

Abstract
Objectives: Bone marrow‐derived mesenchymal stem cells (BM‐MSC) have been widely used for cell therapy and tissue engineering purposes. However, there are still controversies concerning safety of application of these cells after in vitro expansion. Therefore, we aimed to investigate the characteristics of rabbit BM‐MSC during long‐term culture.
Materials and methods: In this study, we have examined growth kinetics, morphological changes, differentiation potential and chromosomal abnormalities, as well as tumour formation potential of rabbit BM‐MSC in long‐term culture.
Results and conclusion: We found that shortly after isolation, proliferation rate of rabbit BM‐MSC decreases until they enter a dormant phase. During this period of quiescence, the cells are large and multinucleate. After some weeks of dormancy we found that several small mononuclear cells originated from each large multinucleate cell. These newly formed cells proliferated rapidly but had inferior differentiation potential. Although they were immortal, they did not have the capability for tumour formation in soft agar assay or in nude mice. This is the first report of spontaneous, non‐tumorigenic immortalization of BM‐MSC in rabbits. The phenomenon raises more concern for meticulous monitoring and quality control for using rabbit BM‐MSC in cell‐based therapies and tissue engineering experiments.
Introduction
Mesenchymal stem cells (MSC) are known to be multipotental cells that can be isolated from different sources such as the bone marrow, adipose tissue or umbilical cord blood (1, 2). They have been widely used in cell therapy experiments. To provide sufficient numbers of cells for transplantation, their in vitro expansion is commonly an unavoidable part of clinical trials and animal studies. Although there are many reports on safe application of MSCs in cell therapy and tissue engineering after in vitro expansion (3, 4, 5, 6, 7, 8), several investigators have recently debated the safety of these cells and expressed the opinion that in vitro expansion of human, mouse and rat MSCs could lead to immortalization and neoplastic changes (9, 10, 11). Although rabbit MSCs have been widely used in animal models of tissue engineering (12, 13, 14), their abnormal alteration during in vitro culture had not been studied up to now.
In this study, we have examined population growth kinetics, morphological changes, differentiation potential and chromosomal modifications as well as tumour formation potential of rabbit bone marrow‐derived mesenchymal stem cells (BM‐MSC) during long‐term ex vivo expansion. We found that culture conditions widely affect characteristics of these cells and lead to immortalization, but not to neoplastic change.
Materials and methods
Isolation and culture of rabbit BM‐MSC
Animal care and all experimentation were approved by the ethical committee of Tarbiat Modares University (Tehran, Iran) and were according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. MSC were isolated from bone marrow aspirates of 10 white New Zealand rabbits as described previously (15). Briefly, aspirates were obtained using 18‐gauge needles on a heparinized 10 ml syringes. Aspirated material was diluted 1:6 with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2.5 μg/ml amphotericin B (all from Gibco‐BRL, Grand Island, NY, USA) and maintained in an incubator. After 24 h, non‐adherent cells were discarded, adherent cells were washed three times in PBS (Gibco) and fresh medium was added. Culture medium was changed every 3 days and at approximately 50% confluence, cells were detached using trypsin–EDTA (Gibco) and were replated; cells were considered as passage 1. Cultures were maintained at 37 °C and 5% CO2 and passaged when reaching confluence of around 80%.
Colony‐forming unit‐fibroblast assay and doubling time (DT) calculation
Colony‐forming unit‐fibroblast (CFU‐F) assay was performed with initial cell density of 100 and 200 cells cultured in a 10 cm plate and medium was changed every 3–4 days. After colony formation, colonies were stained with 0.2% (w/v) violet crystal (Sigma‐Aldrich, St Louis, MO, USA) solution in 20% (v/v) methanol (Merck, Darmstadt, Germany) and counted. DT was calculated by the following formula:
(N 2 indicates the number of cells counted at time, h; N 1, number of cells seeded). All experiments were repeated three times.
Senescence‐associated β‐galactosidase staining
Activity of senescence‐associated β‐galactosidase was detected at pH 6 using senescence β‐galactosidase staining kit (Cell Signalling, Danvers, MA, USA) according the manufacturer’s instructions. Cells were examined using a light microscope for blue stain resulting from β‐galactosidase activity.
Multilineage differentiation
For osteogenic differentiation, cells were cultured in medium containing 10 nm dexamethasone, 0.2 mm ascorbic acid bi‐phosphate and 10 mmβ‐glycerophosphate (all from Sigma‐Aldrich). After 3 weeks induction, cells were stained with alizarin red S to assess mineralization. For adipogenic differentiation, cells were cultured in the presence of 0.5 mm hydrocortisone, 0.5 mm isobutylmethylxanthine and 60 mm indomethacin (all from Sigma‐Aldrich) for 3 weeks and then stained with oil red O solution (Sigma). For differentiation to chondrocytes, 2 × 105 cells were centrifuged to form a pelleted micromass and then treated with TGF‐beta (10 ng/ml; Peprotech, Rocky Hill, NJ, USA), ascorbic acid bi‐phosphate (50 μm), dexamethasone (100 nm) and bFGF (10 ng/ml; Peprotech) for 3 weeks. Chondrocyte differentiation was assessed by alcian blue staining on sections obtained from micromasses.
Chromosomal study
Chromosomal analysis was carried out using a standard GTG banding technique. Mitotic cell division was arrested using 10 μg/ml colcemid solution on a monolayer cell culture; detached cells were treated with hypotonic solution (0.075 m KCl, Merck), and subsequently were fixed in 3:1 (v/v) methanol/acetic acid (Merck). Fifty mitotic cells were studied for each sample.
Transduction of BM‐MSC with lentiviral vector
To track BM‐MSC after transplantation into recipient animals, they were labelled with GFP. Cells were seeded in four‐well plates at a density of 20 000 cells per well and were transduced with lentiviral vector containing e‐GFP gene at multiplicity of infection of 10, in the presence of 8 μg/ml polybrene. Cells were washed in PBS twice after 12 h incubation. After 5 days, cells were visualized using an inverted fluorescence microscope (Nikon TE2000) and transduction efficiency was determined by flow cytometry using PAS flow cytometer and Flomax software (Partec, Münster, Germany).
Soft agar transformation assay
In vitro assessment of tumorigenicity was performed by soft agar transformation assay in six‐well plates. Each well was coated with 1 ml of 0.5% low‐melting‐point agarose (Burlington, ON, Fermentas, Canada), and 2500 cells were suspended in 1 ml of 0.35% agarose dissolved in DMEM with 10% FBS and overlaid. Finally, 500 μl culture medium was added and changed twice a week. After 4 weeks incubation at 37 °C in a humidified atmosphere with 5% CO2, plates were examined using the inverted microscope for formation of colonies. In this study, MKN45 cell line was used as positive control.
In vivo tumour formation assay
To study in vivo tumour formation potential, 0.5 × 106 and 5 × 106 cells were transplanted into nude mice through the tail vein or by subcutaneous injection, respectively. MKN45 cells were used as positive controls. Over 16 weeks, mice were examined grossly for possible tumour development and/or illness. After this period and after being killed, lung, spleen and liver were removed and fixed in phosphate‐buffered formalin and embedded in paraffin wax. Haematoxylin and eosin (H&E) staining and Giemsa staining were performed on 4 μm sections.
Statistical analysis
Data are presented as mean ± SD. Statistical analysis was carried out using spss software (version 11; SPSS Inc., Chicago, IL, USA) and Student’s t‐test was used to compare results. P‐value of <0.05 was considered statistically significant.
Results
Rabbit BM‐MSC underwent several changes in morphology and population growth kinetics during in vitro expansion
Bone marrow aspirated samples were transferred to cell culture flasks and after 24 h, non‐adherent cells were removed and remaining cells were cultured under standard culture conditions. Several days after isolation, expanding colonies were observed. Once these colonies had covered in the region of 50% of the surface of their dish, cells were detached and plated as passage 1 cells (Fig. 1a). These cells proliferated with population doubling time of 33 ± 4 h. After 25 ± 6 days (3.2 ± 1.3 passages), cell morphology was seen to change gradually into large polygonal shapes and their proliferation rate decreased until they stopped dividing and entered a fairly long dormant phase (Fig. 1b). During the period of dormancy, culture medium was changed twice weekly without passaging. Interestingly, we noted that in most of these large cells, there existed several nuclei (Fig. 1c). Thus it seemed that although the cells did not proliferate during this fairly long period, number of nuclei in each cell increased. After 35 ± 7 days quiescence, in seven out of 10 isolations, several small cells were derived from each large multinucleate cell (Fig. 1d,e). These newly formed cells were mononuclear and proliferated continuously over a long time period (more than 1 year; Fig. 1f). In three out of 10 isolations, the large multinucleate cells degraded and detached from the culture surface. Based on these observations, we have categorized the course of in vitro expansion of rabbit BM‐MSC in three parts: (i) Normal phase: period of normal proliferation of isolated cells before they became large and quiescent (25 ± 6 days). (ii) Dormant phase: stage at which the cells became large and multinucleate (35 ± 7 days). (iii) Immortality phase: referring to the long period of proliferation of small mononuclear cells that emerged from the large multinucleate cells.
Figure 1.
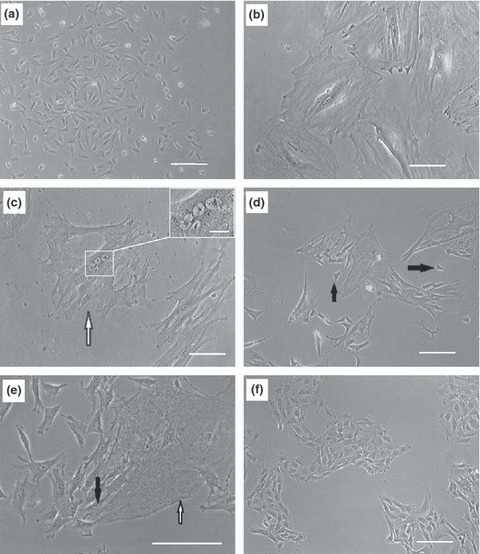
Morphology of rabbit BM‐MSCs in different phases of population growth. Bone marrow contents were transferred to flasks and after 24 h, non‐adherent cells were discarded. Remaining cells were expanded and after reaching 50% confluence, they were detached and replated as passage 1 cells (a). Proliferation rate of the cells gradually descended until they stopped dividing after 3.2 ± 1.3 passages. At this point, cells became large (b) and subsequently, several nuclei were visible in each large cell (arrow head), nuclei at higher magnification (inset scale bar: 20 μm) (c). After 35 ± 7 days in the dormant phase, each large multinucleate cell broke into several small cells (d and e). White arrow shows a large multinucleate cell and black arrows demonstrate newly formed small cells. Small cells proliferated constantly and rapidly over extended periods (f). Scale bars: 100 μm.
DT and CFU‐f remained almost constant during the immortality phase
During writing this manuscript (more than 1 year after isolation), the immortal MSC are still in continuous culture and have exceeded more than 100 passages with almost constant DT equal to 25 ± 1.5 h. These immortal cells still observe contact inhibition and stop dividing as they become confluent. To distinguish between passage numbers of normal and immortal cells, from now on in this report, passage number of cells in normal and immortal phases will be shown as Pn and Pi respectively. Growth kinetics of rabbit BM‐MSC in different phases is shown in Fig. 2.
Figure 2.
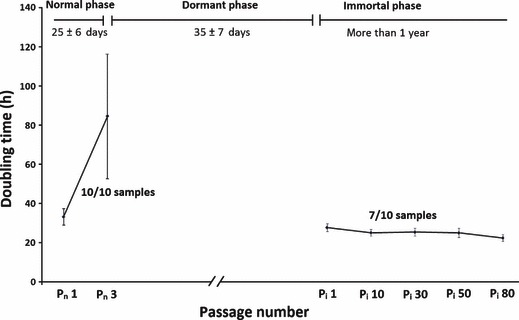
Growth kinetics of rabbit BM‐MSCs. Population doubling time (DT) was calculated for cells at different passages. As cells did not proliferate in dormant phase, DT was not determined for this period.
To compare clonogenic potential of cells in normal and immortality phases, CFU‐F assays were performed. Colony formation potential of cells in Pn 1 was 40 ± 10% and gradually decreased until it was lost at the beginning of the dormant phase. Likewise, during the immortality phase, colony formation potential remained almost constant with a range of 58–63% (Table 1).
Table 1.
Comparison of characteristics of rabbit BM‐MSC in normal and immortality phases
| Phase | Passage number | Differentiation potential | CFU‐F assay (%) | Aneuploidy | Soft agar colony formation | Tumour formation in NOD/SCID mice | ||
|---|---|---|---|---|---|---|---|---|
| Osteocyte | Chondrocyte | Adipocyte | ||||||
| Normal | Pn 1 | 10/10 | 10/10 | 10/10 | 40 ± 10 | 0/10 | 0/10 | 0/10 |
| Immortal | Pi 1 | 7/7 | 0/7 | 0/7 | 58 ± 2 | 0/7 | 0/7 | 0/7 |
| Pi 10 | 7/7 | nd | nd | 57 ± 3 | 0/7 | 0/7 | nd | |
| Pi 30 | 7/7 | nd | nd | 59 ± 3 | 1/7 | 0/7 | 0/7 | |
| Pi 50 | 7/7 | nd | nd | 60 ± 3 | 5/7 | 0/7 | nd | |
| Pi 80 | 7/7 | 0/7 | 0/7 | 63 ± 4 | 7/7 | 0/7 | 0/7 | |
nd, not determined; CFU‐F, colony‐forming unit‐fibroblast.
To study proliferation rate of the cells further, senescence‐associated β‐galactosidase staining was performed; this was strongly positive in cells of the dormant phase. As cells resumed proliferation and entered the immortality phase, they stopped expressing this marker (Fig. 3).
Figure 3.
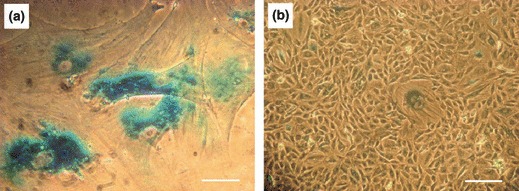
Senescence associated β‐galactosidase staining. The cells in the dormant phase (a) and immortality phase (b). Blue stain indicates activity of senescence‐associated β‐galactosidase at pH 6. Scale bars: 100 μm.
Plasticity of cells decreased in the early immortality phase
To investigate whether the dramatic alterations in MSC during long‐term culture affected functions of the cells, multilineage differentiation potential was examined at different time points. MSC in Pn 1 had the potential to differentiate into adipocytes, osteocytes and chondrocytes in appropriate inductive media (Fig. 4). In contrast, MSC in immortality phase preserved only osteo‐lineage differentiation potential and lost their ability to differentiate into chondrocytes and adipocytes immediately after immortalization (Table 1).
Figure 4.
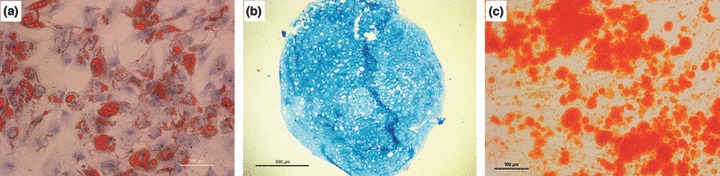
Plasticity of rabbit BM‐MSCs in normal phase examined by differentiation of cells into particular lineages in the presence of appropriate induction media. Cells had potential to differentiate into adipocytes as assessed by oil red O staining (a), Alcian blue staining confirmed differentiation into chondrocytes (b). Cells could also differentiate into the osteoblast lineage as shown by alizarin red S staining (c). Scale Bar: 100 μm (a and c), 500 μm (b).
Chromosomal abnormalities occurred in the late immortality phase
We were interested to determine whether chromosomal number remained intact during long‐term culture. Therefore, cytogenetic analysis was performed on normal phase cells and different passages of immortality phase. These rabbit MSC had normal ploidy with 44 chromosomes per cell in Pn 1, Pi 10 and Pi 20. Six out of seven samples in Pi 30 had normal ploidy and in one sample, a mix of cells with normal (44) and abnormal (45 and 46) chromosome numbers. In all the seven samples examined in P i 80, no population with normal chromosome number was detected and all examined cells were aneuploid with a range of 43–48 chromosomes per cell (Fig. 5).
Figure 5.
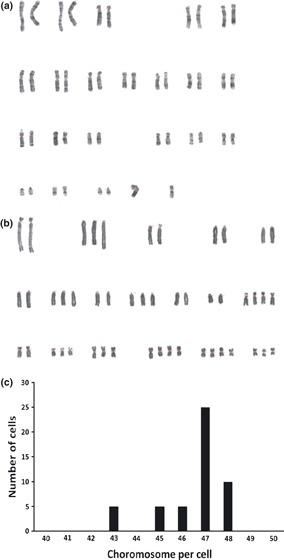
Representative figure of cytogenetic studies on rabbitBM‐MSCs. Cells did not show profound chromosomal aneuploidy in Pi 20 (a), whereas all samples were aneuploid in Pi 80 (b). Graph c shows chromosome numbers per cell for 50 cells in Pi 80.
Rabbit BM‐MSC were not tumorigenic
Soft agar colony formation assay is known to be an in vitro equivalent of tumour formation in nude mice (16). Therefore, we employed this assay to investigate in vitro tumour formation potential of the cells. After repeating the experiment on three occasions, none of the isolations of MSC from Pn 1 and Pi 1, Pi 10, Pi 30, Pi 50 and Pi 80 was observed to form colonies in soft agar after 4 weeks (Fig. 6a). MKN45 cell line, used as positive control in this test, formed several colonies (Fig. 6b).
Figure 6.
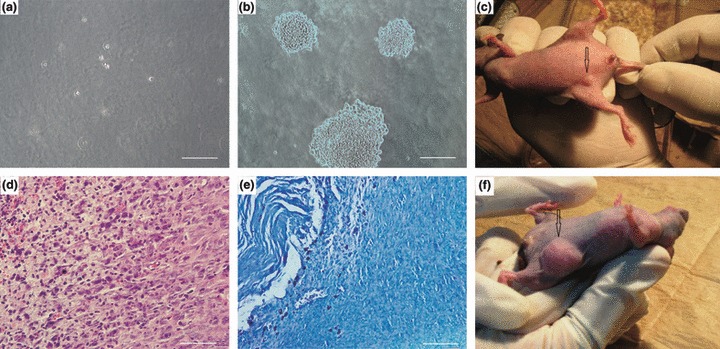
Assessment of in vitro and in vivo tumour formation potential of rabbit BM‐MSCs. MSC could not form colonies in soft agar colony formation assay (a), whereas under the same conditions, MKN45 cells (used as positive controls), formed colonies (b). In mice transplanted with immortal MSCs, within a few days, small nodules became visible at the transplantation site (c). This nodule disappeared by day 8. Histopathological examination of H&E‐stained slides (d) showed that cells were plump to spindle‐shaped with merging eosinophilic cytoplasm. In the centre, cytoplasm of cells showed hydropic clear changes and at the left, necrosis was seen. Apoptotic cells were also noted. Mast cells were arranged at the periphery of the mass in Giemsa‐stained samples (e). No indication of neoplastic change was seen. MKN45 cells formed tumours at the transplantation site (arrowhead) (f). Scale Bars: 100 μm.
In vivo tumorigenic potential was examined by subcutaneous transplantation of normal and immortal cells into nude mice (5 × 106 cells per mouse); mice were followed‐up for 16 weeks after transplantation. In all animals receiving immortal cells, several days after transplantation, a small round nodule (with a maximum diameter of 2 mm on day 5) emerged at the injection site (Fig. 6c). Interestingly, in all experiments, this nodule disappeared after day 8. Histopathological examination of the nodules revealed presence of plump spindle cells with merging eosinophilic cytoplasm and vesicular oval nuclei – but with no signs of neoplasia. These tight aggregates were surrounded by few inflammatory cell infiltrations such as neutrophils and mast cells. The cells showed infiltrative pattern of growth within adjacent soft tissue and muscle (Fig. 6d,e). After 16 weeks of follow‐up, no sign of tumour formation or illness was observed in the animals transplanted with either normal or immortal cells. MKN45 cell line served as positive control in this test and formed progressively growing tumours, detectable after 3 weeks transplantation (Fig. 6f).
To investigate systemic effects of MSC on nude mice, 5 × 105 cells from Pn 1, Pi 1, Pi 30 and Pi 80 were transplanted into each mouse by tail vein injection. No sign of illness was observed in the mice over the 16 weeks follow‐up. Histopathological examination of spleen, lung and liver revealed normal histology with no signs of tumour formation.
Discussion
Here, for the first time in the present study, we have demonstrated that after several passages, proliferation rate of rabbit BM‐MSC declined and cells entered a dormant phase, followed by a phase of immortal proliferation. Immediately after immortalization, cells lost their potential for differentiation into adipocyte and chondrocyte lineages and showed prominent aneuploidy in the late immortality phase. Thus, our findings indicate that spontaneous immortalization is a feature of rabbit BM‐MSCs when they are kept in culture over an appropriate time period. Although immortal MSCs showed chromosomal abnormalities and were not tumorigenic as indicated by in vitro and in vivo assay.
Multinucleation and enlarging of individual cells in the dormant phase were one of the interesting features of these cells during in vitro culture. This phenomenon can also be seen in culture of mouse and rat BM‐MSC (our unpublished data). Several mechanisms could account for this event but nuclear division without cytokinesis (17) is one of the most likely scenarios. After some weeks in the dormant state, each large multinucleate cell produced several small mononuclear cells. Similar observations have been reported on other cell types. Walen et al. described generation of immortalized cells from multinucleate senescent epithelial cells over a microscopically visible process (18). Recently, Gosselin et al. have also demonstrated that transformed and tumorigenic cells are generated from senescent keratinocytes by an unusual budding‐mitosis mechanism (19). The exact reasons of such alterations in the cells during in vitro culture remain to be determined. However, it is generally believed that culture conditions such as the two‐dimensional environment and culture medium ingredients impose various stresses on cells during in vitro expansion (20, 21). In addition, proliferation of the cells in the body is commonly controlled by restrictive mechanisms, which are absent under culture conditions (22).
Similar to our findings, it has been recently reported that rat BM‐MSC undergo spontaneous transformation without tumorigenicity during in vitro expansion (9, 23). Chromosomal abnormality has also been reported as a common finding in cultures of human and mouse BM‐MSC (10, 11). However, in contrast to our data that rabbit BM‐MSC were not tumorigenic, human and mouse BM‐MSCs have been shown to possess the potential of tumour formation after injection to immunodeficient mice (10, 11). In our study, the small nodules that formed in mice a few days after subcutaneous transplantation of immortal cells did not show neoplastic features in histopathological examination and disappeared within 1 week. Formation of this nodule might be caused by high proliferation rate of the immortal cells, which continued to expand for some days after transplantation.
It should be noted that as immortalized rabbit BM‐MSCs are morphologically similar to cells of early normal phase (Fig. 1a,f) perhaps the immortalization phenomenon is ignored by researchers and immortalized cells could be used in experiments instead of normal cells – this could lead to controversial results. To avoid this fault, unusual morphology of large multinucleate cells in the dormant phase could be used as a warning of initiation of a course of striking changes in the cells.
Our data on loss of adipocyte and chondrocyte differentiation potential in the early immortality phase is corroborated with other previous reports indicating that differentiation potential of MSCs isolated from different sources decreases after transformation (10, 24). Therefore, it could be assumed that long‐term expansion of MSCs could be associated with losing of some functions.
The findings of this study and similar studies indicate that extensive culture of BM‐MSC, which is an unavoidable part of most experiments, to provide sufficient cells for in vitro and in vivo studies, not only results in chromosomal abnormalities, but also is probably associated with functional imperfection of the cells. In contrast to rabbit BM‐MSCs, which have a short normal phase (3.2 ± 1.3 passages), according to our previous findings and consistent with the literature (25, 26, 27), human BM‐MSC could be expanded for more than eight passages without any sign of senescence or abnormality, which provides sufficient cell population for further experiments. Manipulation of culture conditions and mimicking in vivo microenvironments could be further investigated to prevent spontaneous immortalization of rabbit BM‐MSCs during ex vivo expansion. Although rapid immortalization of these cells limits their applications for cell therapy and tissue engineering purposes, they could be utilized as a model for basic biology studies on mitosis, multinucleation and immortalization.
Conflict of interest
The authors state that there are no conflicts of interest.
Acknowledgements
This project was supported by a grant from Stem Cell Technology Research Center, Tehran, Iran.
References
- 1. Barry FP, Murphy JM (2004) Mesenchymal stem cells: clinical applications and biological characterization. Int. J. Biochem. Cell Biol. 36, 568–584. [DOI] [PubMed] [Google Scholar]
- 2. Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U et al. (2005) Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp. Hematol. 33, 1402–1416. [DOI] [PubMed] [Google Scholar]
- 3. Fan H, Liu H, Zhu R, Li X, Cui Y, Hu Y et al. (2007) Comparison of chondral defects repair with in vitro and in vivo differentiated mesenchymal stem cells. Cell Transplant. 16, 823–832. [DOI] [PubMed] [Google Scholar]
- 4. Leo AJ, Grande DA (2006) Mesenchymal stem cells in tissue engineering. Cells Tissues Organs 183, 112–122. [DOI] [PubMed] [Google Scholar]
- 5. Masuoka K, Asazuma T, Hattori H, Yoshihara Y, Sato M, Matsumura K et al. (2006) Tissue engineering of articular cartilage with autologous cultured adipose tissue‐derived stromal cells using atelocollagen honeycomb‐shaped scaffold with a membrane sealing in rabbits. J. Biomed. Mater. Res. B Appl. Biomater. 79, 25–34. [DOI] [PubMed] [Google Scholar]
- 6. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B et al. (2001) Bone marrow cells regenerate infarcted myocardium. Nature 410, 701–705. [DOI] [PubMed] [Google Scholar]
- 7. Semont A, Francois S, Mouiseddine M, Francois A, Sache A, Frick J et al. (2006) Mesenchymal stem cells increase self‐renewal of small intestinal epithelium and accelerate structural recovery after radiation injury. Adv. Exp. Med. Biol. 585, 19–30. [DOI] [PubMed] [Google Scholar]
- 8. Wakitani S, Goto T, Pineda SJ, Young RG, Mansour JM, Caplan AI et al. (1994) Mesenchymal cell‐based repair of large, full‐thickness defects of articular cartilage. J. Bone Joint Surg. Am. 76, 579–592. [DOI] [PubMed] [Google Scholar]
- 9. Furlani D, Li W, Pittermann E, Klopsch C, Wang L, Knopp A et al. (2009) A transformed cell population derived from cultured mesenchymal stem cells has no functional effect after transplantation into the injured heart. Cell Transplant. 18, 319–331. [DOI] [PubMed] [Google Scholar]
- 10. Rosland GV, Svendsen A, Torsvik A, Sobala E, McCormack E, Immervoll H et al. (2009) Long‐term cultures of bone marrow‐derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res. 69, 5331–5339. [DOI] [PubMed] [Google Scholar]
- 11. Zhou YF, Bosch‐Marce M, Okuyama H, Krishnamachary B, Kimura H, Zhang L et al. (2006) Spontaneous transformation of cultured mouse bone marrow‐derived stromal cells. Cancer Res. 66, 10849–10854. [DOI] [PubMed] [Google Scholar]
- 12. Amiel D, Kleiner JB, Roux RD, Harwood FL, Akeson WH (1986) The phenomenon of “ligamentization”: anterior cruciate ligament reconstruction with autogenous patellar tendon. J. Orthop. Res. 4, 162–172. [DOI] [PubMed] [Google Scholar]
- 13. Bae JY, Matsumura K, Wakitani S, Kawaguchi A, Tsutsumi S, Hyon SH (2009) Beneficial storage effects of epigallocatechin‐3‐o‐gallate on the articular cartilage of rabbit osteochondral allografts. Cell Transplant. 18, 505–512. [DOI] [PubMed] [Google Scholar]
- 14. Huang JI, Durbhakula MM, Angele P, Johnstone B, Yoo JU (2006) Lunate arthroplasty with autologous mesenchymal stem cells in a rabbit model. J. Bone Joint Surg. Am. 88, 744–752. [DOI] [PubMed] [Google Scholar]
- 15. Harris MT, Butler DL, Boivin GP, Florer JB, Schantz EJ, Wenstrup RJ (2004) Mesenchymal stem cells used for rabbit tendon repair can form ectopic bone and express alkaline phosphatase activity in constructs. J. Orthop. Res. 22, 998–1003. [DOI] [PubMed] [Google Scholar]
- 16. Freedman VH, Shin SI (1974) Cellular tumorigenicity in nude mice: correlation with cell growth in semi‐solid medium. Cell 3, 355–359. [DOI] [PubMed] [Google Scholar]
- 17. Edgar B, Orr‐Weaver T (2001) Endoreplication cell cycles more for less. Cell 105, 297–306. [DOI] [PubMed] [Google Scholar]
- 18. Walen KH (2004) Spontaneous cell transformation: karyoplasts derived from multinucleated cells produce new cell growth in senescent human epithelial cell cultures. In Vitro Cell. Dev. Biol. Anim. 40, 150–158. [DOI] [PubMed] [Google Scholar]
- 19. Gosselin K, Martien S, Pourtier A, Vercamer C, Ostoich P, Morat L et al. (2009) Senescence‐associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res. 69, 7917–7925. [DOI] [PubMed] [Google Scholar]
- 20. Passos JF, von Zglinicki T (2005) Mitochondria, telomeres and cell senescence. Exp. Gerontol. 40, 466–472. [DOI] [PubMed] [Google Scholar]
- 21. von Zglinicki T, Saretzki G, Docke W, Lotze C (1995) Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp. Cell Res. 220, 186–193. [DOI] [PubMed] [Google Scholar]
- 22. Campisi J (1996) Replicative senescence: an old lives’ tale? Cell 84, 497–500. [DOI] [PubMed] [Google Scholar]
- 23. Foudah D, Redaelli S, Donzelli E, Bentivegna A, Miloso M, Dalprà L et al. (2009) Monitoring the genomic stability of in vitro cultured rat bone‐marrow‐derived mesenchymal stem cells. Chromosome Res. 17, 1025–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Huso DL, Harrington J, Kellner J, Jeong DK, Turney J et al. (2005) Outgrowth of a transformed cell population derived from normal human BM mesenchymal stem cell culture. Cytotherapy 7, 509–519. [DOI] [PubMed] [Google Scholar]
- 25. Bernardo ME, Zaffaroni N, Novara F, Cometa AM, Avanzini MA, Moretta A et al. (2007) Human bone marrow derived mesenchymal stem cells do not undergo transformation after long‐term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res. 67, 9142–9149. [DOI] [PubMed] [Google Scholar]
- 26. Bonab MM, Alimoghaddam K, Talebian F, Ghaffari SH, Ghavamzadeh A, Nikbin B (2006) Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shayesteh Y, Khojasteh A, Soleimani M, Alikhasi M, Khoshzaban A, Ahmadbeigi N (2008) Sinus augmentation using human mesenchymal stem cells loaded into a [beta]‐tricalcium phosphate/hydroxyapatite scaffold. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 106, 203. [DOI] [PubMed] [Google Scholar]