

Abstract
The transthyretin (TTR) amyloid diseases are of keen interest, because there are >80 mutations that cause, and a few mutations that suppress, disease. The V122I variant is the most common amyloidogenic mutation worldwide, producing familial amyloidotic cardiomyopathy primarily in individuals of African descent. The substitution shifts the tetramer-folded monomer equilibrium toward monomer (lowers tetramer stability) and lowers the kinetic barrier associated with rate-limiting tetramer dissociation (pH 7; relative to wild-type TTR) required for amyloid fibril formation. Fibril formation is also accelerated because the folded monomer resulting from the tetramer-folded monomer equilibrium rapidly undergoes partial denaturation and self-assembles into amyloid (in vitro) when subjected to a mild denaturation stress (e.g., pH 4.8). Incorporation of the V122I mutation into a folded monomeric variant of transthyretin reveals that this mutation does not destabilize the tertiary structure or alter the rate of amyloidogenesis relative to the wild-type monomer. The increase in the velocity of rate-limiting tetramer dissociation coupled with the lowered tetramer stability (increasing the mol fraction of folded monomer present at equilibrium) may explain why V122I confers an apparent absolute anatomic risk for cardiac amyloid deposition.
The amyloidoses are a large group of protein misfolding diseases that are accelerated by certain point mutations and suppressed by others (1–10). Familial amyloidotic cardiomyopathy (FAC) does not result from loss of transthyretin (TTR) function (because of insolubility); instead, it appears to be caused by tissue-selective TTR amyloid deposition in the heart (11–14). Wild-type (WT) TTR can also deposit as fibrils in the cardiovascular system in the late-onset disease senile systemic amyloidosis, affecting as much as 25% of the population over the age of 80 (15). In addition, there are >80 TTR variants associated with early onset amyloid diseases, including the V30M variant, which preferentially deposits in the peripheral nervous system (16–18).
TTR is a 55-kDa homotetrameric protein, comprised of 127-residue β-sheet rich subunits, which is present in the cerebral spinal fluid and serum. In blood, TTR serves as the secondary thyroxine carrier protein (by using ≈10% of its capacity) and transports retinol via binding ≤1 equivalent of holo retinol-binding protein (RBP) (19). It appears that all of the TTR deposited as amyloid is derived from plasma. Rate-limiting tetramer dissociation of TTR into monomers is necessary but not sufficient for TTR fibril formation, as tertiary structural changes within the monomer are also required (20–23).
We created a TTR variant (WT M-TTR), which proved to be monomeric, normally folded, and nonamyloidogenic under physiological conditions, through the introduction of a mutation in each of the quaternary structural interfaces (F87M/L110M) (20). A denaturing stress (such as acidic pH) induces a conformational change in WT M-TTR facilitating its self-assembly into amyloid fibrils at a rate >100 times faster than the tetrameric WT protein because of the rate-limiting dissociation of the WT tetramer required for fibril formation (20).
It is estimated that ≈4% of African Americans (1.3 million people) are heterozygous for the V122I allele (12). Although the age of onset (typically >60 years of age) is similar for senile systemic amyloidosis (WT) and FAC (V122I) patients, the latter are much more likely to suffer cardiac failure (especially in the case of V122I homozygotes) (12, 13, 24). Here we compare the V122I homotetramer to the WT TTR homotetramer using a biophysical approach in an attempt to define the mechanism of the amyloidogenicity associated with the pathology of FAC. The monomeric WT and V122I M-TTR variants were also compared focusing on the influence of the V122I cardiac mutation on tertiary structural stability and amyloidogenicity. Here we show that the V122I FAC variant destabilizes the TTR tetramer and lowers the kinetic barrier associated with tetramer dissociation, resulting in a greater extent and faster rate of folded monomer formation, a structure that rapidly undergoes partial denaturation and self-assembles into amyloid fibrils (in vitro).
Materials and Methods
Protein Expression and Purification.
Recombinant WT, V122I, F87M/L110M (WT M-TTR), and V122I/F87M/L110M (V122I M-TTR) TTR proteins were expressed and purified as described previously (25). All proteins were further purified by using gel filtration chromatography on a Superdex-75 column (Amersham Pharmacia). Protein concentrations were determined by measuring UV absorbance at 280 nm, by using an extinction coefficient of 7.76 × 104 M−1⋅cm−1. The pH 7 buffer used contains 50 mM sodium phosphate, 100 mM KCl, and 1 mM EDTA.
Fibril Formation Assay.
TTR (0.4 mg/ml) in 10 mM phosphate buffer with 100 mM KCl (pH 7) was diluted 1:1 with 200 mM buffer (100 mM KCl and 1 mM EDTA) to jump to the desired pH (sodium citrate for pH 3.2, sodium acetate for pH 3.6–5.2, and phosphate buffer for pH >5.2). The solutions subjected to a denaturation stress were incubated at 37°C for 72 h, after which the suspensions were vortexed and optical density measured at 400 nm. For time course experiments, a 0.3- to 0.4-mg/ml TTR sample was quickly diluted 1:1 with pH 4.4 buffer while monitoring turbidity at 400 nm (37°C) by using a UV spectrometer equipped with a Peltier temperature control unit.
Fibril formation was also assessed by thioflavin T (ThT) binding, where a 25-μl aliquot of sample (vortexed to achieve homogeneity) was mixed with 173 μl of 50 mM Tris buffer (100 mM KCl, pH 8.0) and 2 μl of ThT stock solution (1 mM) in 10 mM phosphate (pH 7.4). The mixed sample was then excited at 440 nm, and emission at 482 nm was recorded.
Gel Assay-Monitored Quaternary Structure Changes.
Samples prepared identically to those (containing 0.2 mg/ml of TTR) used for the fibril formation assay were incubated at 25°C (instead of 37°C) for 40 h to evaluate the extent of pH-induced tetramer dissociation. A 1,200 μl sample at each pH was then mixed with 24 μl of a 25 mg/ml zwitterionic detergent (Z 3–14) stock solution, which was immediately combined with 350 μl of a 1 M sodium phosphate buffer (pH 7) solution containing 0.5 mg/ml of Z 3–14 to neutralize the pH for gel loading. [We have previously established that the concentration of Z 3–14 used does not allow reconstitution of TTR monomers to tetramers or dissociate the tetramer present in the mixture (26).] Ten microliters of the mixture was mixed with the same volume of 4% nonreducing SDS gel-loading buffer and, without boiling, the samples were loaded on a 12% SDS acrylamide gel. The gel was stained with Coomassie staining reagent (Pierce) and, after drying, the lanes were analyzed by densitometry and quantified using the program scion image.
Trp Fluorescence Monitored Tertiary Structural Changes.
Urea denaturation studies were carried out by diluting a TTR stock solution (0.4 mg/ml) with varying concentrations of chaotrope at pH 7, obtaining a final protein concentration of 0.02 mg/ml. Samples were incubated at room temperature for 24 and 96 h before fluorescence measurements were made. Refolding samples were prepared by diluting 0.4 mg/ml of TTR denatured in 8 M urea (incubated at 4°C for 96 h) into 50 mM phosphate buffer (100 mM KCl, pH 7), obtaining a final protein concentration of 0.02 mg/ml. Refolded samples were incubated at room temperature for 24 h before fluorescence measurements were made. Concentrations of the urea stock solutions were determined by refractive index measurements (27). Tryptophan fluorescence was used to monitor TTR tertiary structural changes. Samples (25°C) were excited at 295 nm and the emission measured from 310 to 410 nm on an Aviv Model ATF105 spectrofluorometer (Aviv Associates, Lakewood, NJ). Fraction unfolded was calculated by using the F355/F335 value at each protein concentration, knowing the folded minimum (F355/F335 = 0.81) and the denatured maximum (F355/F335 = 1.35) fluorescence ratios, assuming a linear dependence.
Rate of Tetramer Dissociation Measured by Linking Tertiary Structural Changes.
The evaluation of tetramer dissociation rates was carried out by removing 200-μl aliquots from a denaturing TTR (0.02 mg/ml) sample (10 ml) in 4.5 M urea [50 mM phosphate buffer (100 mM KCl, pH 7, 25°C)]. The Trp fluorescence emission ratio (F355/F335) as a function of time (25°C) was measured.
Results
The V122I Homotetramer Dissociates to Folded Monomers 3-Fold Faster than WT Homotetramers.
Mounting evidence suggests that the intact TTR tetramer is not directly amenable to denaturation by urea (20, 28). Instead, the tetramer has to dissociate to folded monomers before it can be denatured by urea. The slow dissociation rate of the native tetramer explains the slow approach to equilibrium observed during urea denaturation (Fig. 1A), which is slower for WT than V122I TTR homotetramers. The tryptophan fluorescence senses tertiary structural changes, not quaternary structural changes, on the basis of the fact that the tetramer and recently introduced folded monomeric version of TTR (WT M-TTR) have indistinguishable fluorescence spectra (20).
Figure 1.

(A) Urea induced denaturation of V122I (circles) and WT
(triangles) TTR, after incubation for 24 (open symbols) and 96 h
(closed symbols), as detected by Trp fluorescence. Lines through the
V122I (solid) and WT (dashed) data are smoothing curves to guide the
eye. The lower value of WT plateau (at >3 M urea, 96 h) indicates
this protein has not completely reached equilibrium. (B)
Urea denaturation curves of monomeric V122I M-TTR
(●) and WT M-TTR (▴) (20) (0.02
mg/ml 24 h incubation). That tetrameric V122I TTR (○) (96
h incubation) denaturation occurs via the monomer is demonstrated by
the indistinguishable denaturation curves (● and
○, within error). The solid line through the V122I M-TTR
data are fitted to a two-state model yielding a
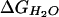 value of 4.7 ± 0.2
kcal⋅mol−1 and m value of 1.4 ± 0.1
kcal⋅mol−1⋅M−1. (C)
The rate of urea- (4.5 M) induced V122I (○) and WT
(●) TTR tetramer dissociation detected by very
fast linked tertiary structural changes. Solid lines are fitted to a
first order single exponential function.
value of 4.7 ± 0.2
kcal⋅mol−1 and m value of 1.4 ± 0.1
kcal⋅mol−1⋅M−1. (C)
The rate of urea- (4.5 M) induced V122I (○) and WT
(●) TTR tetramer dissociation detected by very
fast linked tertiary structural changes. Solid lines are fitted to a
first order single exponential function.
The identical midpoints (Cm) of the urea denaturation curves exhibited by WT and V122I TTR (0.02 mg/ml; 25°C) reflect identical tertiary structural stabilities (Fig. 1A). This interpretation is verified by comparing the denaturation curves of WT and V122I M-TTR monomers, which are identical in terms of Cm and amplitude (Fig. 1B) (20). Moreover, the Cms exhibited by WT and V122I TTR (Fig. 1A) are identical to the Cms displayed by the monomeric versions of these two sequences (Fig. 1B), consistent with the fact that tertiary structural changes are being monitored. The differences in amplitude as a function of time for the data displayed in Fig. 1A reflect the different rates of tetramer dissociation monitored by linked tertiary structural changes of WT and V122I TTR. A slow approach to equilibrium is not observed for the M-TTR constructs (Fig. 1B), because the monomers denature on the millisecond timescale (20). The rate of dissociation of the WT and V122I homotetramers can be monitored by coupling the slow (hour timescale) quaternary structural changes (not detectable by fluorescence) to the fast (millisecond timescale) tertiary structural changes monitored by Trp fluorescence, provided the urea concentration used is in the posttransition region for tertiary structural changes (4.5 M urea). Under these conditions, the monomeric subunits resulting from tetramer dissociation unfold in a few milliseconds and remain unfolded, allowing the rate of tetramer dissociation to be measured (Fig. 1C). The WT tetramer exhibits a dissociation half-life of ≈37.7 h, which is 3-fold longer than that exhibited by V122I TTR (t1/2 ≈11.5 h). These half-lives are referred to as approximate, because there appears to be a minor slower phase (<10%) that interferes with a perfect fit of the data to a single exponential function, possibly derived from an anion stabilized tetramer (28). Nevertheless, it is clear the V122I cardiac mutation increases the velocity of the rate-determining step for amyloidosis 3-fold relative to WT TTR at 4.5 M urea. The dependence of the rates of WT and V122I tetramer dissociation on urea concentration is nearly identical, as revealed by identical slopes in a ln kdissociation vs. urea concentration plot (P. Hammarström, X.J., and J.W.K., unpublished data).
The V122I Homotetramer Dissociates to the Amyloidogenic Intermediate Faster than WT TTR Under a Mild Acid Denaturation Stress.
WT TTR is converted into amyloid by pH-mediated tetramer dissociation linked to tertiary structural changes, resulting in the formation of a so-called monomeric amyloidogenic intermediate that self-assembles into amyloid fibrils (20–23). The V122I FAC variant forms fibrils over a pH range very similar to WT TTR (Fig. 2A). The yield of pH-induced fibril formation from the V122I homotetramer is higher than that of WT TTR at all pH values tested (pH 3.2–6.8; Fig. 2A). The ionic strength in these experiments is high enough (>0.2 M) that significant changes in amyloidogenicity are not expected because of ionic strength increases over the pH range of 3.6–5.2 (acetate buffer, ionic strength increase ≈0.09 M). At the optimal denaturation stress for maximal V122I fibril formation (pH 4.2), V122I forms ≥20% more fibrils than WT, as judged by turbidity and 1.7-fold more, as judged by ThT binding (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). Moreover, the extent of V122I fibril formation is ≥20% that afforded by WT TTR (pH 4.2) at all time points after the initial period up to and beyond the half-life of TTR in plasma (8–18 h; Fig. 2B; ref. 29). The FAC variant formed amyloid fibrils ≈2-fold faster than WT TTR at pH 4.2, as discerned by turbidity data (Fig. 2B). The 2-fold faster rate of V122I TTR fibril formation from the homotetramer relative to WT implies that the V122I tetramer is less stable than the WT tetramer (see below). That tetramer dissociation is rate limiting for TTR amyloidosis was demonstrated by comparing the time courses of amyloid fibril formation from monomeric WT and V122I M-TTR (Fig. 2C), which are identical and 50-fold faster than amyloid formation from the V122I homotetramer (Fig. 2B). The pH-dependent yield of fibrils from monomeric and tetrameric V122I TTR was very similar at long incubation times (see Fig. 6, which is published as supporting information on the PNAS web site), consistent with the idea that in both cases a misfolded amyloidogenic monomer is the direct precursor to amyloid.
Figure 2.

(A) Fibril formation of V122I TTR (0.2 mg/ml) monitored by turbidity (dark gray) and ThT binding (light gray) as a function of pH (37°C, 72 h). Fibril formation from WT TTR is shown by white bars, monitored by turbidity as a function of pH (37°C, 72 h). The relative amount of V122I fibril formation observed for the pH 4.2 sample was assigned to be unity (monitored by either ThT fluorescence or turbidity). The WT fibril yield is less than V122I at all pHs. (B) The rate of fibril formation at pH 4.2 (37°C) for V122I (●) and WT (○) TTR (0.2 mg/ml) ascertained by turbidity at 400 nm. (C) Kinetics of fibril formation from monomeric V122I M-TTR (●) and WT M-TTR (○) at 37°C (0.15 mg/ml, pH 4.4). The lines through the data points are smoothing curves to guide the eye.
The V122I Tetramer Is Destabilized Relative to WT.
The hypothesis that the V122I mutation alters tetramer stability
was probed by comparative pH-dependent tetramer–monomer equilibrium
measurements and concentration-dependent equilibrium constant
measurements at a fixed chaotrope concentration. We followed the
acid-induced quaternary structure changes as a function of pH by using
an SDS/PAGE method previously validated by analytical
ultracentrifugation (23, 26). After TTR was incubated at the desired
pH, the zwitterionic detergent Z3–14 was added before neutralizing the
protein solution to prevent refolding while preserving the TTR
quaternary structure (Fig.
3A). The FAC variant
dissociates to the monomeric amyloidogenic intermediate over nearly the
same pH range as the WT protein (Fig. 3B;
pHm 4.4), the major difference being that V122I
TTR (0.2 mg/ml) is about 10% monomeric at neutral pH, whereas
dissociated WT monomers are nearly undetectable (Fig. 3 A
and B). The extent of V122I monomer formation at a given pH
is increased by 10–20% relative to WT TTR (Fig. 3B). These
data imply that the native V122I tetramer is destabilized by <1
kcal/mol [ΔΔG = −RT
ln(K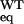 /K
/K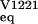 ]
relative to the WT tetramer under these conditions.
]
relative to the WT tetramer under these conditions.
Figure 3.

(A) SDS/PAGE analysis of TTR quaternary structure as a function of pH. (Upper) V122I TTR; (Lower) WT TTR. (B) Mol fraction of monomer as a function of pH calculated by using densitometry from the gels shown in A. A smoothing curve applied to the V122I (●) and WT (○) data guides the eye. (C) Tetramer stability as a function of TTR concentration for V122I (●) and WT (○) in 4 M urea. Samples were incubated for 96 h at 25°C before analysis.
It is possible that in vivo the presence of one equivalent of RBP complexed to TTR might eliminate the differences in the tetramer-folded monomer equilibrium observed in Fig. 3 A and B because of tetramer binding (300 nM Kd for WT) and stabilization (30). However, the gel assay still identifies ≈5–10% monomeric V122I at neutral pH (see Fig. 7, which is published as supporting information on the PNAS web site) in the presence of holo RBP, consistent with rapid on/off rates of RBP binding, validating the physiologic relevance of this study.
The mol fraction of TTR tetramer observed in solution under denaturing conditions (4 M urea; 75% denatured at 0.02 mg/ml, 96 h, 25°C) should be concentration dependent. The dissociation constant (Kd) for a tetrameric protein = 256⋅C3⋅(1 − α)4/α, where α is the percentage of tetrameric protein at a given denaturant concentration, and C is the protein concentration. A comparison of the protein concentration dependence of the dissociation constants characterizing the V122I and WT TTR tetramer-unfolded monomer equilibrium is a direct reflection of the stability of the tetramers. This experiment is not complicated by monomer stability differences, because V122I and WT M-TTR have identical stabilities (Figs. 1B and 2C). The WT protein displayed a dramatic increase in the mol fraction of tetramer observed over the concentration range of 0.1–0.7 mg/ml (96-h incubation, 25°C; Fig. 3C), whereas the fraction of tetramer formed by V122I TTR over the same concentration range is dramatically reduced. An estimation of the difference in free energy between the WT and V122I tetramers (pH 7) at physiological protein concentration (0.25 mg/ml) is ≈0.5 kcal/mol in 4 M urea. Although we are close to equilibrium in the experiments shown in Fig. 3C, it could be that the slower dissociation rate of WT TTR has a contribution.
We have recently shown that anion binding to the TTR tetramer stabilizes this quaternary structure (slowing tetramer dissociation dramatically) inhibiting amyloid fibril formation (28). Although it is not yet known whether there may be a physiologically relevant anion, we studied whether the WT and V122I tetramers were equally susceptible to anion stabilization by Cl− ion. The WT protein is much more susceptible to concentration-dependent anion stabilization and inhibition of amyloid fibril formation than is the V122I cardiac variant (Fig. 4), a feature that may contribute to the amyloidogenicity of V122I TTR.
Figure 4.

Inhibition of WT (○) and V122I (●) fibril formation as a function of Cl− ion concentration. At 0.6 M Cl−, 50% of WT fibrilization is inhibited, whereas V122I amyloidogenecity is reduced only by 10%.
Discussion
TTR is a very interesting protein in that tetramer dissociation [measured by subunit exchange (31) or the data in Fig. 1C] is quite slow, exhibiting a half life of hours to days. Pathogenic mutations like V122I that destabilize the TTR quaternary structure increase its rate of tetramer dissociation and amyloidosis (Fig. 1C), whereas a mutation like T119M, which protects compound heterozygotes from disease by interallelic trans-suppression stabilizes the TTR tetramer dramatically, increasing the barrier for tetramer dissociation, almost completely inhibiting dissociation (32). These observations imply that the transition state for tetramer dissociation does not resemble the tetrameric ground state in structure or energy, because the mutations do not affect the transition state nearly as much as the ground state. Hence changes in stability translate into similar changes in activation barriers. Because the kinetic barrier(s) are already high for WT TTR, stabilizing mutations make them even higher, whereas destabilizing mutations lower the kinetic barrier(s). Thus both kinetics and thermodynamics have to be considered when explaining the influence of a given TTR mutation on amyloidogenicity.
On the basis of estimates of the free energy difference between WT and V122I TTR (≈0.5 kcal/mol), one would expect less than a 5-fold increase in the rate of dissociation for the V122I tetramer, consistent with the 3-fold increase observed. The 3-fold increase in tetramer dissociation rate translates into a 2-fold increase in the rate of V122I TTR fibril formation. The V122I mutation has no detectable effect on the stability of the TTR tertiary structure nor does it alter the amyloid fibril formation rate from the engineered monomeric version of TTR (identical in both respects to WT M-TTR). The V122I mutation selectively acts by lowering the stability of the tetramer (Fig. 3) and consequently lowering the kinetic barrier for tetramer dissociation compared with WT TTR (Figs. 1C and 2B). The 5–10% folded monomeric TTR in equilibrium with the V122I tetramer is a significant risk factor for FAC, because the folded monomer forms amyloid on the time scale of minutes (Fig. 2C) (20), whereas the WT tetramer has to dissociate first, which occurs on a tens of hours time scale (Fig. 2B). In contrast, monomeric WT TTR is hard to detect at neutral pH (Fig. 3A). The physiological relevance of the lowered ability of the V122I tetramer to be stabilized by anions relative to WT is unclear, although this could play a role in FAC by making a greater fraction of the tetramer amenable to dissociation and amyloid formation.
Of the >80 disease-associated TTR variants known, less than 10 have been studied by biophysical methods thus far (14, 23, 26, 33). From emerging and published data, the majority of the pathogenic mutations destabilize the tetramer and lower the kinetic barrier for tetramer dissociation required for amyloidosis (26). The two mutations characterized to date that suppress the onset of amyloid disease have the opposite influence on kinetics and thermodynamics (26, 32, 33). We predict that the uncharacterized variants will also fall into these categories. What is not yet clear because of lack of data is whether the disease-associated mutations will generally alter tertiary structural stability in addition to quaternary structural stability (14). It is interesting that the V122I FAC mutation does not alter the tertiary structural stability relative to WT TTR. Ongoing studies should clarify this issue. Although there are other examples of mutations that appear to alter quaternary structure stability without altering tertiary structure stability, rarely have the isolated tertiary structures been studied carefully (34–37). In addition to familial TTR amyloid diseases, familial amyotrophic lateral sclerosis and several cancers appear to be associated with the quaternary structural destabilization (misfolding) of superoxide dismutase and P53 tumor suppressor, respectively (38–42).
In retrospect, the V122I selective destabilization of the tetramer is not surprising, as this mutation is in the β-sheet mediated quaternary structural interface. Ile-122 is located on the periphery of the H strand, which makes an antiparallel β-sheet interaction with strand H′ of another monomer, stabilizing the dimer interface. The side chain of Ile-122 packs against the side chains of Phe-87′ and Tyr-114′ of the neighboring subunit. The packing between the Ile-122 and Tyr-114′ side chains is slightly altered relative to the Val-122/Tyr-114′ interaction. The subtle movement of the Tyr-114′ side chain in the V122I homotetramer alters its interactions with the AB loop of a second dimer at the face-to-face dimer–dimer interface, rationalizing the observed tetramer destabilization (43). A previously described mutation in the AB loop (V20I) similarly destabilizes the TTR tetramer by altering the interaction of this loop with Tyr-114′ (the AB–AB′ loop interaction is also changed), destabilizing the face-to-face dimer interface, a perturbation resulting in cardiomyopathy (14).
There is strong genetic, medical, and biochemical evidence for the hypothesis that amyloid fibrils are the causative agent of TTR amyloid diseases, including FAC (32, 44–46). The V122I mutation causing familial amyloidotic cardiomyopathy shifts the tetramer-folded monomer equilibrium toward monomer (lowers tetramer stability) and lowers the kinetic barrier for tetramer dissociation, which increases the extent and rate of amyloid fibril formation relative to WT TTR. The increase in the velocity of rate-limiting tetramer dissociation required for amyloid fibril formation, coupled with the presence of folded monomer under physiological conditions, may explain why the V122I cardiac disease penetrance approaches 100%, whereas senile systemic amyloidosis, involving WT TTR amyloid deposition in the heart (similar age of onset), affects less than 25% of the population above age 80.
Supplementary Material
Acknowledgments
We thank Kristina Berecic for help in the initial stages of this project, Joleen White (The Scripps Research Institute) for providing the RBP, and Dr. Per Hammarström for helpful discussions. We are grateful for primary financial support from the National Institutes of Health [R01 DK46335 (J.K.) and R01 AG15916 (J.B.)] and secondary support from The Skaggs Institute of Chemical Biology and The Lita Annenberg Hazen Foundation.
Abbreviations
- FAC
familial amyloidotic cardiomyopathy
- TTR
transthyretin
- WT M-TTR
F87M/L110M variant
- ThT
thioflavin T
- V122I M-TTR
V122I/F87M/L110M variant
- WT
wild type
- RBP
retinol-binding protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 14757.
References
- 1.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 2.Buxbaum J N, Tagoe C E. Annu Rev Med. 2000;51:543–569. doi: 10.1146/annurev.med.51.1.543. [DOI] [PubMed] [Google Scholar]
- 3.Dobson C M. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 4.Fink A L. Folding Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 5.Cohen A S, Connors L H. J Pathol. 1987;151:1–10. doi: 10.1002/path.1711510102. [DOI] [PubMed] [Google Scholar]
- 6.Sipe J D. Annu Rev Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 7.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinsion W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C F, Pepys M B. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 8.Stevens F J, Kisilevsky R. Cell Mol Life Sci. 2000;57:441–449. doi: 10.1007/PL00000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCutchen S L, Colon W, Kelly J W. Biochemistry. 1993;32:12119–12127. doi: 10.1021/bi00096a024. [DOI] [PubMed] [Google Scholar]
- 10.Bellotti V, Mangione P, Merlini G. J Struct Biol. 2000;130:280–289. doi: 10.1006/jsbi.2000.4248. [DOI] [PubMed] [Google Scholar]
- 11.Gorevic P D, Prelli F C, Wright J, Pras M, Frangione B. J Clin Invest. 1989;83:836–843. doi: 10.1172/JCI113966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson D R, Pastore R D, Yaghoubian R, Kane I, Gallo G, Buck F S, Buxbaum J N. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 13.Afolabi I, Asl K H, Nakamura M, Jacobs P, Hendrie H, Benson M D. Amyloid. 2000;7:121–125. doi: 10.3109/13506120009146249. [DOI] [PubMed] [Google Scholar]
- 14.Jenne D E, Denzel K, Blatzinger P, Winter P, Obermaier B, Linke R P, Altland K. Proc Natl Acad Sci USA. 1996;93:6302–6307. doi: 10.1073/pnas.93.13.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westermark P, Sletten K, Johansson B, Cornwell G G., III Proc Natl Acad Sci USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saraiva M J M, Birken S, Costa P P, Goodman D S. J Clin Invest. 1984;74:104–119. doi: 10.1172/JCI111390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Misu K, Hattori N, Nagamatsu M, Ikeda S, Ando Y, Nakazato M, Takei Y, Hanyu N, Usui Y, Tanaka F, et al. Brain. 1999;122:1951–1962. doi: 10.1093/brain/122.10.1951. [DOI] [PubMed] [Google Scholar]
- 18.Connors L H, Richardson A M, Theberge R, Costello C E. Amyloid. 2000;7:54–69. doi: 10.3109/13506120009146826. [DOI] [PubMed] [Google Scholar]
- 19.Monaco H L, Rizzi M, Coda A. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 20.Jiang X, Smith C S, Petrassi H M, Hammarstrom P, White J T, Sacchettini J C, Kelly J W. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 21.Liu K, Cho H S, Lashuel H A, Kelly J W, Wemmer D E. Nat Struct Biol. 2000;7:754–757. doi: 10.1038/78980. [DOI] [PubMed] [Google Scholar]
- 22.Colon W, Kelly J W. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 23.Lai Z, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson D R, Pastore R, Pool S, Malendowicz S, Kane I, Shivji A, Embury S H, Ballas S K, Buxbaum J N. Hum Genet. 1996;98:236–238. doi: 10.1007/s004390050199. [DOI] [PubMed] [Google Scholar]
- 25.Lashuel H A, Wurth C, Woo L, Kelly J W. Biochemistry. 1999;38:13560–13573. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- 26.McCutchen S L, Lai A, Miroy G J, Kelly J W, Colon W. Biochemistry. 1995;34:13527–13536. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 27.Krivacic J R, Urry D W. Anal Chem. 1971;43:1508–1510. doi: 10.1021/ac60305a031. [DOI] [PubMed] [Google Scholar]
- 28.Hammarstrom P, Jiang X, Deechongkit S, Kelly J W. Biochemistry. 2001;40:11453–11459. doi: 10.1021/bi010673+. [DOI] [PubMed] [Google Scholar]
- 29.Benson M D, Kluve-Beckerman B, Liepnieks J J, Murrell J R, Hanes D, Uemichi T. In: The Nature and Origin of Amyloid Fibrils. Bock G, Goode J, Costa P, editors. New York: Wiley; 1996. p. 266. [Google Scholar]
- 30.White J T, Kelly J W. Proc Natl Acad Sci USA. 2001;98:13019–13024. doi: 10.1073/pnas.241406698. . (First Published October 30, 2001; 10.1073/pnas.241406698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider F, Hammarstrom P, Kelly J W. Protein Sci. 2001;10:1606–1613. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammarstrom P, Schneider F, Kelly J W. Science. 2001;293:2462–2465. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 33.Almeida M R, Alves I L, Terazaki H, Ando Y, Saraiva M J. Biochem Biophys Res Commun. 2000;270:1024–1028. doi: 10.1006/bbrc.2000.2554. [DOI] [PubMed] [Google Scholar]
- 34.Lavulo L T, Sossong T M, Brigham-Burke M R, Doyle M L, Cox J D, Christianson D W, Ash D E. J Biol Chem. 2001;276:14242–14248. doi: 10.1074/jbc.M010575200. [DOI] [PubMed] [Google Scholar]
- 35.Srinvas V R, Reddy G B, Ahmad N, Swaminathan C P, Mitra N, Surolia A. Biochem Biophys Acta. 2001;1527:102–111. doi: 10.1016/s0304-4165(01)00153-2. [DOI] [PubMed] [Google Scholar]
- 36.Mullen C A, Jennings P A. J Mol Biol. 1998;276:819–827. doi: 10.1006/jmbi.1997.1530. [DOI] [PubMed] [Google Scholar]
- 37.Swint-kruse L, Elam C R, Lin J W, Wycuff D R, Matthews K S. Protein Sci. 2001;10:262–276. doi: 10.1110/ps.35801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng H X, Hentati A, Tainer J A, Iqbal Z, Cayabyab A, Hung W Y, Getzoff E D, Hu P, Herzfeldt B, Roos B P, et al. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 39.Phillips J, Tainer J, Getzoff E D, Boulianne G L, Kirby K, Hilliker A J. Proc Natl Acad Sci USA. 1995;92:8574–8578. doi: 10.1073/pnas.92.19.8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pramatarova A, Figlewicz D A, Krizus A, Han F Y, Ceballos-Picot I, Nicole A, Dib M, Meininger V, Brown R H, Rouleau G A. Am J Hum Genet. 1995;56:592–596. [PMC free article] [PubMed] [Google Scholar]
- 41.Jeffrey P D, Gorina S, Pavletich N P. Science. 1995;267:1498–1502. doi: 10.1126/science.7878469. [DOI] [PubMed] [Google Scholar]
- 42.Chene P. Oncogene. 2001;20:2611–2617. doi: 10.1038/sj.onc.1204373. [DOI] [PubMed] [Google Scholar]
- 43.Damas A M, Ribeiro S, Lamzin V S, Palha J A, Saraiva M J. Acta Crystallogr. 1996;D52:966–972. doi: 10.1107/S0907444996003307. [DOI] [PubMed] [Google Scholar]
- 44.Jacobson D R, Buxbaum J N. Adv Hum Genet. 1991;20:69–123. doi: 10.1007/978-1-4684-5958-6_2. [DOI] [PubMed] [Google Scholar]
- 45.Kelly J W, Lansbury P T. Amyloid. 1994;1:186–205. [Google Scholar]
- 46.Selkoe D J. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}