

Abstract
Objectives
Oxygen deficiency caused by occlusal trauma and smoking can be present in patients with periodontitis. However, biochemical events important in periodontal tissues during hypoxia remain unclear. The aim of this study was to investigate effects of hypoxia on apoptosis and autophagy of human periodontal ligament cells (PDLCs) in vitro.
Materials and methods
Human PDLCs were obtained and cultured in vitro. Cell viability, apoptosis, autophagy and gene and protein expression were measured in presence and absence of cobalt chloride (CoCl 2).
Results
CoCl 2 induced cytotoxicity of human PDLCs in a concentration‐dependent manner dependent on macromolecular synthesis, and resulted in apoptosis and mitochondrial dysfunction. CoCl 2 also induced redistribution of autophagy marker LC3, increased ratio of LC3‐IIto LC3‐Iand function of lysosomes. Furthermore, CoCl 2 promoted expression of HIF‐1α following upregulation of expressions of Bnip3. Significant increases in expression of IL‐1β and MMP‐8 were also observed. All these results were reversed by pre‐treatment with antioxidant N‐acetylcysteine.
Conclusions
Our data showed that CoCl 2 could induce cytotoxicity through mitochondria‐ apoptotic and autophagic pathways involved in HIF‐1α. CoCl 2‐treated PDLCs may serve as an in vitro model for studies of molecular mechanisms in periodontitis.
Introduction
Periodontitis is one of the most common oral infectious diseases in humans and is characterized by inflammation and destruction of attachment apparatus supporting local and teeth. It is the most prevalent cause of tooth loss and is difficult to treat 1, 2. A number of systemic and local risk factors have been associated with initiation and progression of periodontitis 3 and occlusal trauma has been considered to be a factor that accelerates inflammatory alveolar bone resorption, where periodontitis is present 4, 5. It is well known that physiological blood flow in the periodontal ligament is slightly lower than that of either pulpal or cerebral blood flow 6, 7, and blood flow is easily reduced by occlusal forces. Furthermore, histopathological study in an experimental occlusal trauma model in the dog (beagles) has demonstrated temporal obstruction of periodontal ligament (PDL) capillaries by mechanical compression, thrombus and subsequent reconstruction of blood vessels, resulting in marked resorption of alveolar bone 8. Thus, it is highly possible that hypoxia occurs in such traumatized PDL tissue.
Moreover, in cases of tooth transplantation or replantation, periodontal tissues containing osteogenic fibroblasts, which may differentiate into either cementoblasts or osteoblasts, decrease during severe ischaemia. If the periodontal ligament is exposed to a dry environment for more than 30 min after tooth extraction, its cells' ability to differentiate and to proliferate is lost 9, 10. In addition, one epidemiological study has reported that oxygen deficiency caused by smoking can be associated with periodontal tissue breakdown. Furthermore, growth of anaerobic bacterial biofilm may further lower oxygen tension in the vicinity of periodontal tissues 11. However, little is known of the behaviour of human periodontal ligament cells (PDLCs) under conditions of hypoxia in vitro.
Hypoxia‐inducible factor (HIF)‐1, a heterodimeric transcriptional factor, plays a central role in regulation of gene expression to maintain oxygen homeostasis 12. Under normoxic conditions, the HIF‐1α subunit is rapidly degraded through ubiquitinylation by pVHL and proteosomes. This is triggered through post‐translational hydroxylation at specific proline residues 13. However, under hypoxic conditions, HIF‐1α evades hydroxylation and is translocated to the nucleus where it heterodimerizes with HIF‐1α 14. Some metals are known to be hypoxic mimicking agents and these include cobalt chloride, nickel chloride and desferrioxamine 15. Recently, it has been reported that CoCl2 treatment induces HIF‐1α expression by binding to the PAS domain, resulting in blockage of HIF‐1α‐pVHL binding and thereby HIF‐1α stability 16.
There are at least two types of programmed cell death, apoptosis and autophagic cell death 17 being the most well recognised. Apoptosis is characterized by a series of biochemical events that lead to a variety of morphological changes, such as loss of cell membrane asymmetry accompanied by phosphatidylserine translocation from the inner cell membrane to the cell surface, cell shrinkage, nuclear fragmentation, chromatin condensation and chromosomal DNA fragmentation 18, 19. Autophagy on the other hand is a self‐degradative process that is important for balancing sources of energy at critical times. Autophagy also plays a housekeeping role in removing misfolded or aggregated proteins and clearing damaged organelles (such as mitochondria, endoplasmic reticulum and peroxisomes) as well as eliminating intracellular pathogens. Thus, autophagy is generally regarded as an overall survival mechanism 20.
Hypoxia and CoCl2 treatment have been reported to correlate with apoptotic and pro‐apoptotic factors 21, 22 and hypoxia also activates autophagy through effects that are dependent on target genes induced by HIF 23. The relationship between apoptosis and autophagy is now the subject of considerable research but effects of hypoxia on apoptosis and autophagy of periodontal ligament cells still largely remain unknown.
As an important component of the periodontal attachment apparatus, human PDLCs are significant in progress of periodontitis. In our previous study, we found that human PDL cell viability decreased in a dose‐dependent manner after treatment with CoCl2 for 48 h 24. Bearing this in mind, expression of HIF‐1α and its correlation with apoptosis and autophagy in human PDLCs might be of great importance in development of periodontitis. In the present study, we have examined effects of hypoxia induced by CoCl2 on human PDLCs and present evidence that hypoxia induces apoptosis and autophagic cell death.
Materials and methods
Human PDLC culture and CoCl 2‐mimic hypoxic treatment
Human PDLCs were prepared from explants of periodontal ligaments of teeth extracted for orthodontic treatment, from healthy humans with their informed consent. PDLCs were isolated and cultured according to the method of Sun et al. 2 with minor modification. Periodontal ligament fragments were removed from the mid‐third of the root and cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 20% foetal bovine serum (Gibco Biocult, Paisley, UK). Culture medium was changed every 7 days, for 10–20 days until confluent cell monolayers were formed. After four or five subcultures, homogeneous, slim, spindle‐shaped cells were obtained.
For hypoxia treatment, PDLCs were seeded on plates by incubation with various concentrations of CoCl2. In a proportion of experiments, PDLCs were pre‐treated with N‐acetylcysteine for 2 h prior to CoCl2 exposure for 48 h.
Cell viability assay
Cell viability assays were performed using Cell Counting Kit‐8 (CCK‐8) (WST‐8; Dojindo, Kumamoto, Japan) according to the manufacturer's instructions. PDLCs were plated into 96‐well plates. After treatment, CCK‐8 was added to culture media at 37 °C for 3 h. Absorbance was measured at 450 nm using an auto‐microplate reader (Bio‐Rad, Hercules, CA , USA). Cell viability was expressed as percentage of viable cells relative to untreated cells, using absorbance at 450 nm. All experiments were performed in five wells in three separate experiments.
Measurement of apoptosis and mitochondrial membrane potential
Percentage of cells actively undergoing apoptosis was determined by flow cytometry using the Annexin V‐FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) according to manufacturer's instructions. Briefly, after incubation with CoCl2 for 48 h, PDLCs were harvested and resuspended in binding buffer. Cells were mixed with annexin VFITC and PI. After incubating for 15 min in the dark, analysis was performed by flow cytometry.
High‐content screening was used to examine mitochondrial membrane potential (MMP) of the PDLCs. MitoTracker/Hoechst solution (Cellomics Inc, Pittsburgh, PA, USA) was added to wells of the 96‐well plates, then, fixation solution was added to each well. Wells were aspirated, and each plate was washed once in wash buffer‐M. Then plates were sealed and run on the ArrayScan HCS System. Intensity of red fluorescence represented MMP of the cells.
Detection of autophagy
PDLCs were plated in 12‐well plates and treated with CoCl2 for 48 h. After exposure, cells were fixed in 4% paraformaldehyde and permeabilized with 0.2% (v/v)Triton X‐100, for 15 min. After blocking with 2% (w/v) bovine serum albumin, fixed cells were incubated overnight at 4 °C with anti‐LC3 antibody (dilution 1:100, CST). Cultures were then washed and incubated for 2 h at room temperature in fluorescence‐conjugated secondary antibody (Alexa Fluor 555‐donkey anti‐rabbit IgG, 1:200; Invitrogen Life Technologies, Carlsbad, CA, USA). Prevalence of autophagic cells was determined by LC3 antibody location, viewed by confocal microscopy (LSM‐510; Carl Zeiss AG, Oberkochen, Germany) at excitation wavelength of 488 nm, and were photographed.
Induction of autophagy was assessed by detecting expression of Beclin1 or by increased ratio between LC‐II and LC3‐I, an indicator of autophagy induction, by immunoblotting.
Quantitative reverse transcription polymerase chain reaction
PDLCs were detached and homogenized using TRIZOL (Invitrogen, Carlsbad, CA, USA) and total RNA extracted. Real‐time PCR was performed with ABI7500 instrumentation (Applied Biosystems, Foster City, CA, USA) in a total volume of 50 μl per glass capillary. Gene‐specific primers used were: forward GTGCCACATCATCACCATA and reverse CAAAGCGACAGATAACACG for HIF‐1α; forward TCCTTCCATCTCTGCTGCTCTC and reverse AAGGTGCTGGTGGAGGTTGTC for Bnip3; forward TTGAAGCTGATGGCCCTAAA and reverse ACAGGTGCATCGTGCACATA for interleukin‐1β (IL‐1β); forward GCATTCAGGCCATCTATGGAC and reverse TCCACGGAGTGTGGTGATAGC for matrix metalloproteinase 8 (MMP‐8); forward CCTGTACGCCAACACAGTGC and reverse ATACTCCTGCTTGCTGATCC for β‐actin. Cycling protocols for all were identical. mRNA levels relative to that of housekeeping gene β‐actin, were calculated by ΔΔCt method. Data were averaged from three dishes in each group.
Western blot analysis
PDLCs were washed in PBS and lysed in ice‐cold SDS buffer; lysates were sonicated and protein concentration was determined using a BCA assay kit (Pierce, Rockford, IL, USA). Proteins were separated using SDS–PAGE and transferred into PVDF membranes (Millipore, Billerica, MA, USA) which were incubated overnight at 4 °C, with the following primary antibodies, anti‐: HIF‐1α (Novus Biologicals, Inc, Littleton, CO, USA), Bnip3 (Abcam, Cambridge, MA, USA), and β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After incubation in appropriate horseradish peroxidase conjugated secondary antibody (Zhongshan Biotechnology, Beijing, China), membranes were incubated with ECL reagents for chemiluminescence then exposed to X‐ray autoradiography film (Fuji, Tokyo, Japan).
Measurement of cytokines
Concentrations of cytokines IL‐1β and MMP‐8 in the cell culture supernatant were determined by enzyme‐linked immunosorbent assay (ELISA) from commercial kits for determination of human IL‐1β or MMP‐8. Cell culture supernatants, samples or standard solutions were added to 96‐well plates coated with murine monoclonal antibody for 2 h. After subsequent treatment for 1–2 h with alkaline phosphatase‐conjugated polyclonal antibody, treation with p‐nitrophenyl phosophate was carried out for 20 min. Trisodium phosphate solution was added to terminate the reaction. Absorbance of each well was measured at 450 nm, concentration was calculated from a standard curve and analysed statistically using Student's t‐testing.
Method and antiserum specificity controls were included in each assay. Plate‐ to‐plate variation controls were also employed when appropriate. IL‐1β and MMP‐8 concentrations were expressed as ng/ml.
Statistical analysis
Statistical analyses of the data were performed using analysis of variance (ANOVA) and Student–Newman–Keuls tests. Level of significance was set at P < 0.05.
Results
Cell viability assay
Our previous studies have shown that PDL cell viability decreases after treatment with CoCl2 from 100 to 800 μm for 48 h 24. EC50 of CoCl2 was found to be 169 μm (Fig. 1a). CoCl2 at concentrations of 200 and 400 μm, for 48 h, which markedly reduced cell viability compared to controls, were used to induce PDLC injury in the subsequent experiments.
Figure 1.
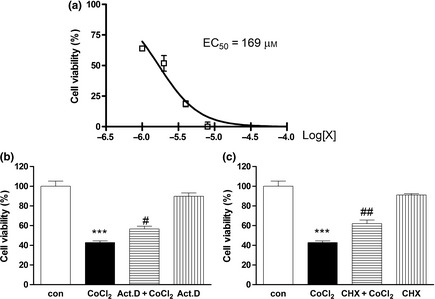
Effects of hypoxia induced by CoCl 2 on human PDL cell viability. PDLCs were cultured in complete media, with various concentration of CoCl 2, for 48 h. Cell viability was determined using the CCK‐8 kit. Half maximal inhibitory concentration (EC50) of hypoxia induced by CoCl 2, on cell viability of human PDLCs (X, concentration of CoCl 2) (a). Viability of control cells without CoCl 2 treatment defined as 100% versus Control group. Protective effects of actinomycin D (b) and cycloheximide (c) against hypoxia‐induced cytotoxicity (Act. D, actinomycin D; CHX, cycloheximide). Data presented as mean ± SEM. ***P < 0.001 versus control, #P < 0.05 versus CoCl 2 group, ##P versus CoCl 2 group.
To determine transcription dependence of CoCl2‐induced cell death, inhibitors of macromolecular synthesis were applied. After pre‐treatment with actinomycin D or cycloheximide for 2 h, cultures were exposed to 400 μm CoCl2 for 48 h. Both RNA synthesis inhibitor actinomycin D and protein synthesis inhibitor cycloheximide, inhibited CoCl2‐induced cytotoxicity. Thus, it would seem that mRNA and protein syntheses are indeed required for induction of cytotoxicity by CoCl2 (Fig. 1b,c).
Apoptosis and mitochondrial membrane potential assay
Flow cytometric analysis was used to present levels of apoptosis induced by CoCl2. Levels of apoptosis were 3.805 ± 0.325%, 6.74 ± 0.12%, 11.88 ± 0.95%, of control cells and 200, 400 μm CoCl2‐treated cells for 48 h, respectively. When pre‐treated with NAC, apoptosis was 8.47 ± 0.27% (Fig. 2a,b). These results demonstrate that CoCl2 increased levels of apoptosis in concentration and time‐dependent manner, which was abolished by treatment with an antioxidant.
Figure 2.
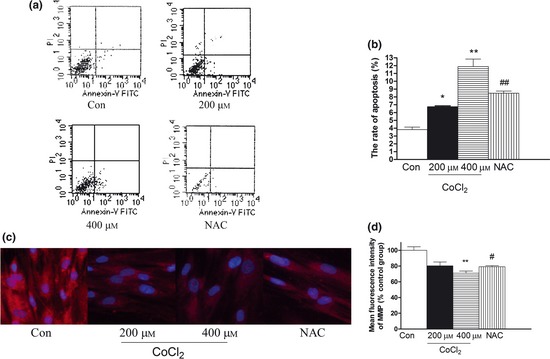
Effect of CoCl 2 ‐induced hypoxia on apoptosis and MMP (mitochondrial membrane potential) in human PDLC s. Apoptosis was detected by flow cytometry using Annexin V‐FITC Apoptosis Detection Kit. (a) Photomicrograph representative of three different sets of experiments. After pre‐treatment with NAC for 2 h, cultures were exposed to 400 μm CoCl 2 for 48 h. CoCl 2 increased level of apoptosis in a concentration‐and time‐dependent manner. (b) Data represent mean ± SEM of three experiments. ***P < 0.001 versus control, ## P < 0.05 versus CoCl 2 group. (c) By application of MitoTracker Red, red fluorescence occurred surrounding mitochondrial membranes; using ArrayScan HCS System (×20). (d) Data represent mean ± SEM of four individual variations. **P < 0.01 versus control; # P < 0.05 versus 400 μm CoCl 2 group.
To confirm the presence of apoptosis, we then measured loss of the mitochondrial membrane potential (Δψm), which is a common early event in apoptotic processes induced by a variety of stimuli 25. Integrity of Δψm during apoptosis caused by CoCl2 was monitored using MitoTracker red staining through the ArrayScan HCS System. Red fluorescence selectively accumulated in mitochondria and subsequently aggregated as a function of mitochondrial potential. Compared to control cells, CoCl2 treatment substantially attenuated fluorescence of mitochondrial membranes and reduced membrane potential. After NAC pre‐treatment, mitochondrial membrane potential recovered (Fig. 2c,d). These results indicate that CoCl2 had induced disruption of Δψm and caused mitochondrial dysfunction, by the mitochondrial apoptosis pathway.
Autophagy assay
To investigate the role of CoCl2‐induced hypoxia in autophagy, we measured effects of CoCl2 on the following autophagy markers: autophagic vesicles (AVs)/autophagosomal compartments, amphisomes and autolysosomes 26. Lysosomal mass/pH dye is a weak base that accumulates in lysosomes (acidic organelles) and determines changes to lysosomal physiology. We used the ArrayScan HCS System to detect locations of lysosomal mass/pH dye. Significant fluorescence intensity enhancement indicated increase in vesicular acidification after CoCl2 treatment. When pre‐treated with NAC, fluorescence intensity was lower (Fig. 3a,b). These results indicate that CoCl2 caused lysosome dysfunction in the cells.
Figure 3.
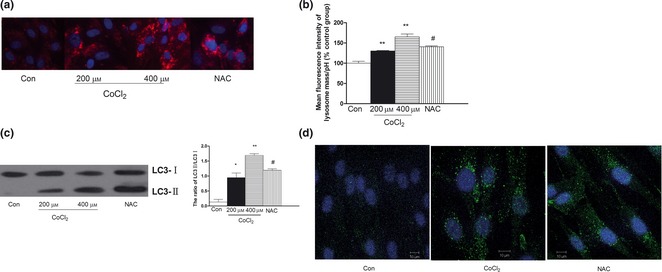
Effect of CoCl 2 ‐induced hypoxia on lysosome function and expression of LC 3 in human PDLC s. After pre‐treatment with NAC for 2 h, cultures were exposed to 400 μm CoCl 2 for 48 h. (a) Lysosomal mass/pH dye was used to determine the changes in lysosomal physiology using the ArrayScan HCS System (×20). (b) Data represent mean ± SEM of four individual procedures. Significant fluorescence intensity increase indicated increase in vesicular acidification after CoCl 2 treatment. (c) Detection of LC3 by western blotting and densitometric quantification ratio of LC3 normalized to β‐actin expression. There is increase in ratio of LC3‐II to LC3‐I. (d) Detection of LC3 was measured by confocal microscopy. Cells were stained with DAPI (blue) and induction of autophagy was visualized by LC3‐punctuated cells (green) under confocal microscopy. Scale bar, 5 μm. Con, control. *P < 0.05 versus control, **P < 0.01 versus control, # P < 0.05 versus CoCl 2 group.
Synthesis and processing of LC3 (also called ATG8) increase during autophagy, making it a key readout of autophagy levels 27. Induction of autophagy was visualized as LC3‐punctuated cells (green), by confocal microscopy. When PDLCs were treated with CoCl2, protein redistributed into distinct puncta, a characteristic of cells undergoing autophagy. In addition, to obtain additional confirmation of CoCl2‐induced autophagic cell death, we examined conversion of LC3‐I to LC3‐II using western blot analysis. Accumulation of LC3‐II, which is associated with autophagosome membranes, can occur due to increased autophagosome formation. As shown in Fig. 6, following CoCl2 treatment, there was increase in ratio of LC3‐II to LC3‐I and reduction in this ratio after NAC pre‐treatment (Fig. 3c,d).
Figure 6.
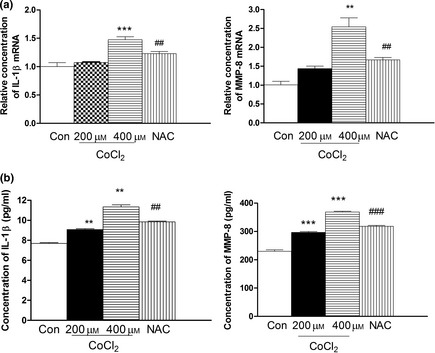
Effect of CoCl 2 ‐induced hypoxia on expression of IL ‐1β and MMP 8 in human PDLC s. After pre‐treatment with NAC for 2 h, cultures were exposed to 400 μm CoCl 2 for 48 h. (a) Expressions of IL‐1β mRNA and MMP8 mRNA were examined by real‐time PCR in cells treated with 200 and 400 μm CoCl 2 for 48 h. (b) Effect of hypoxia on production of IL‐1β and MMP8 was evaluated by ELISA. Significantly higher concentrations of IL‐1β and MMP‐8 were detected in the hypoxia group than in controls at 48 h. ** P < 0.05 versus control, ***P < 0.01 versus control, ## P < 0.01 versus 400 μm CoCl 2 group, ### P < 0.01 versus 400 μm CoCl 2 group.
To confirm induction of autophagy by molecular means, we measured expression of Beclin 1, an autophagy‐related protein 28. Beclin 1 expression increased significantly in 200 and 400 μm CoCl2‐treated cells (Fig. 4) and could be attenuated by NAC. These data suggest that hypoxia had induced autophagy by upregulating Beclin1 expression.
Figure 4.
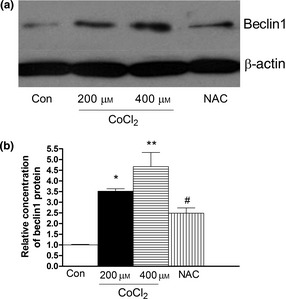
Effect of CoCl 2 ‐induced hypoxia on expression of Beclin in human PDLC s. After pre‐treatment with NAC for 2 h, cultures were exposed to 400 μm CoCl 2 for 48 h. (a) Expression of Beclin was examined by western blot analysis. At 48 h, treatment with 400 μm CoCl 2 increased expression of Beclin. (b) Beclin expression increased significantly in CoCl 2 treated cells at 48 h. *P < 0.05 versus control, **P < 0.01 versus control, # P < 0.05 versus CoCl 2 group.
HIF‐1α and BNIP3 expression
HIF‐1α, a key transcription factor, plays pivotal roles in hypoxia. Expressions of HIF‐1α and its related gene were observed using RT‐PCR and western blot analysis. As shown in Fig. 5, CoCl2 did not promote levels of HIF‐1α mRNA expression, but did increased levels of HIF‐1α protein expression. However, expression of Bnip3 mRNA, and protein levels were significantly higher after CoCl2 treatment compared to controls (P < 0.01). Furthermore, NAC attenuated upregulation of HIF‐1α and downstream BNIP3 gene after CoCl2, which prevented apoptosis.
Figure 5.
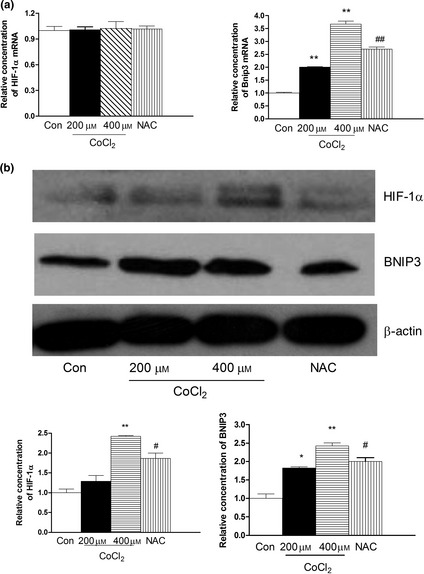
Effect of CoCl 2 ‐induced hypoxia on expression of HIF ‐1α, Bnip3 in human PDLC s. After pre‐treatment with NAC for 2 h, cultures were exposed to 400 μm CoCl 2 for 48 h. (a) Expressions of HIF‐1α mRNA and BNIP3 mRNA were examined by real‐time PCR in cells incubated with 200 and 400 μm CoCl 2 for 48 h. CoCl 2 did not promote level of HIF‐1α mRNA expression. However, CoCl 2 increased level of Bnip3 mRNA expression. (b) Expression of HIF‐1α and BNIP3 protein were detected by western blotting. β‐actin was used as a control. Levels of HIF‐1α and BNIP3 protein were significantly increased by CoCl 2 at 48 h. *P < 0.05 versus control, **P < 0.01 versus control, # P < 0.05 versus CoCl 2 group.
IL‐1β and MMP‐8 expression
We evaluated effects of hypoxia on production of IL‐1β and MMP8 cytokines by PDLCs, by quantification of their extracellular concentrations using QPCR and ELISA. Significantly higher concentrations of IL‐1β and MMP‐8 were detected in hypoxia groups than in controls, at 48 h (Fig. 6). Expression of IL‐1β and MMP‐8 recovered with NAC treatment.
Discussion
To our knowledge, this is the first study to report effects of CoCl2‐induced hypoxia, on apoptosis and autophagic cell death in primary human PDLCs. The periodontal ligament is a dense soft connective tissue, situated between tooth‐root cementum and its adjacent alveolar bone, which provides physiological tooth mobility under functional loading 29. Compression of the PDL induces changes in blood vessel morphology characterized by ischaemia and reduced blood flow. These changes cause local hypoxia, which may have an effect on PDLCs and cause release of chemical mediators, resulting in adaptive responses to maintain homoeostasis of periodontal tissue. Hypoxia‐induced cell death is a major concern in various clinical settings such as hypoxic/ischaemic disease, organ transplantation, and others 21, 30, 31, 32. To elucidate the mechanisms by which human PDLCs respond to hypoxia, we used treating them with CoCl2 as a model, and demonstrated that CoCl2 triggered both apoptosis and autophagy in PDLCs involving the HIF‐1α pathway.
Goldberg et al. 33 have reported that 1% O2/5% CO2/94% N2, or in the presence of CoCl2, can mimic hypoxic conditions experimentally. CoCl2 has been widely used as a hypoxia mimic both in vitro and in vivo studies 34, 35. CoCl2 can mimic hypoxic responses of cultured cells 36, including transcriptional changes of some genes, such as that of HIF‐1α 37, 38, which are potent transcription factors activating target genes that initiate cell death or cause population growth arrest, in response to stress or DNA damage. In this study, viability of human PDLCs after CoCl2 treatment reduced with concentrations of CoCl2, consistent with amounts of CoCl2‐induced expression of HIF‐1α protein. The results strongly indicate that CoCl2‐induced hypoxia may result in cytotoxicity of human PDLCs.
When cells are exposed to chronic or extreme hypoxia, initiation by HIF‐1α results in apoptosis. As a transcription factor, HIF‐1α can participate in cell death by activating pro‐death genes, including Bnip3 39. The gene for Bnip3 (Bcl‐2/adenovirus E1B 19‐kDa interacting protein 3) is a primary pro‐death gene of hypoxia. Bnip3 expression can be upregulated under hypoxia in cell lines derived from carcinomas, fibroblasts and macrophages 40. We have also found that expression of Bnip3 in human PDLCs after CoCl2 treatment was significantly elevated both in mRNA and protein levels.
Previous studies have shown that overexpression of Bnip3 results in its integration into the outer mitochondrial membrane with specific orientation – its N‐terminus in the cytoplasm and C‐terminus in the membrane 41. After integration into the mitochondrial membrane, Bnip3 can interact with components of MPT pores to induce their rapid opening and dissipation of ΔΨm, followed by apoptotic cell death 42. We have also proven that CoCl2 promoted activation of HIF‐1α, upregulated expression of HIF‐1α and Bnip3, and ultimately resulted in apoptosis of the cells; this could be attenuated by use of NAC, an inhibitor of HIF‐1 α. This indicated that CoCl2‐induced hypoxic insults can be mediated in the HIF‐1α pathway.
It has been illustrated that autophagy resulting the total devastation of the cell is one of several types of programmed cell death (PCD). Autophagy plays key physiological cellular functions and also has pathophysiological roles. Too much autophagy or too little autophagy is linked to pathogen infection, neurodegeneration, muscular disorders, liver disease and cancer 43. Accumulating evidence has suggested that HIF‐1 plays an important role in induction of autophagy via enhanced expression of its target gene Bnip3 44, 45 or by inhibition of the mammalian target of rapamycin (mTOR). In our study, we found CoCl2 caused lysosome dysfunction in human PDLCs. Following CoCl2 treatment, there was increase in ratio of LC3‐II to LC3‐I, an indicator of autophagy. Expression of Beclin 1, an autophagy‐related protein, also is upregulated in human PDLCs treated with CoCl2. When pre‐treated with NAC, CoCl2‐induced autophagy was attenuated. Here, we report for the first time that CoCl2 can induce autophagy of human PDLCs through enhanced protein levels of HIF‐1α.
Hypoxia and inflammation often occur simultaneously in numerous diseases, including in ischaemic diseases, chronic inflammatory diseases and growing tumours 46, 47. Recent studies have demonstrated that hypoxia can activate macrophages, dendritic cells and monocytes, which are involved in the inflammatory response, by altering gene expression and cytokine secretion 48, 49. HIF‐1α is considered important in inflammatory processes as it also regulates phagocytosis of mononuclear cells 50. Hypoxia stimulates expression of several cytokines, from various cells, such as human PDLCs, gingival fibroblasts and synovial fibroblasts 5, 51. Pro‐inflammatory cytokines including IL‐β and MMP‐8 are pivotal in pathogensis of periodontal disease. One important effect of IL‐1β is to stimulate fibroblasts to produce destructive metalloproteinases, such as collagenase and stromelysin, which degrade components of the extracellular matrix in periodontitis. IL‐1β has been confirmed to be a potent inducer of bone resorption and of connective tissue degradation via induction of MMPs 52, 53. In addition, pathologically excessive MMP plays a significant role in periodontal destruction 54. MMP‐8 (collagenase 2) is a collagenolytic enzyme that can initiate digestion of type I collagen, the most dominant interstitial collagen type, in periodontal tissues. Collagen degradation is regarded to be one of the key factors in uncontrolled tissue destruction in periodontitis 55. Our data show that expression and release of IL‐1β and MMP‐8 in CoCl2 treated groups were statistically higher than in control groups. However, NAC can abolish these changes. The results also suggest that hypoxia and HIF‐1α may play a role in periodontal diseases.
In conclusion, exposure of human PDLCs to CoCl2‐induced hypoxia results in hypoxic cell death with apoptosis and autophagy, due to activation of the HIF‐1α pathway. Further investigation to elucidate mechanisms of hypoxia‐induced apoptosis and autophagic cell death will be useful in developing methods for prevention of periodontal diseases.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (Project No. 30801292), Shanghai Leading Academic Discipline Project (Project No. S30206), Shanghai Municipal Science and Technology Commission (Project No. 11ZR1419300), Shanghai Education Commission (Project No. 12YZ037), and the National Defense Science and Technology Development Fund of Shanghai JiaoTong University (Project No. 11GFF50).
References
- 1. Oliver RC, Brown LJ, Loe H (1998) Periodontal diseases in the United States population. J. Periodontol. 69, 9–13. [DOI] [PubMed] [Google Scholar]
- 2. Sun Y, Shu R, Zhang MZ, Wu AP (2008) Toll‐like receptor 4 signaling plays a role in triggering periodontal infection. FEMS Immunol. Med. Microbiol. 52, 362–369. [DOI] [PubMed] [Google Scholar]
- 3. Williams RC (1990) Medical progress, periodontal disease. N. Engl. J. Med. 322, 373–382. [DOI] [PubMed] [Google Scholar]
- 4. Polson AM, Zander AH (1983) Effect of periodontal trauma upon intrabony pockets. J. Periodontol. 54, 586–591. [DOI] [PubMed] [Google Scholar]
- 5. Motohira H, Hayashi J, Tatsumi J, Tajima M, Sakagami H, Shin K (2007) Hypoxia and reoxygenation augment bone‐resorbing factor production from human periodontal ligament cells. J. Periodontol. 78, 1803–1809. [DOI] [PubMed] [Google Scholar]
- 6. Ohuchi M, Fujimura A (2004) Changes of blood flow in pressure side of periodontal ligament tooth movement. Dent. J. Iwate Med. University 29, 15–23. [Google Scholar]
- 7. Kim S (1985) Regulation of pulpal blood flow. J. Dent. Res. 64, 590–596. [DOI] [PubMed] [Google Scholar]
- 8. Matsuo M, Tsutomi K, Terada H, Hori T, Takahashi K (1991) The changes in periodontal ligament in the case of thrombus caused by the malocclusal forces. Microcirc. Annu. 7, 129–130. [Google Scholar]
- 9. Amemiya H, Matsuzaka K, Kokubu E, Ohta S, Inoue T (2008) Cellular responses of rat periodontal ligament cells under hypoxia and re‐oxygenation conditions in vitro. J. Periodont. Res. 43, 322–327. [DOI] [PubMed] [Google Scholar]
- 10. Takizawa H (1991) An experimental study of tooth preservation for tooth transplantation and replantation especially, biological activity of periodontal ligament. J. Jpn. Prosthodont. Soc. 35, 723–737. [Google Scholar]
- 11. Mettraux GR, Gusberti FA, Graf H (1984) Oxygen tension (pO2) in untreated human periodontal pockets. J. Periodontol. 55, 516–521. [DOI] [PubMed] [Google Scholar]
- 12. Semenza GL (2001) HIF‐1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 13, 167–171. [DOI] [PubMed] [Google Scholar]
- 13. Bruick RK, McKnight SL (2001) A conserved family of prolyl‐4‐hydroxylation that modify HIF. Science 294, 1337–1340. [DOI] [PubMed] [Google Scholar]
- 14. Ardyanto TD, Osaki M, Tokuyasu N, Nagahama Y, Ito H (2006) CoCl2‐induced HIF‐1alpha expression correlates with proliferation and apoptosis in MKN‐1 cells: a possible role for the PI3K/Akt pathway. Int. J. Oncol. 29, 549–555. [PubMed] [Google Scholar]
- 15. Goldberg MA, Dunning SP, Bunn HF (1998) Regulation of erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science 242, 1412–1414. [DOI] [PubMed] [Google Scholar]
- 16. Kanaya K, Kamitani T (2003) pVHL‐independent ubiquitination of HIF1α and its stabilization by cobalt ion. Biochem. Biophys. Res. Commun. 306, 750–755. [DOI] [PubMed] [Google Scholar]
- 17. Lockshin RA, Zakeri Z (2004) Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 36, 2405–2419. [DOI] [PubMed] [Google Scholar]
- 18. Wyllie AH, Kerr JF, Currie AR (1980) Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251–306. [DOI] [PubMed] [Google Scholar]
- 19. Earnshaw WC (1995) Nuclear changes in apoptosis. Curr. Opin. Cell Biol. 7, 337–343. [DOI] [PubMed] [Google Scholar]
- 20. Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volm M, Koomagi R (2000) Hypoxia‐inducible factor (HIF‐1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer Res. 20(3A), 1527–1533. [PubMed] [Google Scholar]
- 22. Piret J, Lecocl C, Toffoli S, Ninane N, Raes M, Michiels C (2004) Hypoxia and CoCl2 protect HepG2 cell against serum deprivation and t‐BHP‐induced apoptosis: a possible anti‐apoptotic role for HIF‐1. Exp. Cell Res. 295, 340–349. [DOI] [PubMed] [Google Scholar]
- 23. Semenza GL (2010) HIF‐1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song ZC, Shu R, Li XT, Hu JC, Zhang XL (2009) Influence of cobalt chloride‐induced hypoxia on the proliferation and apoptosis of periodontal ligament cells. Shanghai Kou Qiang Yi Xue 18, 489–492. [PubMed] [Google Scholar]
- 25. Smaili SS, Hsu YT, Carvalho AC, Rosenstock TR, Sharpe JC, Youle RJ (2003) Mitochondria, calcium and pro‐apoptotic proteins as mediators in cell death signaling. Braz. J. Med. Biol. Res. 36, 183–190. [DOI] [PubMed] [Google Scholar]
- 26. Zhaorigetu S, Yang Z, Toma I, McCarffrey TA, Hu CA (2011) Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks beclin 1‐dependent autophagy in atherosclerotic cells. J. Biol. Chem. 286, 27389–27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barth S, Glick D, Macleod KF (2010) Autophagy: assays and artifacts. J. Pathol. 221, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sinha S, Levine B (2008) The autophagy effector Beclin 1: a novel BH3‐only protein. Oncogene 27(Suppl. 1), S137–S148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuda N, Yokoyama K, Takeshita S, Watanabe M (1998) Role of epidermal growth factor and its receptor in mechanical stress‐induced differentiation of human periodontal ligament cells in vitro. Arch. Oral Biol. 43, 987–997. [DOI] [PubMed] [Google Scholar]
- 30. Rayner BS, Duong TT, Myers SJ, Witting PK (2006) Protective effect of a synthetic anti‐oxidant on neuronal cell apoptosis resulting from experimental hypoxia re‐oxygenation injury. J. Neurochem. 97, 211–221. [DOI] [PubMed] [Google Scholar]
- 31. Bhogal RH, Weston CJ, Curbishley SM, Bhatt AN, Adams DH, Afford SC (2011) Variable responses of small and large human hepatocytes to hypoxia and hypoxia/reoxygenation (H‐R). FEBS Lett. 585, 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Härtel FV, Holl M, Arshad M, Aslam M, Gündüz D, Weyand M et al (2010) Transient hypoxia induces ERK‐dependent anti‐apoptotic cell survival in endothelial cells. Am. J. Physiol. Cell Physiol. 298, C1501–C1509. [DOI] [PubMed] [Google Scholar]
- 33. Goldberg MA, Dunning SP, Bunn HF (1988) Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science 242, 1412–1415. [DOI] [PubMed] [Google Scholar]
- 34. Merelli A, Caltana L, Girimonti P, Ramos AJ, Lazarowski A, Brusco A (2011) Recovery of motor spontaneous activity after intranasal delivery of human recombinant erythropoietin in a focal brain hypoxia model induced by CoCl2 in rats. Neurotox Res. 20, 182–192. [DOI] [PubMed] [Google Scholar]
- 35. Badr GA, Zhang JZ, Tang J, Kern TS, Ismail‐Beigi F (1999) Glut1 and glut3 expression, but not capillary density, is increased by cobalt chloride in rat cerebrum and retina. Brain Res. Mol. Brain Res. 64, 24–33. [DOI] [PubMed] [Google Scholar]
- 36. Ji Z, Yang G, Shahzidi S, Tkacz‐Stachowska K, Suo Z, Nesland JM et al (2006) Induction of hypoxia‐inducible factor‐1alpha overexpression by cobalt chloride enhances cellular resistance to photodynamic therapy. Cancer Lett. 244, 182–189. [DOI] [PubMed] [Google Scholar]
- 37. Zou W, Yan M, Xu W, Huo H, Sun L, Zheng Z et al (2001) Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP‐1 activation. J. Neurosci. Res. 64, 646–653. [DOI] [PubMed] [Google Scholar]
- 38. Zhu X, Zhou W, Cui Y, Zhu L, Li J, Feng X et al (2010) Pilocarpine protects cobalt chloride‐induced apoptosis of RGC‐5 cells: involvement of muscarinic receptors and HIF‐1 alpha pathway. Cell. Mol. Neurobiol. 30, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL (2001) HIF‐1‐dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673. [PubMed] [Google Scholar]
- 40. Vengellur A, LaPres JJ (2004) The role of hypoxia inducible factor 1alpha in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol. Sci. 82, 638–646. [DOI] [PubMed] [Google Scholar]
- 41. Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S et al (2000) BNIP3 and genetic control of necrosis‐like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 20, 5454–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang L, Wei L, Jiang D, Wang J, Cong X, Fei R (2007) SARS‐CoV nucleocapsid protein induced apoptosis of COS‐1 mediated by the mitochondrial pathway. Artif. Cells Blood Substit. Immobil. Biotechnol. 35, 237–253. [DOI] [PubMed] [Google Scholar]
- 43. Mazure NM, Pouysségur J (2010) Hypoxia‐induced autophagy: cell death or cell survival? Curr. Opin. Cell Biol. 22, 177–180. [DOI] [PubMed] [Google Scholar]
- 44. Zhang H, Bosch‐Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB et al (2008) Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Wu Y, Li X, Xie W, Jankovic J, Le W, Pan T (2010) Neuroprotection of deferoxamine on rotenone‐induced injury via accumulation of HIF‐1 alpha and induction of autophagy in SH‐SY5Y cells. Neurochem. Int. 57, 198–205. [DOI] [PubMed] [Google Scholar]
- 46. Karhausen J, Haase VH, Colgan SP (2005) Inflammatory hypoxia: role of hypoxia‐inducible factor. Cell Cycle 4, 256–258. [PubMed] [Google Scholar]
- 47. Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote‐Garcia JC et al (2009) Hypoxia‐inducible factor‐dependent induction of netrin‐1 dampens inflammation caused by hypoxia. Nat. Immunol. 10, 195–202. [DOI] [PubMed] [Google Scholar]
- 48. Bosco MC, Puppo M, Blengio F, Fraone T, Cappello P, Giovarelli M et al (2008) Monocytes and dendritic cells in a hypoxic environment: spotlights on chemotaxis and migration. Immunobiology 213, 733–749. [DOI] [PubMed] [Google Scholar]
- 49. Safronova O, Pluemsampant S, Nakahama K, Morita I (2009) Regulation of chemokine gene expression by hypoxia via cooperative activation of NFkappaB and histone deacetylase. Int. J. Biochem. Cell Biol. 41, 2270–2280. [DOI] [PubMed] [Google Scholar]
- 50. Strieter RM (2003) Mastering innate immunity. Nat. Med. 9, 512–513. [DOI] [PubMed] [Google Scholar]
- 51. Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E (2000) Interleukin 1 induces hypoxia‐inducible factor 1 in human gingival and synovial fibroblasts. Biochem. J. 350(pt1), 307–312. [PMC free article] [PubMed] [Google Scholar]
- 52. Birkedal‐Hansen H (1993) Role of matrix metalloproteinases in human periodontal diseases. J. Periodontol. 64(5 Suppl.), 474–484. [DOI] [PubMed] [Google Scholar]
- 53. Preshaw PM, Taylor JJ (2011) How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? J. Clin. Periodontol. 38(Suppl. 11), 60–84. [DOI] [PubMed] [Google Scholar]
- 54. Sorsa T, Tjäderhane L, Konttinen YT, Lauhio A, Salo T, Lee HM et al (2006) Matrix metalloproteinases: contribution to pathogenesis, diagnosis and treatment of periodontal inflammation. Ann. Med. 38, 306–321. [DOI] [PubMed] [Google Scholar]
- 55. Sorsa T, Tjäderhane L, Salo T (2004) Matrix metalloproteinases (MMPs) in oral diseases. Oral Dis. 10, 311–318. [DOI] [PubMed] [Google Scholar]