproliferative effect of 76% as compared to 45% and 63%, respectively, for roscovitine and IR alone following 72‐h treatments (Fig. 4b). No decrease in cell viability was measured by Trypan blue assay. The same antiproliferative effect of the combination roscovitine and IR was also observed in MRC5V1 and Hacat cells (data not shown). Furthermore, HMEC‐1 cell cycle analysis showed radiation induced G2 cell cycle arrest, which was potentiated in presence of roscovitine and was not associated with an induction of apoptosis. No increase in sub‐G1 peaks and annexin V binding were measured in cells treated with roscovitine, IR alone, or with the combination after 24 h (Fig. 5).
Figure 5.

Kinetic study of HMEC‐1 cell cycle distribution were performed at the indicated time on sham control cells and samples treated with IR and/or roscovitine by using flow cytometry analysis. Apoptosis level was detected by annexin V binding assay at 24 h. A representative experiment from two independent experiments is shown.
Discussion
The regulation of VEGF in the extracellular microenvironment was implicated in the angiogenic switch, promoting the progression of premalignant lesions to an invasive cancer (1, 3). The use of different approaches to inhibit VEGF signalling, including neutralizing antibodies against VEGF or VEGFR‐2, provided evidence that the growth of some tumours is dependent on VEGF‐induced angiogenesis (17, 18, 19). The radiation sensitivity of the tumour vasculature was proposed as a major determinant of the overall response to radiotherapy (10, 11, 12). However, the variable therapeutic responses notified the importance of an optimal scheduling of radiation therapy in combination with the anti‐angiogenic therapies (12, 15).
In the present study, we reported a differential response to radiation therapy modulated by CDK inhibition with roscovitine that changes VEGF levels within the tumour and its microenvironment. The in vivo experiments showed the antitumoral effect of the combination roscovitine and IR (Table 1) associated with a reduction of angiogenesis level within the tumour. As evidence of the marked effect of the combined treatment, we observed a reduction in microvessel density and angiogenic hot spots which were correlated with a decrease in PECAM‐1 and VEGF mRNA levels in tumour samples. These findings might be related to a vessel destabilization and a change in vessel morphology. Early studies reported that VEGF/VEGFR‐2 inhibitions not only block angiogenesis in tumours but also change by reducing the diameter, tortuosity, and permeability of vessel tumours and even transform the surviving tumour vessels into a more normal phenotype (15, 17, 20). We found that this reduction of PECAM‐1 mRNA level was correlated with the highest level of Ang‐2 and the lowest level of VEGF in tumours treated with the combination roscovitine and IR. Holash et al. (9) proposed a new model of angiogenesis that involves a dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. The presence or the absence of VEGF is considered as the switch that determines the outcome of Ang‐2 stimulation in the angiogenic remodelling process. The vessel destabilization was correlated with a high autocrine level of Ang‐2 expression leading to vessel regression in the absence of tumour‐derived VEGF. Ang‐2 destabilizes vessels by competing with Ang‐1 for binding to Tie‐2 and thus inducing pericyte loss. Ang‐1 is described as a transcriptionally regulated molecule in some tumours that acts in a paracrine manner between endothelial cells and the surrounding mesenchyme to maintain vascular integrity. Lund et al. (21) showed in human glioblastoma no change in the angiopoietin levels after IR, whereas they reported an increase of VEGF released in a dose‐dependent manner following irradiation. Moreover, the VEGF up‐regulation by IR was found to be independent of HIF‐1 transactivation. On the other hand, Winkler et al. (15) studied the vascularization changes after VEGFR‐2 blockade and its responses to IR in human brain tumour. It was found that VEGFR‐2 blockade creates a ‘normalization window’ that improves radiation tumour response. This effect was correlated with transient pericyte recruitment by up‐regulation of Ang‐1 just following the VEGFR‐2 blockade treatment. Interestingly, our results show that the combined treatment of roscovitine and IR induces vessels destabilization involving changes in the dynamic balance between Ang‐1 and its functional antagonist Ang‐2 and, might reflect a late stage of a normalization window. Furthermore, in vitro capillary‐like tube formation was significantly impaired after 24‐h treatment with the combination of roscovitine and IR. Although some inter‐cell contact was established, no mature network was generated. These in vitro findings suggest that the combination roscovitine and IR might have an early effect on the control of the angiogenic switch and thus result 30 days after the treatment as vessel destabilization within the tumour.
In the tumour angiogenesis, it was shown that VEGF stimulates neovascularization through autocrine and paracrine secretions from endothelial and tumour cells (1, 3, 8). The release of VEGF selectively induces endothelial cell proliferation and migration, increasing permeability of microvessels and activating proteolytic enzymes involved in tumour invasiveness. Thus, we investigated in vitro whether the treatment roscovitine and IR might influence the endothelial cell invasion by Matrigel invasion chamber assay. Consistent with previous reports, our results show IR‐induced VEGF expression (13, 21). The highest level of VEGF secreted was observed in IR‐conditioned medium of MDA‐MB 231 breast cancer cells. When roscovitine was added to the irradiated cells, we found that the combination induced a decrease in VEGF bioavailability suggesting a modulation of pro‐angiogenic paracrine stimulation from breast cancer cells.
In the present study, we found no enhancement of radiation response by CDK inhibition in fibroblast, keratynocyte, and endothelial cells. We previously demonstrated a radiosensitization effect of roscovitine in the MDA‐MB 231 cell lines (16) and other breast and prostate cancer cell lines. Thus, far to our knowledge, these compelling findings indicate for the first time that roscovitine induces a differential radiation response between normal and cancer cells. On one hand, Mauceri et al. (22) showed different effects on clonogenic survival induced by angiostatin plus IR combination resulting in endothelial cell radiosensitization but not on cancer cells. However, angiostatin inhibits endothelial cell proliferation and combining angiostatin with IR does not induce apoptosis and normal tissue toxicity, although it improves tumour growth inhibition. On the other hand, studies showed that VEGF mediates tumour radioresistance by rescuing the tumour endothelial cells from radiation‐induced cell death (13, 14). By using neutralizing antibodies against VEGF, it was demonstrated that the enhancement of endothelial cell radioresistance depends on the VEGF‐stimulated activation of MAPK and not the PI3‐K/Akt signalling pathways, which is involved in the apoptotic response of endothelial cells (14, 23, 24). The MAPK signalling pathway was found to be mainly activated by IR and to have a role in survival and proliferation. Roscovitine and other CDK inhibitors were shown to target MAPK signalling pathways by inhibition of the kinase activity of ERK1 and ERK2 (25). Little is known about endothelial cell sensitivity to CDK inhibitors. Yet, one study showed that flavopiridol‐induced apoptosis in human umbilical vein endothelial cells was independent of the cell cycle progression (26) suggesting that CDKs may not represent the only critical target enzymes. Induction of apoptosis by CDK inhibitors was found to be cell type‐specific but also dose‐dependent of drug. In the present study, we used a cytostatic concentration of roscovitine and no cytotoxicity was found in normal cells as well as tumour cells. Our data showed that the antiproliferative effect of roscovitine was potentiated by IR and not attributed to apoptosis. In addition to IR, environmental stimuli, such as hypoxia, were found to activate angiogenesis and VEGF expression, but through activation of PI3‐K/Akt pathways (12). However, it was demonstrated that flavopiridol down‐regulates hypoxia‐induced VEGF in human monocytes (27). Because VEGF induction by IR and hypoxia uses different signalling pathways, it still remains to be determined by which mechanism roscovitine might induce modulation of VEGF level stimulated by IR.
In conclusion, we demonstrated that the combination of roscovitine and IR leads to the inhibition of endothelial cell proliferation, invasion and, is associated to a decrease in VEGF bioavailability. Furthermore, in vitro capillary‐like tube formation was impaired. Interestingly, no enhancement in endothelial cell killing was observed by contrast with strategies using anti‐angiogenic agents combined with IR (13, 20, 28). In our xenograft model, we observe a marked decrease in microvascular density induced by the combination associated with a change in VEGF expression and Angs/Tie‐2 pathways. Evidences on the relationship between tumour and its vasculature in tumour growth led us to hypothesize that the combination roscovitine and IR might play a crucial role in the early tumour stage during the angiogenic switch and result in the destabilization of tumour vasculature. Therefore, the combination of roscovitine and IR appears to be an important strategy that combines antitumoral activity and anti‐angiogenic effect without increasing normal cell damages.
Acknowledgements
We thank Patrice Ardouin, Valérie Frascogna, and the animal facility staff for animal care at the Institut Gustave Roussy, Villejuif, France. We thank Arlette Vervisch for flow cytometry at the CNRS UPR 9079, Institut Andre Lwoff, Villejuif, France.
References
- 1. Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 3, 353–364. [DOI] [PubMed] [Google Scholar]
- 2. Linderholm BK, Lindahl T, Holmberg L, Klaar S, Lennerstrand J, Henriksson R et al (2001) The expression of vascular endothelial growth factor correlates with mutant p53 and poor prognosis in human breast cancer. Cancer Res. 5, 2256–2260. [PubMed] [Google Scholar]
- 3. Ferrara N, Gerber HP, Lecouter J (2003) The biology of VEGF and its receptors. Nat. Med. 6, 669–676. [DOI] [PubMed] [Google Scholar]
- 4. Sledge GW Jr (2002) Vascular endothelial growth factor in breast cancer: biologic and therapeutic aspects. Semin. Oncol. 11, 104–110. [DOI] [PubMed] [Google Scholar]
- 5. Foekens JA, Peters HA, Grebenchtchikov N, Look MP, Meijer‐Van Gelder ME, Geurts‐Moespot A et al (2001) High tumor levels of vascular endothelial growth factor predict poor response to systemic therapy in advanced breast cancer. Cancer Res. 14, 5407–5414. [PubMed] [Google Scholar]
- 6. Berra E, Pages G, Pouyssegur J (2000) MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 1, 139–145. [DOI] [PubMed] [Google Scholar]
- 7. Thakker GD, Hajja DP, Muller WA, Rosengart TK (1999) The role of phosphatidylinositol 3‐kinase in vascular endothelial growth factor signalling. J. Biol. Chem. 15, 10002–10007. [DOI] [PubMed] [Google Scholar]
- 8. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J (2000) Vascular‐specific growth factors and blood vessel formation. Nature 470, 242–248. [DOI] [PubMed] [Google Scholar]
- 9. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D et al (1999) Vessel cooption, regression and growth in tumors mediated by angiopoietins and VEGF. Science 284, 1994–1998. [DOI] [PubMed] [Google Scholar]
- 10. Garcia‐Barros M, Paris F, Cordon‐Cardo C, Lyden D, Rafii S, Haimovitz‐Freidman A et al (2003) Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 300, 1155–1159. [DOI] [PubMed] [Google Scholar]
- 11. Moeller BJ, Cao Y, Li CY, Dewhirst MW (2004) Radiation activates HIF‐1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals and stress granules. Cancer Cell 5, 429–441. [DOI] [PubMed] [Google Scholar]
- 12. Wachsberger P, Burd R, Dicker AP (2003) Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin. Cancer Res. 6, 1957–1971. [PubMed] [Google Scholar]
- 13. Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM et al (1999) Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 14, 3374–3378. [PubMed] [Google Scholar]
- 14. Gupta VK, Jaskowiak NT, Beckett MA, Mauceri HJ, Grunstein J, Johnson RS et al (2002) Vascular endothelial growth factor enhances endothelial cell survival and tumor radioresistance. Cancer J. 1, 47–54. [DOI] [PubMed] [Google Scholar]
- 15. Winkler F, Kozin SV, Tong RT, Chae S, Booth MF, Garkavtsev I et al (2004) Kinetics of vascular normalisation by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin‐1, and matrix metalloproteinases. Cancer Cell 6, 553–563. [DOI] [PubMed] [Google Scholar]
- 16. Maggiorella L, Deutsch E, Frascogna V, Chavaudra N, Jeanson L, Milliat F et al (2003) Enhancement of radiation response by roscovitine in human breast carcinoma in vitro and in vivo . Cancer Res. 10, 2513–2517. [PubMed] [Google Scholar]
- 17. Huang J, Frischer JS, Serur A, Kadenhe A, Yokoi A, McCrudden KW et al (2003) Regression of established tumors and metastases by potent vascular endothelial growth factor blockade. Proc. Natl. Acad. Sci. USA 100, 7785–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang W, Ran S, Sambade M, Huang X, Thorpe PE (2002) A monoclonal antibody that blocks VEGF binding to VEGFR2 (KDR/Flk‐1) inhibits vascular expression of Flk‐1 and tumor growth in an orthotopic human breast cancer model. Angiogenesis 5, 35–44. [DOI] [PubMed] [Google Scholar]
- 19. Kim KJ, Li B, Armanini M, Gillett N, Phillips HS, Ferrara N (1993) Inhibition of vascular endothelial growth factor‐induced angiogenesis suppresses tumour growth in vivo . Nature 362, 841–844. [DOI] [PubMed] [Google Scholar]
- 20. Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E (1999) Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Invest. 103, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lund EL, Bastholm L, Kristjansen PEG (2000) Therapeutic synergy of TNP‐470 and ionizing radiation: effects on tumor growth, vessel morphology, and angiogenesis in human glioblastoma multiforme xenografts. Clin. Cancer Res. 6, 971–978. [PubMed] [Google Scholar]
- 22. Mauceri HJ, Hana NN, Beckett MA, Gorski DH, Staba M, Stellato KA et al (1998) Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature 394, 287–291. [DOI] [PubMed] [Google Scholar]
- 23. Tan J, Hallahan DE (2003) Growth factor‐independent activation of protein kinase B contributes to the inherent resistance of vascular endothelium to radiation‐induced apoptotic response. Cancer Res. 22, 7663–7667. [PubMed] [Google Scholar]
- 24. Edwards E, Geng L, Tan J, Onishko H, Donnelly E, Hallahan DE (2002) Phosphatidylinositol 3‐kinase/Akt signaling in the response of vascular endothelium to ionizing radiation. Cancer Res. 16, 4671–4677. [PubMed] [Google Scholar]
- 25. Knockaert M, Lenormand P, Gray N, Schultz P, Pouyssegur J, Meijer L (2002) p42/p44 MAPKs are intracellular targets of the CDK inhibitor purvalanol. Oncogene 42, 6413–6424. [DOI] [PubMed] [Google Scholar]
- 26. Brusselbach S, Nettelbeck DM, Sedlacek HH, Muller R (1998) Cell cycle‐independent induction of apoptosis by the anti‐ tumor drug Flavopiridol in endothelial cells. Int. J. Cancer 1, 146–152. [DOI] [PubMed] [Google Scholar]
- 27. Melillo G, Sausville EA, Cloud K, Lahusen T, Varesio L, Senderowicz AM (1999) Flavopiridol, a protein kinase inhibitor, down‐regulates hypoxic induction of vascular endothelial growth factor expression in human monocytes. Cancer Res. 21, 5433–5437. [PubMed] [Google Scholar]
- 28. Rofstad EK, Henriksen K, Galappathi K, Mathiesen B (2003) Antiangiogenic treatment with thrombospondin‐1 enhances primary tumor radiation response and prevents growth of dormant pulmonary micrometastases after curative radiation therapy in human melanoma xenografts. Cancer Res. 14, 4055–4061. [PubMed] [Google Scholar]