

Abstract
Objective
3‐hydroxy‐3‐methylglutaryl‐coenzyme A (HMG‐CoA) reductase inhibitors (statins) can affect post‐translational processes, thus being responsible for decreased farnesylation and geranylgeranylation of intracellular small G proteins such as Ras, Rho and Rac, essential for cell survival and proliferation. In this regard, recent in vitro and in vivo studies suggest a possible role for both statins and farnesyl transferase inhibitors in the treatment of malignancies. Within such a context, the aim of our study was to investigate effects of either simvastatin (at concentrations of 1, 15, and 30 μm) or the farnesyl transferase inhibitor R115777 (at concentrations of 0.1, 1, and 10 μm), on two cultures of human non‐small lung cancer cells, adenocarcinoma (GLC‐82) and squamous (CALU‐1) cell lines. In particular, we evaluated actions of these two drugs on phosphorylation of the ERK1/2 group of mitogen‐activated protein kinases and on apoptosis, plus on cell numbers and morphology.
Materials and Methods
Western blotting was used to detect ERK phosphorylation, and to assess apoptosis by evaluating caspase‐3 activation; apoptosis was also further assessed by terminal deoxynucleotidyl‐mediated dUTP nick end labelling (TUNEL) assay. Cell counting was performed after trypan blue staining.
Results and conclusion
In both GLC‐82 and CALU‐1 cell lines, simvastatin and R115777 significantly reduced ERK phosphorylation; this effect, which reached the greatest intensity after 36 h treatment, was paralleled by a concomitant induction of apoptosis, documented by significant increase in both caspase‐3 activation and TUNEL‐positive cells, associated with a reduction in cell numbers. Our results thus suggest that simvastatin and R115777 may exert, in susceptible lung cancer cell phenotypes, a pro‐apoptotic and anti‐proliferative activity, which appears to be mediated by inhibition of the Ras/Raf/MEK/ERK signalling cascade.
Introduction
Lung cancer is the leading cause of cancer death worldwide. In particular, non‐small cell lung cancer (NSCLC), including adenocarcinoma, squamous cell carcinoma and large cell carcinoma, accounts for more than 80% of total pulmonary malignancies 1. Current therapeutic strategies have only able partial efficacy, and therefore concerted efforts are widely under way to develop new pharmacological approaches directed against molecular targets involved in NSCLC pathogenesis. A key role in the oncogenic process is played by the Ras family of guanine nucleotide binding, small G‐proteins 2, 3. Indeed, Ras proteins work within a multicomponent signalling network that can be activated by a variety of growth factor receptors, whose stimulation leads to transcription of several different genes implicated in cell differentiation, proliferation and apoptosis. Ras family members, including HRas, KRas and NRas, activate Raf1, which in turn triggers a phosphorylation cascade leading to subsequent activation of MEK1/2 and ERK (extracellular signal‐regulated kinases) subgroups of mitogen‐activated protein kinases (MAPK) 4. Upon Ras‐dependent phosphorylation, ERK becomes active and also is able to translocate to the cell nucleus where many of its substrates are located.
Carcinogenic mutations of KRas that stabilize the GTP‐bound active conformation of this G‐protein (thus being responsible for its constitutive activation), have been detected in about 20% of lung cancers, with relevant predominance occurring in the adenocarcinoma subtype of NSCLC 5. Experimental evidence suggests that Ras mutations could be caused at least in part by cigarette smoking 6, a very well‐known risk factor for lung cancer. Moreover, an important contribution to cancer development can also arise from post‐translational modification of Ras proteins, essential for their subcellular location and biological function. In particular, Ras activation requires covalent attachment of a non‐sterol isoprenoid consisting of either farnesyl pyrophosphate (FPP) or geranyl geranyl pyrophosphate (GGPP), which directs Ras family members towards the endoplasmic reticulum, where further post‐translational changes provide target signals for translocation to plasma membrane 7.
This basic understanding has led to evaluation of potential anti‐cancer activity of drugs that are capable of interfering with post‐translational modification of Ras proteins 8. For these reasons, increasing attention is currently being paid to statins. The latter inhibit 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase, which is the rate limiting enzyme of the mevalonate pathway, which leads to synthesis of downstream mevalonate derivatives, FPP and GGPP 9. Thus, it is noteworthy that simvastatin can inhibit cell population expansion of SCLC (small cell lung cancer) cell lines; this effect is likely to be due to interference with Ras‐dependent activation of the MEK/ERK signalling complex 10. Addition of pravastatin to standard chemotherapeutic agents has elicited significant increase in survival of patients with hepatocellular carcinoma 11. Cerivastatin has been shown to exert inhibition of metastatic activity of invasive breast cancer cell lines 12 and lovastatin demonstrates induction of cell death in several different cancer cell lines, including those of prostate cancer, squamous cell carcinoma and myeloma plasma cells 13, 14, 15.
Anti‐tumour effects can also be caused by other compounds that target post‐translational modifications of the Ras superfamily, such as some inhibitors of farnesyl transferase, the enzyme that catalyses covalent attachment of FPP (farnesylation) to the cysteine residue of so‐called CAAX carboxyl‐terminal motif 7. Farnesyl transferase inhibitors can be effective as anti‐proliferative and pro‐apoptotic agents 8; among these drugs, imidazole compound R115777 (tipifarnib) is characterized, with regard to Ras farnesylation, by competitive inhibitory activity, probably due to interaction of the imidazole group with the coordination structure of the zinc catalytic site of farnesyl transferase 16, 17. R115777 is able to exert relevant anti‐proliferative action on human pancreatic tumour and bladder cancer cell lines, as well as to induce marked pro‐apoptotic effects on human melanoma cells; in contrast, cells of the human lung adenocarcinoma A549 line appear to be relatively insensitive to R115777 16.
On the basis of all the above‐mentioned considerations, the aim of our study was to investigate, in human NSCLC cells, effects on ERK phosphorylation of either simvastatin or R115777. Furthermore, to assess potential pro‐apoptotic action of each of these two drugs, caspase‐3 activation, TUNEL assay and cell viability were also assessed. We decided to use simvastatin as it is a powerful HMG‐CoA reductase inhibitor and also exhibits marked lipophilic features, thus being able to permeate cell membranes. Our findings are on to two human NSCLC cell lines, adenocarcinoma cell line (GLC‐82) and squamous cell line (CALU‐1).
Materials and methods
Reagents
Anti‐phospho‐ERK1/2 monoclonal antibody was purchased from New England Biolabs (Beverly, MA, USA); anti‐(total)‐ERK1/2 polyclonal antibody was commercially provided by Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti‐caspase‐3 monoclonal antibody E83‐77 was purchased from Abcam (Cambridge, UK) and trypan blue was purchased from Sigma (St. Louis, MO, USA). Purified crystalline powders of both simvastatin and farnesyl transferase inhibitor R115777 were commercially obtained from Sigma, then dissolved into refrigerated and light‐protected DMSO stock solutions.
Culture and treatment of human lung cancer cells
GLC‐82 is a lung adenocarcinoma cell line 18 and CALU‐1 is a squamous carcinoma cell line 19. Both lung cancer cell lines were cultured at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS, penicillin 100 U/ml, streptomycin 100 μg/ml, and fungizone 25 μg/ml. All cells were maintained at 37 °C in a humidified 5% CO2 atmosphere and were plated in a 100‐mm polystyrene dish (Falcon, Becton‐Dickinson, Lincoln Park, NJ, USA) and ten ml supplemented DMEM was added, as previously described 20. When cells had achieved ~70% confluence they were treated with either simvastatin or R115777; there was no change of medium after treatment. All cells were then incubated for 36 h with different concentrations of simvastatin (1, 15, 30 μm) or R115777 (0.1, 1, 10 μm). Solvent alone, as employed above to dissolve the drugs, was used as control. After treatment, cells were processed for protein extraction and immunoblotting.
Protein extraction and immunoblot analysis
Following treatment with simvastatin or R1157777, cells were lysed for western blotting, in radioimmunoprecipitation assay (RIPA) buffer (150 mm NaCl, 1.5 mm MgCl2, 10 mm NaF, 10% glycerol, 4 mm EDTA, 1% Triton X‐100, 0.1% SDS, 1% deoxycholate, 50 mm Hepes, pH 7.4, plus PPIM, 25 mm β‐glycerophosphate, 1 mm Na3VO4, 1 mm PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin). Protein extracts were then separated on a 12.5% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Amersham Pharmacia, Little Chalfont, UK), as previously described 21. Immunoblotting was performed using anti‐phospho‐ERK1/2 monoclonal antibody. After being ‘stripped’, membranes were re‐probed with polyclonal antibody against total (phosphorylated and unphosphorylated) ERK1/2. Blots were also incubated with monoclonal antibody directed against active caspase‐3, a common enzymatic marker of apoptosis 22. Antibody binding was visualized by enhanced chemiluminescence (ECL‐Plus; Amersham Pharmacia); intensities of experimental bands were analysed by computer‐assisted densitometry and expressed as arbitrary units (AU; control levels set at 100). These experiments were performed in triplicate.
TUNEL assay
To perform terminal deoxynucleotidyl‐mediated dUTP nick end labelling (TUNEL) assay, cells of each line were plated on 24 × 24 mm cover slips (Carlo Erba Reagenti, Milan, Italy) placed in six‐well microtitre plates (Corning Incorporated, Corning, NY, USA) in DMEM supplemented with 10% FCS, penicillin 100 U/ml, streptomycin 100 mg/ml, and fungizone 25 mg/ml; 24 h after plating, when cells had reached 50–60% confluence, cells of each line were treated with either simvastatin (30 μm) or R115777 (10 μm); incubation medium was not changed. Following treatment, TUNEL assay was performed using MEBSTAIN Apoptosis TUNEL Kit Direct (MBL, Woburn, MA, USA), strictly following instructions furnished by the manufacturer. Photographs were acquired using a Leica GRDM confocal microscope (Leica, Wetzlar, Germany).
Cell viability
Cell viability was assessed by light microscopy and trypan blue dye exclusion. Cell numbers were evaluated by direct counting, using a haemocytometer. All experiments were performed in triplicate.
Statistical analysis
All data are expressed as mean ± SEM. Statistical evaluation of results was performed by analysis of variance (ANOVA). Differences identified by ANOVA were pinpointed by unpaired Student's t‐test and threshold of statistical significance was set at P < 0.05.
Results
ERK phosphorylation
After 36 h, significant reduction in ERK1/2 phosphorylation was detected in both CALU‐1 (Fig. 1, panels a and b) and GLC‐82 (Fig. 1, panels c and d) cell lines, as a consequence of treatment with either simvastatin or R115777. This effect was observed at each drug concentration (Fig. 1; Table 1). As the monoclonal antibody (anti‐phospho‐ERK1/2) used here specifically recognizes the phosphorylated, active form of ERK1/2, the remarkable reduction in phosphorylation pattern of this MAPK subgroup can be considered as a reliable marker of ERK functional inhibition, elicited by both simvastatin and R115777. These two drugs exerted their effects uniquely on phosphorylation‐dependent activation of ERK1/2, without affecting its total expression, as demonstrated by the unchanged binding patterns of anti‐(total) ERK polyclonal antibody (Fig. 1).
Figure 1.

Effects of different concentrations of simvastatin and R115777 on phosphorylated ERK1/2 (p‐ERK) and total ERK1/2 expression, evaluated by western blotting, on CALU‐1 cells (Panels a and b) and GLC‐82 cells (Panels c and d).
Table 1.
Effects of different concentrations of simvastatin and R115777 on phosphorylated ERK1/2 (p‐ERK)
| GLC p‐ERK | Control | Simvastatin 1 μm | Simvastatin 15 μm | Simvastatin 30 μm |
| 100 ± 1.17 | 71 ± 1.15a | 68 ± 1.0a | 49 ± 1.12a | |
| Control | R115777 0.1 μm | R115777 1 μm | R115777 10 μm | |
| 100 ± 1.13 | 55 ± 0.98a | 54 ± 0.99a | 55 ± 1.02a | |
| CALU p‐ERK | Control | Simvastatin 1 μm | Simvastatin 15 μm | Simvastatin 30 μm |
| 100 ± 1.08 | 78 ± 1.04a | 54 ± 0.99a | 51 ± 1.11a | |
| Control | R115777 0.1 μm | R115777 1 μm | R115777 10 μm | |
| 100 ± 1.10 | 47 ± 1.02a | 41 ± 1.01a | 44 ± 1.14a |
Data expressed as arbitrary units.
P < 0.01.
Caspase‐3 activation, TUNEL assay and cell viability
After 36 h, both simvastatin and R115777 induced significant increase in caspase‐3 activation, detectable at each drug concentration (Fig. 2; Table 2) and paralleled by concomitant increase in the number of TUNEL‐positive cells (Fig. 3). Furthermore, after 12, 24 and 36 h, simvastatin elicited a time‐ and concentration‐dependent reduction in viability of CALU‐1 and GLC‐82 cells, as detected by cell counts and expressed as confluence percentages (Figs 4 and 5). In particular, this effect was more prominent after 36 h incubation of both cell lines with simvastatin. Similarly, R115777 caused a significant decrease in cell numbers, especially at highest concentrations (Figs 4 and 5). Moreover, light microscopy (magnification: 40×) indicated that both drugs induced morphological changes, as well as a reduction in cell adhesion (Figs 6 and 7). With regard to changes in cell shape, these were already detectable after 12 h, and reached their most relevant expression at the 36th hour, especially after simvastatin treatment.
Figure 2.
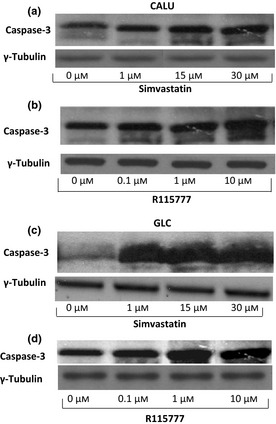
Effects of different concentrations of simvastatin and R115777 on active caspase‐3 expression, evaluated by western blotting on CALU‐1 cells (Panels a and b) and GLC‐82 cells (Panels c and d).
Table 2.
Effects of different concentrations of simvastatin and R115777 on active caspase‐3 expression
| GLC caspase‐3 | Control | Simvastatin 1 μm | Simvastatin 15 μm | Simvastatin 30 μm |
| 26 ± 1.02 | 90 ± 1.15a | 91 ± 1.11a | 100 ± 1.08a | |
| Control | R115777 0.1 μm | R115777 1 μm | R115777 10 μm | |
| 28 ± 1.05 | 68 ± 1.12a | 110 ± 1.14a | 105 ± 1.19a | |
| CALU caspase‐3 | Control | Simvastatin 1 μm | Simvastatin 15 μm | Simvastatin 30 μm |
| 38 ± 1.11 | 60 ± 1.05a | 96 ± 1.08a | 100 ± 1.04a | |
| Control | R115777 0.1 μm | R115777 1 μm | R115777 10 μm | |
| 33 ± 1.17 | 69 ± 1.01a | 71 ± 1.15a | 100 ± 1.12a |
Data expressed as arbitrary units.
P < 0.01.
Figure 3.
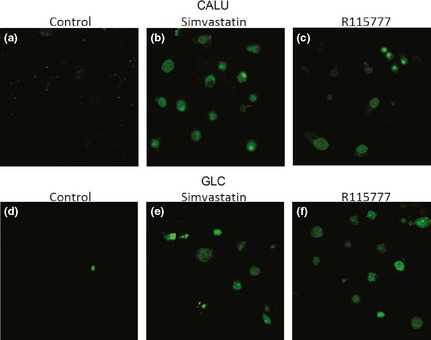
TUNEL assay on CALU‐1 cells (Panels a–c) and on GLC‐82 cells (Panels d–f) treated with simvastatin 30 μm (Panels b and e) or with R115777 10 μm (Panels c and f).
Figure 4.
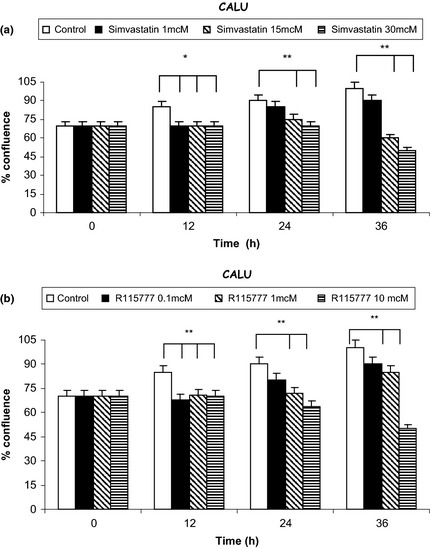
Effects of different concentrations of simvastatin (Panel a) and R115777 (Panel b) on CALU‐1 cell counts, expressed as confluence percentages. *P < 0.05; **P < 0.01.
Figure 5.
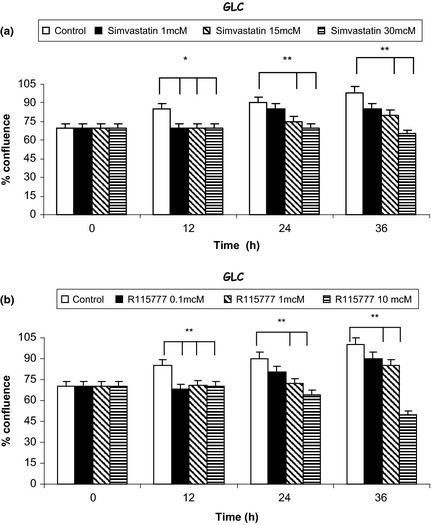
Effects of different concentrations of simvastatin (Panel a) and R115777 (Panel b) on GLC‐82 cell counts, expressed as confluence percentages. *P < 0.05; **P < 0.01.
Figure 6.
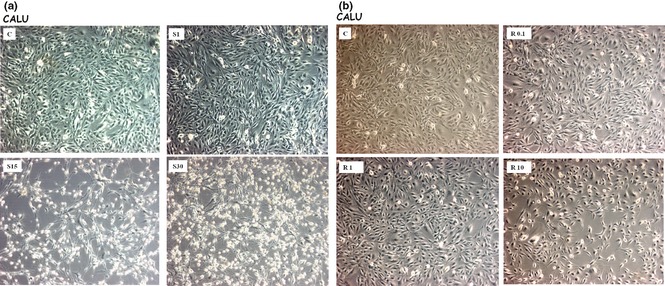
Effects of different concentrations of simvastatin (Panel a) and R115777 (Panel b) on CALU‐1 cell viability and morphology, visualized by light microscopy (40×); C: control.
Figure 7.
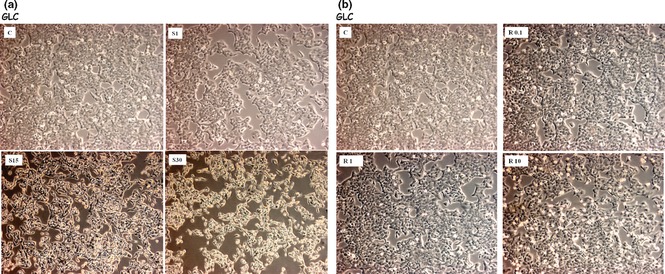
Effects of different concentrations of simvastatin (Panel a) and R115777 (Panel b) on GLC‐82 cell viability and morphology, visualized by light microscopy (40×); C: control.
Discussion and conclusions
Our results show that in both CALU‐1 and GLC‐82 lung cancer cell lines, simvastatin and R115777 significantly inhibited ERK1/2 phosphorylation and induced caspase‐3 activated apoptosis; these effects, which reached greatest intensity after 36 h treatment, were paralleled by concomitant reduction in cell numbers. Time‐ and concentration‐dependent reductions in cell viability elicited by both these drugs were also associated with cell structural changes, possibly caused by cytoskeletal rearrangements, which could have been responsible for loss of cell survival signals. This implies that cell death was probably preceded by relevant cellular alterations affecting cell survival and proliferation. Moreover, it is reasonable to suppose that the cause of drug‐induced cell death was apoptosis, as suggested by activation of caspase‐3 cystein protease, acting as a common effector pathway for apoptotic processes originating at both cell membrane and mitochondrial levels 23. This was confirmed by TUNEL analysis.
Simvastatin and R115777‐dependent reductions in ERK phosphorylation were paralleled by increases in caspase‐3 activation and TUNEL positivity, as well as by concomitant decreases in cell viability. Taken together, our findings thereby suggest that inhibition of ERK activation plays a key role in the context of molecular mechanisms underlying pro‐apoptotic and cytotoxic properties exerted by simvastatin and R115777 in NSCLC cells. Indeed, in other cell lines, anti‐apoptotic effects have been reported as a consequence of ERK1/2 stimulation by Ki‐Ras 24. In particular, Raf/MEK/ERK pathway can result in phosphorylation of pro‐apoptotic Bad protein, thus contributing to its inactivation and sequestration, triggering subsequent anti‐apoptotic responses mediated by homodimerization of Bcl‐2 protein 25, 26. Furthermore, stimulation of the Raf/MEK/ERK cascade can also result in phosphorylation‐dependent activation of anti‐apoptotic Mcl‐1 protein, as well as in phosphorylation‐dependent degradation of pro‐apoptotic Bim protein 27, 28, 29.
By inhibiting HMG‐CoA reductase, statins can suppress Ras‐dependent activation of ERK via impairment of protein prenylation, as well as by interference with formation of cholesterol‐rich lipid microdomains (lipid rafts) in plasma membranes 30. These two processes are crucial for activity of several signalling proteins related to Ras 31. Thus, it is very interesting to point out that lovastatin is capable of downregulating Ras and inhibiting phosphorylation of Raf and ERK1/2 in NSCLC cells 32. In these ways, all the mechanisms can significantly contribute to pro‐apoptotic action of simvastatin, observed in our present study. Similarly, by preventing post‐translational changes required for Ras activation, farnesyl transferase inhibitor R115777 could have exerted its pro‐apoptotic effect on CALU‐1 and GLC‐82 cells via blockade of Ras‐dependent signalling pathways. Moreover, cancer cells usually express high levels of HMG‐CoA reductase, which appear to be required by NSCLC cells to satisfy their increased need for isoprenoids and lipids; these biological features can also contribute to explain, at least in part, the high sensitivity of CALU‐1 and GLC‐82 cells to isoprenoid‐depleting effects of statins and farnesyl transferase inhibitors.
However, current data suggest that both statins and farnesyl transferase inhibitors are characterized by only modest anti‐cancer properties, when used as monotherapies 8; in cancer treatment, statins are mainly only considered to be potentially useful drugs when combined with conventional chemotherapeutic agents 33. With regard to lung cancer, as 40–80% of NSCLC subtypes overexpress epidermal growth factor receptor (EGFR) (whose activation triggers downstream signalling networks involving the Ras‐Raf‐MAPK pathway), ongoing experimental approaches evaluate therapeutic potential of pharmacological associations including statins and EGFR tyrosine kinase inhibitors such as gefitinib 3, 34, 35. In particular, some in vitro studies have shown that combination of EGFR tyrosine kinase inhibitor gefitinib with lovastatin produces synergistic cytotoxicity in gefitinib‐resistant NSCLC cells 32. A recent randomized phase II trial has demonstrated that, when compared with gefitinib alone, in patients with advanced NSCLC, drug association of gefitinib–simvastatin is capable of improving both response rate and progression‐free survival 36. Although no positive results in NSCLC have been obtained either in vitro or in vivo using the farnesyl transferase inhibitor lonafarnib in combination with carboplatin and paclitaxel, as well as testing R115777 alone 16, 37, the latter elicited significant anti‐tumour effects in our present study. This implies that the variety of cell responses induced by treatment with R115777 probably depends on specific biological features characterizing distinct malignant phenotypes. It is thus possible that both cultures of CALU‐1 and GLC‐82 cells could be phenotypically susceptible to pro‐apoptotic and anti‐proliferative actions of R115777.
In conclusion, our findings concerning anti‐tumour activities of simvastatin and R115777, as detected in two NSCLC cell lines CALU‐1 and GLC‐82, further corroborate potential usefulness of in vitro and in vivo studies aimed at evaluating effects of statins and farnesyl transferase inhibitors on lung cancer, eventually to be associated with other anti‐cancer drugs. Thus, future development of the present investigation could be aimed at assessing activity of drug combinations including a statin or a farnesyl transferase inhibitor, and a chemotherapeutic agent commonly used in treatment of lung cancer, such as gemcitabine.
References
- 1. Zalcman G, Bergot E, Lechapt E (2010) Update on nonsmall cell lung cancer. Eur. Respir. Rev. 19, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fernandez‐Medarde A, Santos E (2011) Ras in cancer and developmental diseases. Genes Cancer 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lawson MH, Elsen T (2009) The biology of lung cancer. Eur. Respir. Mon. 44, 88–105. [Google Scholar]
- 4. Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40. [DOI] [PubMed] [Google Scholar]
- 5. Wistuba II, Gazdar AF (2006) Lung cancer preneoplasia. Annu. Rev. Pathol. 1, 331–348. [DOI] [PubMed] [Google Scholar]
- 6. Ahrendt SA, Decker PA, Alawi EA, Zhu YR, Sanchez‐Cespedes M, Yang SC et al (2001) Cigarette smoking is strongly associated with mutation of the K‐ras gene in patients with primary adenocarcinoma of the lung. Cancer 92, 1525–1530. [DOI] [PubMed] [Google Scholar]
- 7. Zhang FL, Casey PJ (1996) Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 65, 241–269. [DOI] [PubMed] [Google Scholar]
- 8. Kostantinopolous PA, Karamouzis MV, Papavassiliou AG (2007) Post‐translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555. [DOI] [PubMed] [Google Scholar]
- 9. Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM (2005) Statins and cancer prevention. Nat. Rev. Cancer 5, 930–942. [DOI] [PubMed] [Google Scholar]
- 10. Khanzada UK, Pardo OE, Meier C, Downward J, Seckl MJ, Arcaro A (2006) Potent inhibition of small‐cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signaling. Oncogene 25, 877–887. [DOI] [PubMed] [Google Scholar]
- 11. Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y et al (2001) Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br. J. Cancer 84, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denoyelle C, Vasse M, Korner M, Mishal Z, Ganné F, Vannier JP et al (2001) Cerivastatin, an inhibitor of HMG‐CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis 22, 1139–1148. [DOI] [PubMed] [Google Scholar]
- 13. Dimitrolaukos J, Ye LY, Benzaquen M, Moore MJ, Karnel‐Reid S, Freedman MH et al (2001) Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin‐induced apoptosis: therapeutic implications. Clin. Cancer Res. 7, 158–167. [PubMed] [Google Scholar]
- 14. Marcelli M, Cunningham GR, Haidacher SJ, Padayatty SJ, Sturgis L, Kagan C et al (1998) Caspase‐7 is activated during lovastatin‐induced apoptosis of the prostate cancer line LNCaP. Cancer Res. 58, 76–83. [PubMed] [Google Scholar]
- 15. van de Donk NW, Kamphuis MM, Lokhorst HM, Bloem AC (2002) The cholesterol lowering drug lovastatin induces cell death in myeloma plasma cells. Leukaemia 16, 1362–1371. [DOI] [PubMed] [Google Scholar]
- 16. End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P et al (2001) Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro . Cancer Res. 61, 131–137. [PubMed] [Google Scholar]
- 17. Huang C‐C, Casey P, Fierke CA (1997) Evidence for a catalytic role of zinc in protein farnesyltransferase. J. Biol. Chem. 272, 20–23. [DOI] [PubMed] [Google Scholar]
- 18. Fan YS, Li P (1987) Cytogenetic studies of four human lung adenocarcinoma cell lines. Cancer Genet. Cytogenet. 26, 317–325. [DOI] [PubMed] [Google Scholar]
- 19. Kaufmann SH, Mabry M, Jasti R, Shaper JH (1991) Differential expression of nuclear envelope lamins A and C in human lung cancer cell lines. Cancer Res. 51, 581–586. [PubMed] [Google Scholar]
- 20. Gallelli L, Pelaia G, Fratto D, Muto V, Falcone D, Vatrella A et al (2010) Effects of budesonide on p38 MAPK activation, apoptosis and IL‐8 secretion, induced by TNF‐α and Haemophilus influenzae in human bronchial epithelial cells. Int. J. Immunopathol. Pharmacol. 23, 471–479. [DOI] [PubMed] [Google Scholar]
- 21. Gallelli L, Falcone D, Pelaia G, Renda T, Terracciano R, Malara N et al (2008) IL‐6 receptor superantagonist SANT7 inhibits TGF‐β‐induced proliferation of human lung fibroblasts. Cell Prolif. 41, 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nicholson DW, Thornberry NA (1997) Caspases: killer proteases. Trends Biochem. Sci. 22, 299–306. [DOI] [PubMed] [Google Scholar]
- 23. O'Sullivan MP, Tyner JW, Holtzman MJ (2003) Apoptosis in the airways: another balancing act in the epithelial program. Am. J. Respir. Cell Mol. Biol. 29, 3–7. [DOI] [PubMed] [Google Scholar]
- 24. Cuda G, Paternò R, Ceravolo R, Candigliota M, Perrotti N, Perticone F et al (2002) Protection of human endothelial cells from oxidative stress: role of RAS‐ERK1/2 signaling. Circulation 105, 968–974. [DOI] [PubMed] [Google Scholar]
- 25. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EWT, Chang F et al (2007) Roles of the RAF/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 1773, 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine‐phosphorylation of death agonist BAD in response to survival factor results in binding to 14–3‐3 not Bcl‐XL. Cell 87, 589–592. [DOI] [PubMed] [Google Scholar]
- 27. Harada H, Quearry B, Ruiz‐Vela A, Korsmeyer SJ (2004) Survival factor‐induced extracellular signal‐regulated kinase phosphorylation BIM, inhibiting its association with BAX and proapoptotic activity. Proc. Natl. Acad. Sci. USA 101, 15313–15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ley R, Balmanno K, Chalmers C, Hadfield K, Weston C, Cook SJ (2003) Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome‐dependent degradation of the BH3‐only protein Bim. J. Biol. Chem. 278, 18811–18816. [DOI] [PubMed] [Google Scholar]
- 29. Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R et al (2003) Activation of ERK1/2 by delta Raf‐1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene 22, 1288–1293. [DOI] [PubMed] [Google Scholar]
- 30. Nowakowski GS, Maurer MJ, Habermann TM, Ansell SM, Macon WR, Ristow KM et al (2010) Statin use and prognosis in patients with diffuse large B‐cell lymphoma and follicular lymphoma in the rituximab era. J. Clin. Oncol. 28, 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jakobisiak M, Golab J (2003) Potential antitumor effects of statins. Int. J. Oncol. 23, 1055–1069. [PubMed] [Google Scholar]
- 32. Park IH, Kim JY, Jung JI, Han JY (2010) Lovastatin overcomes gefitinib resistance in human non‐small cell lung cancer cells with K‐Ras mutations. Invest. New Drugs 28, 791–799. [DOI] [PubMed] [Google Scholar]
- 33. Hindler K, Cleeland CS, Rivera E, Collard CD (2006) The role of statins in cancer therapy. Oncologist 11, 306–315. [DOI] [PubMed] [Google Scholar]
- 34. Dimitrolaukos J, Lorimer IA, Goss G (2006) Strategies to enhance epidermal growth factor inhibition: targeting the mevalonate pathway. Clin. Cancer Res. 12, 4426s–4431s. [DOI] [PubMed] [Google Scholar]
- 35. Zhang X, Chang A (2007) Somatic mutations of the epidermal growth factor receptor and non‐small cell lung cancer. J. Med. Genet. 44, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han JY, Lee SH, Yoo NJ, Lee SH, Moon YJ, Yun T et al (2011) A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non‐small cell lung cancer. Clin. Cancer Res. 17, 1553–1560. [DOI] [PubMed] [Google Scholar]
- 37. Basso AD, Kirschmeier P, Bishop WR (2006) Lipid post‐translational modifications. Farnesyl transferase inhibitors. J. Lipid Res. 47, 15–31. [DOI] [PubMed] [Google Scholar]