

Abstract
The cytoskeleton undergoes dramatic changes during apoptosis and many cytoskeletal proteins are known to be degraded during this process. The number of proteases found to be involved in apoptosis is growing but the role of the proteolysis they cause remains poorly understood. This report describes for the first time that myosin heavy chain is cleaved in aortic endothelial cell apoptosis induced either by tumour necrosis factor‐α or okadaic acid. The cleavage was specific since a well‐defined major 97 kDa fragment of myosin heavy chain was produced. The intermediate filament component vimentin was also cleaved into well‐defined fragments (31, 28 and 23 kDa). Kinetic studies showed that proteolysis occurred concomitantly with the morphological changes associated with apoptosis, i.e. cellular condensation and fragmentation in apoptotic bodies. These data suggest that the degradation of myosin and vimentin could be involved in the execution of the morphological alteratins observed during apoptotic cell death.
INTRODUCTION
Apoptosis is an active physiological cell death which plays an essential role in the physiology of pluricellular eukaryotes. It mediates the renewal of tissues in adults, the maturation and the homeostasis of the immune system, and takes place during the normal terminal differentiation of cells such as keratinocytes (for review see Raff 1996). A loss or impairment of the apoptotic programme contributes to pathological situations such as cancer development and abnormal growth, whilst inappropriate activation of apoptosis also provokes diseases as is the case in acquired immune deficiency syndrome (AIDS) or in neurodegenerative diseases ( Williams 1991, Orrenius 1995).
Apoptosis was originally defined by morphological criteria ( Kerr, Wyllie ,amp; Currie 1972). Classically, the cells undergoing apoptosis present cytoplasmic and chromatin condensation and undergo fragmentation in membrane‐bound vesicles or blebs: the apoptotic bodies. Concomitant to these morphological characteristics, specific biochemical events develop during apoptosis. These include internucleosomal DNA degradation, specific ribosomal 28S RNA cleavage and proteolysis of specific proteins ( Wyllie 1980, Houge et al. 1995 , Patel, Gores amp; Kaufmann 1996). All these effects are currently believed to be mediated directly or indirectly by activation of members of the caspase family of proteases (for review see Thornberry amp; Lazebnik 1998).
A prior study reported that the tumour necrosis factor (TNF)‐α; induces, in bovine aortic endothelial (BAE) cells, the morphological modifications and internucleosomal DNA degradation characteristic of apoptosis ( Robaye et al. 1991 ). The protein synthesis inhibitor, cycloheximide (CHX), acts synergistically with TNF‐α; to induce apoptosis in these cells ( Polunovsky et al. 1994 , Robaye et al. 1994 ). This indicates that, in these cells, the apoptotic machinery is constitutively present and that apoptosis development depends on the modification of existing proteins.
The present work was aimed at characterizing these modifications, using the two‐dimensional electrophoresis of proteins on polyacrylamide gel (2D‐PAGE). The most striking differences were observed in cytoskeletal proteins, i.e. that myosin heavy chain and vimentin undergo proteolysis. The cytoskeleton is known to be a target in apoptosis, and degradation of vimentin by apoptotic caspases has already be described ( van England et al. 1997 , Hashimoto et al. 1998 ). The study presented here indicates that the myosin heavy chain appears to be a novel target of proteolysis during the apoptotic cell death.
MATERIALS AND METHODS
Materials
Recombinant human TNF‐α; was a generous gift from Boehringer Ingelheim (Ingelheim, Germany). Culture medium, fetal calf serum (FCS) and agarose (electrophoresis grade) were from Gibco BRL Life Technologies (Paisley, UK). Trypsin was from Flow Laboratories (Bioggio, Switzerland). Reagent‐grade chemicals and biochemicals were purchased from Merck (Overijse, Belgium) and UCB (Brussels, Belgium). Pefablocreg; was from Pentapharm (Basel, Switzerland). Collagenase (type I), cycloheximide, okadaic acid, TEMED and Brillant blue R250 were purchased from Sigma‐Aldrich (St Louis, MO, USA). The sequencing grade modified trypsin was from Promega (Leiden, The Netherlands). Anti‐myosin antibodies (65ndash;791) were from ICN Biomedicals, Inc. (Costa Mesa, CA, USA). Glycine was a National Diagnostics (UK) product, and acetonitrile (HPLC far UV) was from LAB SCAN (Dublin, Ireland). CHAPS, dithiothrietol (DTT), servalytes, acrylamide and bis‐acrylamide were from SERVA (Poly lab, Antwerpen, Belgium). Triton X‐100 was from Boehringer Mannheim (Manheim, Germany). Ampholytes and agarose IEF were purchased from Pharmacia LKB (Uppsala, Sweden). Biolytes and ammonium persulphate were from Bio‐Rad (Hercules, CA, USA).[35S] Methionine, [3H]thymidine, [32P]orthophosphate, molecular weight markers‐[14C]‐methylated protein, anti‐vimentin antibodies (RPN 1102) and Hyperfilm MP were purchased from Amersham (Little Chalfont, UK). En3Hance was a NEN (Boston, MA, USA) product. Z‐Val‐Ala‐Asp(Ome)‐CH2F was from Calbiochemreg;(Euro Biochem, Bierges Belgium).
Bovine aortic endothelial (BAE) cells culture
Endothelial cells were obtained from aortas of freshly slaughtered cows and cultivated as previously described ( Van Colvorden amp; Boeynaems 1984). Briefly, cells harvested after a collagenase (2509 U/ml) treatment were cultured in 9cm Petri dishes for 3 days in minimum essential medium (MEM)‐ d‐valine supplemented with FCS (10%, v/v), 100 units/ml penicillin, 100micro;g/ml streptomycin and 2.5micro;g/ml amphotericin B. This MEM‐D‐ valine prevents growth of smooth muscle cells ( Marcum et al. 1986 ). The culture is pursued in Dulbecco's modified Eagle medium (DMEM) supplemented with FCS (10%, v/v), Hamapos;s F‐12 (20%, v/v) and antibiotics as described above (= complete culture medium). When cells were confluent, they were submitted to a classical trypsin treatment.
Quantification of DNA degradation
Cells (1.5ndash;2.5times;105) were seeded in 35mm Petri dishes in complete culture medium. After 24h, the cellular DNA was labelled by a 24‐h incubation of the cells with [3H]thymidine (37 kBq/ml, 1.48ndash;2.22 TBq/mmol). After two washes with DMEM, the cells were incubated with or without tested agents. At the end of incubation, the culture medium was collected and the cells were scraped and lysed in 1.3 ml Tris‐HCl 5mM pH8.0, EDTA 20mM and Triton X‐100 0.5%. In order to separate DNA fragments from intact chromatin, the lysates were submitted to a 15 000g centrifugation at 4deg;C. The radioactivity contained in the culture medium (med), the 15 000g pellet and supernatant (sup) was counted by liquid scintillation. The percentage of DNA degradation was calculated as follows:
DNA degradation (%) = (dpm med + dpm 15 000 g sup)/total dpm
where
total dpm (degradation per minute) = dpm med + dpm 15 000 g sup + dpm 15 000 g pellet
Electron microscopic study
BAE cells were seeded in 10 cm Petri dishes (106 cells/dish). Forty‐eight hours later, cells were incubated in the presence or absence of apoptosis‐inducing agents. At the end of the incubation, they were harvested by gentle centrifugation after trypsinization. They were then fixed and processed as previously reported ( Mosselmans et al. 1988 ). Ultrathin sections stained with uranyl acetate and lead citrate were examined in a Philips 301 electron microscope.
Study of proteins pattern by two‐dimensional gel electrophoresis
Cells (1.5ndash;2.5 times; 105) were seeded in 3.5cm Petri dishes as described above. The cellular proteins were labelled 24 h with [35S]methionine (5.55 MBq/ml,gt;37 TBq/mmol) prior to apoptotic stimulation. Cells were then washed twice with DMEM and incubated with or without the studied agents in complete culture medium. At the end of incubation, the cells were scraped and lysed in 200micro;l 2D‐PAGE buffer (9.5 M urea, 2% (w/v) CHAPS, 1.6% (v/v) ampholines pH 5ndash;7, 0.4% (v/v) servalytes pH 2ndash;11, 5% (v/v) beta‐mercaptoethanol). In some experiments, ampholines were replaced by Bio‐Lytes 5ndash;7 or by servalytes 5ndash;7 at the same concentrations. The proteins were then submitted to 2D‐PAGE separation as previously described ( Lecocq, Lamy amp; Dumont 1990). Briefly, isoelectric focusing was performed in 2.5 mmtimes; 14 cm tubes. After pre‐focusing, the proteins were submitted to electrophoresis for 16 h at 500 V, followed by 1 h at 1000 V. The gels were then equilibrated for 30 min in 0.06 M Tris‐HCl, pH 6.8, 2% SDS, 100 mM DTT (or 5% (v/v) beta;‐mercaptoethanol), 10% glycerol. The first‐dimensional gels were then set on 6ndash;16% polyacrylamide gradient gels (1 mm × 20 cm × 16 cm). The second dimensional gels were run at 4°C and started at 200V. During the electrophoresis, the voltage was raised to 360V. After classical fixation, En3Hance treatment and drying, the gels were submitted to autoradiography on preflashed Hyper films MP.
Polypeptides purification by two‐dimensional electrophoresis
Approximately 5 × 108 endothelial cells were cultured on 22 × 22 cm Petri dishes in complete culture medium. The day before confluence, cells were incubated in presence of TNF‐α (30ng/ml) and CHX (2µg/ml) for 6 h. After incubation, cells were scraped and lysed in 2D‐PAGE buffer. Proteins (±28;250µg that applied on the first dimension gel) were submitted to 2D‐PAGE separation as described above. About 50 bi‐dimensional gels were run. Spots of interest were cut out from Coomassie blue stained gels.
Partial amino acid sequence determination
After equilibration (in 12 mM Tris‐HCl pH6.8, 1% (w/v) SDS, 10% (v/v) glycerol, 0.1% (w/v) bromophenol blue, 1% (v/v) β‐mercaptoethanol), the gel pieces were concentrated. Two methods were used. Polypeptides 1 and 4′ were concentrated on agarose gels (1.5% (w/v) agarose, 365 mM Tris‐HCl pH 8.7, 1% (w/v) (SDS) ( Rider et al. 1995 ). At the end of the electrophoresis, the piece of agarose containing the concentrated polypeptides was cut out after staining with 0.1% (w/v) amido black, 1% (v/v) acetic acid, 40% (v/v) methanol. After three water washes, digestion buffer was added (100 mM Tris‐HCl pH 8.5, 2 mM CaCl2, 5% acetonitrile, 2% octylglucoside) and the sample was boiled until the gel was completely dissolved. The tryptic digestion was then started at 37°C by adding 1µg trypsin. After 8 h ofincubation, 1mug fresh trypsin was added again, and the digestion was pursued for 16 h. Finally, the sample was frozen at −80°C for at least 1 h, then centrifuged at 15000g 10min. Supernatants were collected and the resulting peptides were separated by reverse‐phase high‐performance liquid chromatography (HPLC) (C18 column).
Polypeptides 5 and 6 were concentrated on a 10% polyacrylamide gel and proteins were electroblotted onto PVDF membranes using 50mM Tris base, with 50mM boric acid as transfer buffer ( Rasmussen et al. 1991 ). After blotting, the membranes were soaked in methanol for 5min and washed four times with distilled water: proteins were then visualized by staining with 0.1% amido black in 45% methanol, 9% acetic acid. The protein bands were cut out and submitted to a tryptic proteolysis. The digestion was carried out in the presence of 1mug porcine trypsin during 4h at 37°C. Supernatants were collected and the resulting peptides were separated by reverse‐phase HPLC (C18 column).
Peptides were eluted by an acetonitrile gradient, and peaks were collected manually. For polypeptides 1 and 4′, peptides obtained after tryptic digestion were separated twice: first in the presence of heptafluorobutyric acid (0.1%), and, in a second step, the major peaks collected were separated in the presence of trifluoroacetic acid (0.1%).The major peaks were taken for sequence analysis by Edman degradation which was carried out on a protein sequencer 477 A (PE Biosystems, Zaventem, Belgium).
Western blot analysis of proteins
After 2D‐PAGE, proteins were transferred overnight at 60V and 4°C onto a nitrocellulose membrane using 20 mM Tris, 154 mM glycine, 20% (v/v) methanol as transfer buffer. The immunodetection was realized by the ECL Western blotting analysis system (Amersham) using a biotinylated‐secondary mouse antibody (1/5000). The monoclonal primary antibody anti‐vimentin (Amersham) was used at 1/10.
Immunoprecipitation of myosin heavy chain proteins
Cells were seeded as described above (5 × 10 5cells/dish) and incubated 6 h in the presence of [32P]orthophosphate (9.25 MBq/ml) with or without TNF‐α (30 ng/ml)+CHX (2 µg/ml). At the end of the incubation, cells were rinsed twice with NaCl 0.9% and scraped in the lysis buffer (1.5 mM MgCl2, 5 mM KCl, 10 mM Hepes pH 7.5, 0.5% (v/v) NP40, 2.5 mM EDTA, 50mg/l Pefabloc® and 50mug/l leupeptine). After 30 min under gentle agitation at 4°C, the lysates were centrifuged at 8000g at 4°C. The pellet was solubilized in 100µl Bravo buffer (50 mM Tris‐HCl, pH7.5, 0.5% (w/v) sodium dodecyl sulphate (SDS) and 70 mM DTT), boiled 10 min and then 400µl RIPA buffer (50 mM Tris‐HCl pH 7.5, 150 mM NaCl, 1% (v/v) NP40 and 0.5% (w/v) deoxycholate) were added. The samples were incubated 2h at 4°C in the presence of 1/100 normal rabbit serum, followed by a 1½h incubation with Sepharose‐conjugated protein A (10%, w/v). The lysates were centrifuged 30 s at 15 000g, and the supernatants were treated with anti‐myosin antibodies (1/50) for 2 h at 4°C. This incubation was followed by a precipitation with Sepharose‐conjugated protein A and the recovered pellets were then rinsed twice with RIPA buffer, once with 10mM Tris‐HCl pH 7.5, and finally solubilized in Laemmli buffer, boiled 3min and submitted to a SDS‐PAGE. The polyacrylamide gels were then submitted to autoradiography on Hyper film MP after drying.
RESULTS
Apoptosis in BAE cells
When treated with TNF‐α (30ng/ml) in the presence of CHX (2µg/ml), some BAE cells showed morphological characteristics of apoptotic cell death after only 1.5–2.5 h. These included loss of flat morphology by condensation and retraction ( Figure 1a,b), condensation of heterochromatin at the periphery of the nucleus, and fragmentation of the cell into multiple protrusions (or blebs) which later dissociated to form apoptotic bodies ( Figure 1b,d). Once started, these morphological alterations fully developed within minutes. There was a time‐dependent increase in the number of cells exhibiting such morphological changes. The DNA of TNF‐α + CHX‐treated cells presented the classical apoptotic internucleosomal degradation (data not shown). As previously reported, the kinetics of TNF‐α + CHX‐induced DNA fragmentation and morphological changes in BAE cells were not dissociable ( Robaye et al. 1991 ). Therefore, in further studies, DNA fragmentation was used as general marker of apoptotic cell death.
Figure 1.
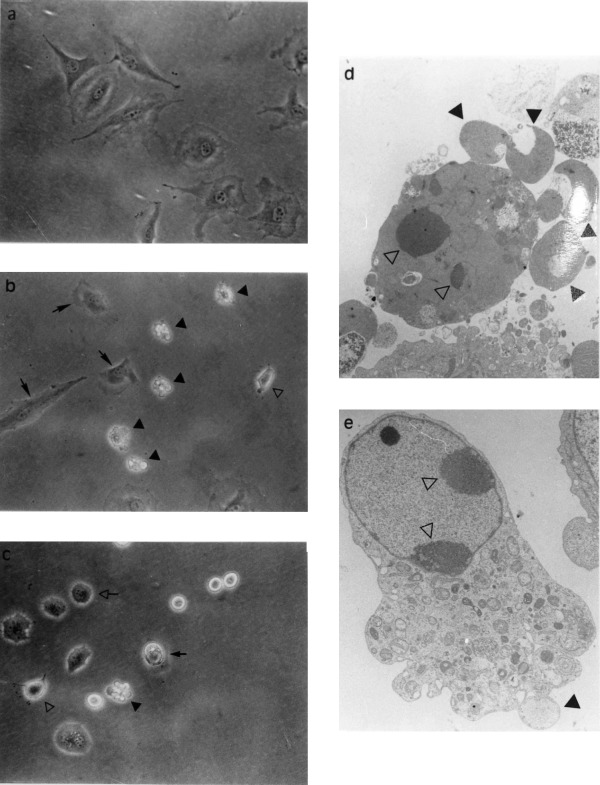
Morphology of TNF‐α + CHX‐and okadaic acid treated BAE cells were treated or not with specified agents for determined periods and examined directly under phased contrast microscopy (a,b,c) or processed for examination by elctron microscopy (d,e) as described. (a) control BAE cells; (b) 3 h TNF‐α (30 ng/ml) + CHX (2 µg/ml)‐treated BAE cells; and (c) 24 h okadaic acid (250nM)‐treated BAE cells: arrow non‐affected cells open arrow, rounded cells; closed arrow, apoptotic cell; open arrowhead, condensing cell; closed arrowhead, fragmented apoptotic cell. (d) 6 h TNF‐α (30 ng/ml) + CHX (2 µg/ml)‐treated BAE cells and (e) 8 h okadaic acid (250 nM)‐treated BAE cells: open arrowhead, condensed chromatin; closed arrowhead, apoptotic bodies.
The incubation of BAE cells with the protein phosphatase inhibitor okadaic acid (250 nM) also led to apoptosis, but the process developed more slowly, the first sign of DNA fragmentation only detected after 5–6h of treatment, i.e. 2–3h later than with the TNF‐α + CHX‐treatment ( see Figure 3a for comparison of time‐courses). At that time, classical morphological manifestations of apoptotic cell death became observable ( Figure 1e). Cell fragmentation into apoptotic bodies was also observed, but still with a lower rate than was seem in the cells exposed to TNF‐α + CHX ( Figure 1c).
Figure 3.
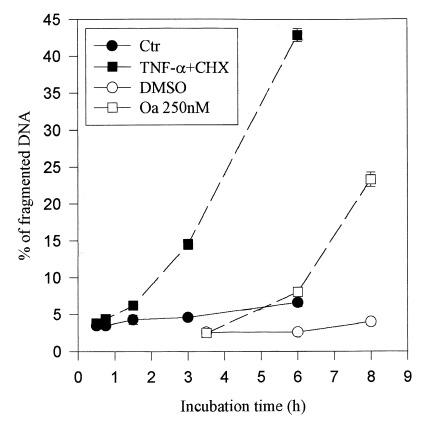
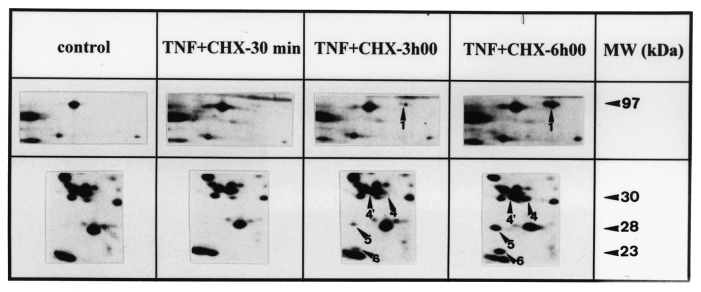
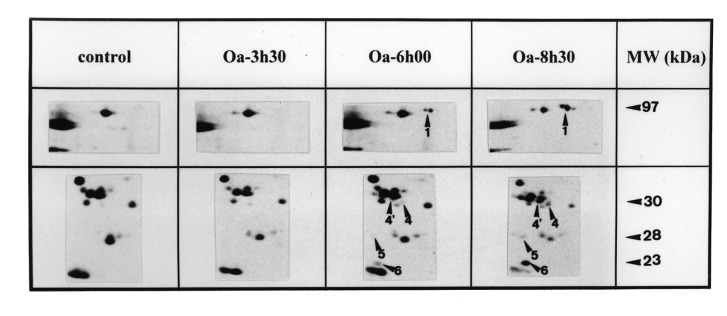
Kinetics of DNA fragmentation and protein modifications in BAE cells. (a) Kinetics of DNA fragmentation. BAE cells were seeded and 24 h later their DNA was prelabelled for 24h with [3H]thymidine (37 kBq/ml). After washing, they were incubated with specified agents for indicated times. At the end of incubation, cells were lysed and the percentage of DNA fragmentation was estimated as reported in Materials and methods. The data are mean of triplicate measurements ±sd of one representative experiment out of two. (b) and (c) Kinetics of protein modifications observed on 2D‐PAGE. The cells were seeded and 24h later, their proteins were prelabelled with [35S]methionine (3.7 MBq/ml). After washing, they were incubated or not with (b) TNF‐α (30ng/ml) + CHX (2µg/ml) or (c) okadaic acid (250nM) for specified times. They were then lysed and their proteins were submitted to 2D‐PAGE as described in Materials and methods. The 2D‐PAGE patterns of controls were obtained from extracts of cells directly harvested after the labelling period. The intensity of spots studied here did not significantly vary during the periods investigated in control conditions (data not shown).The figure reproduces limited regions of the 2D‐PAGE pattern. MW, molecular weight. Data shown are representative of two experiments.
Study of two‐dimensional pattern of proteins in apoptotic BAE cells
The two‐dimensional electrophoretic pattern of proteins from BAE cells prelabelled with [35S]methionine was modified when the cells were submitted to the TNF‐α + CHX apoptotic treatment ( Figure 2). Some spots (open arrowhead on control autoradiography, Figure 2a) present in control conditions were less intense or undetectable in extracts from treated cells, whereas other spots (closed arrowhead on TNF‐α + CHX autoradiography, Figure 2b) detectable in extracts of TNF‐α + CHX‐treated BAE cells, were not present or exhibited much lower intensity in the controls.
Figure 2.
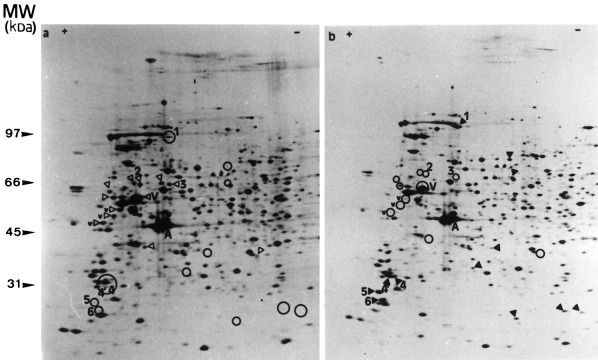
Two‐dimensional pattern of [35S]methionine‐prelabelled proteins extracted from BAE cells. BAE cells were seeded and 24 h later, their proteins were prelabelled with [35S]methionine (3.7 MBq/ml). They were incubated without (a) or with TNF‐α (30 ng/ml) in the presence of CHX (2 µg/ml) (b) for 6h. The cells were then lysed and their proteins were submitted to 2D‐PAGE as described in Materials and methods. V, vimentin; v, high MW degradation products of vimentin (‘staircase’); A, actin; open arrowhead, spots of reduced intensity in treated cells; closed arrowhead, spots observed in treated cells but not in control cells. Spots observed on one pattern gel but not on the other one were localized on the latter by an open circle to facilitate the analysis of the patterns.
In order to see how these modifications at the level of proteins relate to the progression of the apoptotic cell death, the kinetics of appearance of these modifications were compared with apoptotic DNA fragmentation. In the 2D‐PAGE pattern of TNF‐α + CHX‐treated BAE cells, five spots (marked 1, 4, 4′, 5 and 6 in Figure 2) dramatically increased in intensity when DNA fragmentation became detectable ( Figure 3a). Their intensity then further increased in parallel with DNA fragmentation ( Figure 3a,b). Other spots marked in Figure2 exhibited changes in intensity, but these, except for spots 2 and 3, occurred well after the first manifestations of DNA fragmentation, i.e. after 3 h (data not shown).
When apoptosis was triggered by okadaic acid, the kinetics of DNA fragmentation was delayed compared to when apoptosis was induced with TNF‐α + CHX. The first signs of DNA degradation were detectable after 5h of okadaic acid‐treatment, i.e. 3 h later than was observed with TNF‐α + CHX‐treatment. In this case, the appearance of spots 1, 4, 4′, 5 and 6 was also delayed, compared to TNF‐α + CHX‐treatment, but still corresponded to that of DNA fragmentation ( Figure 3c). The intensity of these spots also further increased with the time of treatment, as did DNA fragmentation ( Figure 3a).
Identification of the 97kDa polypeptide (spot 1)
As they were sufficiently abundant, proteins corresponding to spots 1, 4′, 5 and 6 were purified directly by 2D‐PAGE and microsequenced by Edman degradation. ‘Spot number 1’ corresponded to a triad of products with a molecular weight (MW) of approximately 97 kDa ( Figures 2 and 3), of which the two more acidic ones were phosphorylated (data not shown). All three showing parallel time‐course modification in intensity during apoptosis, it was assumed that they correspond to the native and differentially phosphorylated forms of a single protein. The four microsequences obtained from this 97 kDa purified protein are detailed in Table1. A search through the non‐redundant GenBank CDS databank revealed that they all showed 100% identity with internal amino acid sequences of the human non‐muscle type A myosin heavy chain (MHC). All were located in the carboxyterminal domain of the protein ( Table 1). As the MW of the native form of this phosphorylable actin‐binding protein is about 215 kDa, the possibility that the 97 kDa polypeptide might be a carboxyterminal product of its partial proteolysis was considered. In order to test this assumption and to determine whether phosphorylation of the protein plays a role in the degradation of the native MHC, phosphorylated MHC found in BAE cells after (TNF‐α + CHX) incubation was quantified by immunoprecipitation. A single 32P‐labelled band appeared in immunoprecipitates from control cells (Figure4), corresponding to a MW slightly higher than 200 kDa, thus fitting with that of the myosin heavy chain. This band was absent in extracts from TNF‐α + CHX‐treated BAE cells, collected when they were apoptotic ( Figure 4, lane 2). Antibodies used in this experiment did not recognize the 97kDa‐bovine MHC degradation product.
Table 1.
Partial amino acid sequences of four spots purified by 2D‐PAGE
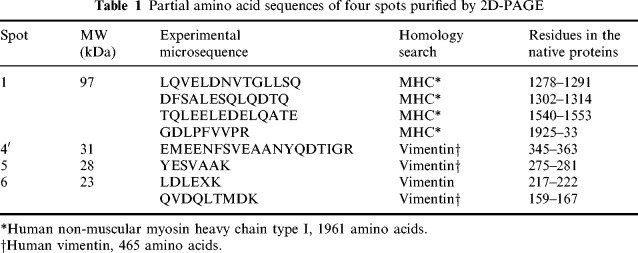
Figure 4.
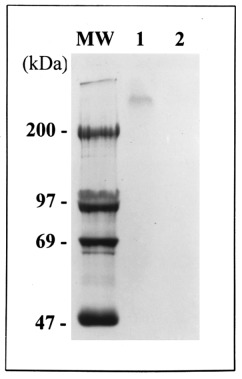
Immunoprecipitation of phosphorylated myosin heavy chain. BAE cells were incubated in the presence of [32P]orthophosphate (9.25MBq/ml) in the presence or not of TNF‐α (30ng/ml) + CHX (2 µg/ml) for 6 h. At the end of the incubation, cells were lysed and proteins were submitted to immunoprecipitation with the anti‐myosin antibodies (1/50) as described in Materials and methods. Immunoprecipited proteins were submitted to electrophoresis on polyacrylamide gel in the presence of SDS. After drying, the gel was submitted to autoradiography. Lane 1, control BAE cells; lane 2, TNF‐α + CHX‐treated cells; MW: molecular weight.
Inhibition of myosin heavy chain proteolysis by caspase inhibitor
The effect of the caspase inhibitor Z‐Val‐Ala‐Asp(Ome)‐Ch2F (Z‐VAD‐fmk) was studied on the MHC proteolysis induced by TNF‐α + CHX. As shown in Figure 5, this caspase inhibitor completely prevented the appearence of the 97 kDa‐MHC degradation product, which indicates that MHC seems to be a new substrate for one of the different caspases activated during apoptosis. Concomitantly with this inhibition, it was observed that the apoptotic cell death induced in BAE cells by the TNF‐α + CHX‐incubation is also present in the presence of Z‐VAD‐fmk (data not shown)
Figure 5.
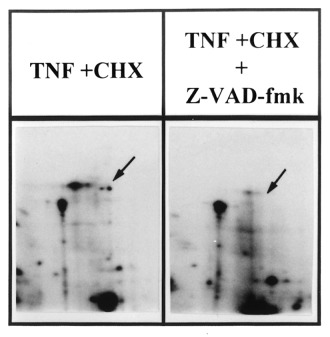
Effect of Z‐VAD‐fmk on the myosin heavy chain proteolysis induced by TNF‐α + CHX. The cells were seeded and 24h later, their proteins were prelabelled with [35S]methionine (37 MBq/ml). After washing, they were incubated with TNF‐α (30 ng/ml) + CHX (2 µg/ml) in the absence (a) or presence (b) of Z‐VAD‐fmk (20 ‐M) for 4 h. They were then lysed and their proteins were submitted to 2D‐PAGE as described in Materials and methods.
Identification of polypeptides corresponding to spots 4′, 5 and 6
Microsequencing of polypeptides corresponding to spots 4′, 5 and 6 purified directly by 2D‐PAGE revealed that the three polypeptides showed 100% identity with amino acid sequences of human vimentin. The different experimental microsequences covered different regions of the sequence of this intermediary filament‐associated protein ( Table 1). This suggested that they were the product of an apoptosis‐associated proteolysis of vimentin. By Western blot immunodetection, the native vimentin and the ‘staircase’ associated degradation products were identified on the 2D‐PAGE pattern of protein extracted from control BAE cells (marked V and v in Figure 2 and Figure 6). Compared with cells treated 24h with TNF‐α + CHX, which exhibited 74.4±2.6% of DNA degradation, it was observed that the spots corresponding to intact and partially degraded vimentin completely disappeared ( Figure 6). The degradation products identified by microsequencing were not recognized by the monoclonal anti‐vimentin antibody used.
Figure 6.
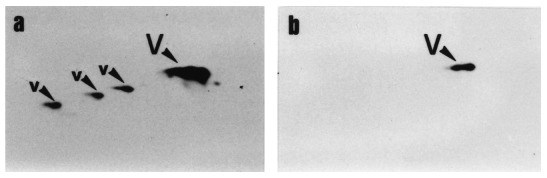
Immunodetection of vimentin on western blot. BAE cells were untreated (a) or treated (b) with TNF‐α (30ng/ml) + CHX (2 µg/ml) for 24 h. At the end of incubation, the cells were scraped, lysed and submitted to a 2D‐PAGE as described in Materials and methods. Western blotting was carried out using a monoclonal anti‐vimentin antibody. V, vimentin; v, high MW degradation products of vimentin (‘staircase’).
DISCUSSION
The data show that, whether induced by okadaic acid or by TNF‐α + CHX, apoptosis of BAE cells displayed the same qualitative characteristics, i.e. internucleosomal DNA fragmentation, condensation and marginalization of chromatin, cell condensation, and formation of apoptotic bodies. However, the kinetics of the process differed with the two stimuli, TNF‐α + CHX‐treated BAE cells becoming apoptotic within ±2 h, okadaic acid‐treated cells within ±6 h; these times could vary depending on the primary culture conditions.
Analysis of the 2D‐PAGE pattern of proteins extracted from BAE cells revealed modifications when cells were triggered to undergo apoptosis. Attention was focused on five modifications among all those scored as they took place concomitantly with DNA and cell fragmentation and followed a similar time‐course evolution relative to that of DNA fragmentation with the two kinetically different treatments, TNF‐α + CHX and okadaic acid. These particularities suggest that these protein modifications could be involved in the control or execution phase of the apoptotic pathway.
As they were sufficiently abundant, the purification of these polypeptides could be directly performed by 2D‐PAGE of proteins extracted from apoptotic BAE cells. Microsequencing by Edman degradation demonstrated that four of these polypeptides were degradation products of known proteins with higher molecular weight. Indeed, spot 1 of 97 kDa is a proteolytical fragment of the non‐muscular myosin heavy chain type A, a cytoskeletal protein of about 220 kDa. Spots 4′, 5 and 6, respectively, 31, 28 and 23 kDa are three degradation products of an intermediate filaments (IFs) protein, vimentin, which has an MW of 58kDa. These results were confirmed by immunodetection experiments that showed that the amount of native vimentin and MHC decreased when cells became apoptotic, and that the respective network completely disappeared in these cells. It was also shown that MHC is a new substrate for a caspase‐like activity.
To our knowledge, the present report is the first to demonstrate the limited degradation of MHC proteins in relation to apoptosis. This observation seems in contradiction with recent data suggesting that myosin contractile activity is stimulated through an increased phosphorylation of myosin light chain in apoptotic blebbing cells ( Mills et al. 1998 ). Indeed, the degradation described here led to the dissociation of the carboxy‐terminal rod end of myosin involved in thick filament formation, and the amino‐terminal head that interacts with actin filaments. It is more than possible that this dissociation inactivates the myosin contractile activity, as both parts of the protein seem to be required to generate a motion between thick and actin filament. However, it still possible that intact and active myosin could be required at an earlier stage of the morphological alterations and is subsequently degraded like other cytoskeletal proteins.
Vimentin has already been described as a proteolytical substrate during apoptosis ( van England et al. 1997 , Hashimoto et al. 1998 ). van England and co‐workers reported that vimentin is cleaved into two degradation products of 50 and 43kDa that do not correspond to the 31, 28 and 23 fragments described here. However, the kinetics of proteolysis reported by these authors seems to correspond to the kinetics of vimentin degradation observed in endothelial cells. This suggests that what is important is the dissolution of the vimentin network, and not the degradation products generated by this dissolution. In the same way, Hashimoto et al. (1998) described during apoptosis of human skin fibroblasts, the appearance of three major large fragments of vimentin (46, 48 and 52 kDa) and several minor bands, i.e. a fragment of 29 kDa.
Various other cytoskeletal proteins are also degraded during the apoptotic process, amongst these the keratin 18, which is a major component of the Ifs, as is vimentin, and is cleaved by a caspase‐like activity, which leads to a reorganization of the IFs ( Caulin, Salvesen & Oshima 1997). Fodrin and the nuclear lamins are two other cytoskeletal substrates for the various proteases induced during apoptosis ( Lazebnik et al. 1995 , Cryns et al. 1996 ). Other than the fact that these degradations leads to the dissolution of the cytoskeletal network, no particular role has been found at present. This is not true for gelsolin, a caspase‐3 substrate, for which the expression of the cleavage product in multiple cell types caused the cells to round up, detach from the plate, and undergo nuclear fragmentation ( Kothakota et al. 1997 ). Regardless, the proteolysis of vimentin and MHC and subsequent dissolution of the cytoskeletal network may be involved in the retraction and condensation observed in cells which become apoptotic. Indeed, it has been shown that the expression of the non‐muscle myosin in BAE cells is associated with the acquisition of a flattened morphology ( Wong, Pollard & Herman 1982, Franke et al. 1984 , Borrione et al. 1990 ). On the other hand, in conjunction with microtubules and microfilaments, IFs seem responsible for anchorage of the nucleus in the cell and for the maintenance of cell shape by binding the lamina network around nuclear pores and the membrane skeleton ( Geiger 1987).
In conclusion, the present study has shown that concomitant with the occurrence of apoptotic morphological modifications, the cytoskeleton is dramatically affected. This is associated with the proteolysis of an increased number of cytoskeletal proteins including myosin heavy chain and vimentin.
Acknowledgements
This work was supported by the Fonds National de la Recherche Scientifique Medicale, Télévie, the Fondation cancérologique de la Caisse d'Epargne, the Association Belge contre le Cancer, the Association Sportive Belge contre le Cancer, and by the Belgian Programme on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister's Office, Federal Service for Sciences, Technology and Culture. N. Suarez‐Huerta was supported by the Fond pour la formation à la Recherche dans l'Industrie et dans l'Agriculture (F.R.I.A.). This work was carried out within the framework of a contract of the ‘Human Capital’ of the European Community.
REFERENCES
- Borrione AC, Zanellato AMC, Giuriato L et al. (1990). Non‐muscle and smooth muscle myosin isoforms in bovine endothelial cells. Exp Cell Res. 190,1. [DOI] [PubMed] [Google Scholar]
- Caulin C, Salvesen GS, Oshima RG.(1997). Caspase cleavage of keratin 18 and reaorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 138,1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns VL, Bergeron L, Zhu H, Li H, Yuan J. (1996). Specific cleavage of a‐fodrin during Fas– and tumor necrosis factor‐induced apoptosis is mediated by an interleukin‐1b‐converting enzyme/ced‐3 protease distinct from the poly (ADP‐ribose) polymerase protease. J. Biol. Chem. 271,31277. [DOI] [PubMed] [Google Scholar]
- Franke RP, Gräfe M, Schnittler H, Seiffge D, Mittermayer C. (1984). Induction of human vascular endothelial stress fibres by fluid shear stress. Nature.307,648. [DOI] [PubMed] [Google Scholar]
- Geiger B. (1987). Intermediate filaments: looking for a function. Nature.329,392. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Inoue S, Ogawa S et al. (1998). Augmentation of vimentin in human skin fibroblasts exposed to tamoxifen: a possible involvement of caspase‐3. Biochem. Biophys. Res. Com. 247,401. [DOI] [PubMed] [Google Scholar]
- Houge G, Robaye B, Eikhom TK et al. (1995). Fine mapping of 28S rRNA sites specifically cleaved in cells undergoing apoptosis. Mol. Cell. Biol. 15,2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AH, Currie AR.(1972). Apoptosis: a basic biological phenomenon with wide‐ranging: implications in tissue kinetics. Br. J. Cancer. 26,239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothakota S, Azuma T, Reinhard C et al. (1997). Caspase‐3‐generated fragment of gelsolin: effector of morphological change in apoptosis. Science.278,294. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Takahashi A, Moir RD et al. (1995). Studies of lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc. Natl. Acad. Sci. USA. 92,9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecocq R, Lamy F, Dumont JE. (1990). Use of two‐dimensional gel electrophoresis and autoradiography as a tool in cell biology: the example of the thyroid and the liver. Electrophoresis.11,200. [DOI] [PubMed] [Google Scholar]
- Marcum JA, Atha GH, Fritze LMS, Nawroth P, Stern D, Rosenberg RD. (1986). Cloned bovine aortic endothelial cells synthesize anticoagulantly active heparan sulfate proteoglycan. J. Biol. Chem. 261,7507. [PubMed] [Google Scholar]
- Mills JC, Stone NL, Erhardt J, Pittman RN. (1998). Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J. Cell Biol. 140,627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosselmans R, Hepburn A, Dumont JE, Fiers W, Galand P. (1988). Endocytic pathway of recombinant murine tumor necrosis factor in L929 cells. J. Immunol. 141,3096. [PubMed] [Google Scholar]
- Orrenius S. (1995). Apoptosis: molecular mechanisms and implications for human diseaseM. J. Int. Med. 237,529. [DOI] [PubMed] [Google Scholar]
- Patel T, Gores GJ, Kaufmann SH. (1996). The role of proteases during apoptosis. FASEB J. 10,587. [DOI] [PubMed] [Google Scholar]
- Polunovsky VA, Wendt CH, Ingbar DH, Peterson MS, Bitterman PB (1994). Induction of endothelial cell apoptosis by TNFα: modulation by inhibitors of protein synthesis. Exp. Cell. Res. 214,584. [DOI] [PubMed] [Google Scholar]
- Raff MC. (1996). Size control: the regulation of cell number in animal development. Cell. 86,173. [DOI] [PubMed] [Google Scholar]
- Rasmussen HH, Van Damme J, Puype M, Gesseries B, Celis JE, Vandekerckhove J. (1991). Microsequencing of proteins recorded in human two‐dimensional gel protein databases. Electrophoresis.12,873. [DOI] [PubMed] [Google Scholar]
- Rider MH, Puype M, Van‐Damme J et al. (1995). An agarose‐based gel‐concentration system for microsequence and mass spectrometric characterization of proteins previously purified in polyacrylamide gels starting at low picomole levels. Eur. J. Biochem. 230,258. [DOI] [PubMed] [Google Scholar]
- Robaye B, Doskeland AP, Suarez‐Huerta N, Doskeland SO, Dumont JE. (1994). Apoptotic cell death analyzed at the molecular level by two‐dimensional gel electrophoresis. Electrophoresis.15,503. [DOI] [PubMed] [Google Scholar]
- Robaye B, Mosselmans R, Fiers W, Dumont JE, Galand P. (1991). Tumor necrosis factor induces apoptosis (programmed cell death) in normal endothelial cells. In Vitro. Am. J. Pathol.138,447. [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA & Lazebnik Y. (1998). Caspases: Enemies within. Science.281,1312. [DOI] [PubMed] [Google Scholar]
- Van Colvorden A & Boeynaems JM. (1984). Physiological concentration of ADP stimulates the release of prostacyclin from bovine aortic endothelial cells. Prostaglandins.27,615. [DOI] [PubMed] [Google Scholar]
- Van England M, Kuijpers HJH, Ramaekers FCS, Reutelingsperger CPM, Schutte B. (1997). Plasma membrane alterations and cytoskeletal changes in apoptosis. Exp. Cell Res. 235 421. [DOI] [PubMed] [Google Scholar]
- Williams GT. (1991). Programmed cell death: apoptosis and oncogenesis. Cell 65,1097. [DOI] [PubMed] [Google Scholar]
- Wong AJ, Pollard TD, Herman IM. (1982). Actin filament stress fibers in vascular endothelial cells in vivo . Science.219,867. [DOI] [PubMed] [Google Scholar]
- Wyllie AH. (1980). Glucocorticoid‐induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature.284,555. [DOI] [PubMed] [Google Scholar]