

Abstract
Objectives
MicroRNAs (miRNAs) are small non‐coding RNAs that post‐transcriptionally regulate gene expression and mediate diverse physiological processes. In this study, we investigated functions of miRNA miR‐34c‐3p in non‐small cell lung cancer (NSCLC).
Materials and methods
miR‐34c‐3p expression was evaluated by qPCR. Cell viability was examined by MTT and proliferation by cell cycle analysis. Cell migration and invasion were tested using Transwells with/without Matrigel coating. Western blot analysis was performed for eIF4E, c‐Myc, Cyclin D1, survivin and Mcl‐1 protein expression.
Results
miR‐34c‐3p expression was significantly reduced in tissues and serum samples from NSCLC patients and in NSCLC cell lines A549, H460, H23, H157 and H1299. Overexpression of miR‐34c‐3p in A549 and H157 cells reduced cell proliferation, migration and invasion, whereas transfection with miR‐34c‐3p inhibitor (miR‐34c‐3p‐in) produced opposite effects. Target analysis using algorithms miRanda, TargetScan and DIANA identified eIF4E as a potential target of miR‐34c‐3p. Luciferase assay using the eIF4E 3′‐UTR reporter carrying a putative miR‐34c‐3p target sequence revealed eIF4E to be a specific target of miR‐34c‐3p. Overexpression of miR‐34c‐3p in NSCLS cell lines led to significant reduction in mRNA and protein levels of eIF4E, whereas inhibition of miR‐34c‐3p resulted in significant increase in eIf4e protein levels, confirming eIF4E to be a direct target of miR‐34c‐3p in NSCLS. Overexpression of eIF4E in A549 cells promoted cell proliferation, migration and invasion, which were partially reversed by miR‐34c‐3p.
Conclusion
miR‐34c‐3p directly targeted eIF4E and reduced miR‐34c‐3p expression in NSCLC, promoting cell cycle progression, proliferation, migration and invasion.
Abbreviations
- 4E‐BP1
4E binding protein 1
- Akt
protein kinase B
- EGFR
epidermal growth factor receptor
- eIF4E
eukaryotic translation initiation factor 4E
- MAP kinase
mitogen‐activated protein kinase
- miRNA
microRNA
- Mnk1
mitogen‐activated kinase interacting kinase
- mTOR
mammalian target of rapamycin
- NSCLC
non‐small cell lung cancer
- PI3K
phosphoinositide‐3‐kinase
- siRNA
small interfering RNA
- UTR
untranslated region
- VEGFR
vascular endothelial growth factor receptor
Introduction
MicroRNAs (miRNAs) are a class of small non‐coding RNAs (20–24 nucleotides) that post‐transcriptionally regulate gene expression and play key roles in diverse biological processes, including development, proliferation, differentiation and apoptosis1. miRNAs bind to complementary sequences at 3′‐untranslated regions (UTR) of target mRNAs resulting either in mRNA degradation or inhibition of translation 1, 2. To date, approximately 2000 miRNAs have been identified in the human genome and this number is rapidly increasing 3. More than 60% of the protein‐coding genes in humans are estimated to include an miRNA target‐binding site in their 3′‐UTR region. As they control gene expression, miRNAs have been extensively investigated in cancer research as therapeutic targets and as biomarkers 4, 5, 6, 7. miR‐34c‐3p is one of the mature miRNAs of miR‐34c. It can inhibit glioma cell proliferation and induce apoptosis 8. miR‐34c‐3p has been found to be down‐regulated in NSCLC, but its function in NSCLC tumourigenesis still remains unknown 9, 10.
Lung cancer is the most common type of malignancy, worldwide. In 2014, an estimated 160,000 deaths in the United States alone were due to lung cancer, accounting for 20% of all cancer‐related deaths 11. Approximately, 85% of lung cancers are classified as non‐small cell carcinomas (NSCLC). Mutations or defects in several cellular kinase‐signalling pathways (PI3K/mTOR/Akt, EGFR, LKB, c‐MET), and genetic and epigenetic alterations have been shown to be associated with development and progression of lung cancer 12. Thus, kinases have been major targets for therapeutic development in this area. However, existing therapeutics (including recently FDA‐approved EGFR inhibitor erlotinob and the VEGFR inhibitor bevacizumab) are not sufficiently effective. This is because of heterogeneity of lung cancer, which involves multiple mechanisms. Thus, improved understanding of the pathways contributing to its pathogenesis are necessary for development of effective therapeutics strategies.
Elevated expression of eukaryotic translation initiation factor 4E (eIF4E) has been found in several tumour types, including NSCLS 13, 14, 15. eIF4E recognizes and binds the 5′ cap structure of an mRNA and delivers it to the eIF4F complex to enable translation 16. It has been found that expression and activity of eIF4E are directly correlated with brief survival of patients with NSCLS 17. eIF4E activity is regulated transcriptionally by c‐myc 11, through phosphorylation by MAP kinase‐interacting kinase Mnk118, and by interaction with translational repressor 4E‐binding proteins (4E‐BP) 19. Overexpression of phospho‐eIF4E has been shown to promote cell proliferation and metastasis through the Akt pathway 20. Interfering with eIF4E expression using siRNA has been found to inhibit NSCLC cell population growth and invasion, and Mnk inhibitors have been found to block tumour extension in experimental models of lung cancer 13, 18.
Accumulating evidence supports the notion that initiation of translation is one of the major targets of miRNAs. miRNAs have been shown to block assembly of the eIF4G complex in humans and in drosophila21, 22. Although studies have found aberrant expression of several miRNAs, including miR‐221, miR‐222, miR‐21, miR‐205, miR125, miR143, miR145, miR96, miR34 and miR30b in NSCLC 23, 24, 25, 26, 27, the precise nature of miRNAs that may target and regulate function of eIF4E has not been well studied. Here, we show that miR‐34c‐3p directly targets eIF4E expression and represses NSCLC cell proliferation, migration and invasion.
Materials and methods
Tissue samples
This study was approved by the Ethics Committee of Harbin Medical University. All NSCLC tissue samples and paired normal lung tissues were collected via surgical resection from patients diagnosed between 2012 and 2013 at the Harbin Medical University Cancer Hospital Department of Chest Surgery. Immediately after collection, the tissue samples were snap‐frozen in liquid nitrogen and stored at −80 °C until use. Both tumour and normal samples were confirmed by pathological examination. Written informed consent was obtained from all the participants.
Cell lines and culture
Human lung cancer cell lines A549, H460, H157, H1299 and H23, and a normal diploid human lung fibroblast cell line (MRC5), were obtained from and maintained as recommended by the American Type Culture Collection (ATCC, Manassas, VA, USA). A549 and MRC‐5 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA, USA), and H460, H157, H1299 and H23 were grown in RPMI‐1640 (Life Technologies). In each case, medium was supplemented with 10% foetal bovine serum (Life Technologies), 100 U/ml penicillin and 100 U/ml streptomycin. All cells were maintained at 37 °C in a humidified incubator containing 5% CO2.
Quantitative real‐time PCR
Total RNA from tissue samples and cultured NSCLC cells was extracted using TRIzol (Life Technologies) and reverse transcribed using a cDNA Library Construction Kit (Takara, Dalian, China). TaqMan quantitative real‐time PCR (qPCR) measurement of miR‐34c‐3p was performed on an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using a microRNA assay kit (Applied Biosystems); U6 snRNA was used for normalization 28. Real‐time PCR detection of eIF4E was performed using SYBR Green Master Mix (Applied Biosystems). Primers for eIF4E were: forward, 5′‐TACTAAGAGCGGCTCCACCAC‐3′, reverse 5′‐TCGATTGCTTGACGCAGTCTCC‐3′; For GAPDH, the primers were: forward, 5′‐ TGCACCACCAACTGCTTAGC‐3′, reverse 5′‐GGCATGGACTGTGGTCATGAG‐3′.
Transfection
miR‐34c‐3p, miR‐NC (negative control), miR‐34c‐3p‐in (miR‐34c‐3p inhibitor) and miR‐in‐NC (negative control for mir‐34c‐3p‐in) were obtained from IBSBIO (Shanghai, China) and diluted to 10 μm concentration in nuclease‐free water. mRNAs were transfected alone or in combination, into NSCLC cells at final total concentration of 15 nm, using Lipofectamine® RNAiMAX Transfection Reagent (Life Technologies) and subsequent measurements performed 48 h later.
MTT assay
Cell viability was measured using 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetra‐zolium (MTT) assay. Transfected A549 and H157 cells were seeded in 96‐well plates at 3 × 103 cells per well. Cells were allowed to grow for 0 to 4 days. MTT (Promega, Madison, WI, USA) solution was then added to each well and cells were incubated at 37 °C for 4 h. Absorbance was measured at 550 nm using a scanning multi‐well spectrophotometer (Biotek, Winooski, VT, USA).
Cell cycle analysis
The NSCLC cells (1 × 106) were fixed in ice‐cold 70% ethanol and stored at 4 °C until analysed. They were washed twice in cold PBS and incubated in 50 μg/ml propidium iodide (PI) and 0.1 mg/ml RNase A in PBS at room temperature in dark for 1 h. The cell cycle was analysed using a flow cytometer (BD Biosciences, San Jose, CA, USA) and FLOWJO software (Treestar, Ashland, OR, USA).
Cell migration and invasion assays
In vitro cell migration and invasion assays were performed using Transwell chambers. Transfected cells (2 × 105) in 100 μl DMEM supplemented with 1% FBS were seeded in tops of chambers, 8 μm pore size Transwells (PengBo Biotech, Changsha, China). For invasion assays, cells were plated in Transwells precoated with Matrigel (BD Biosciences). Inserts of chambers were deposited on 24‐well plates containing 400 μl DMEM supplemented with 10% FBS in the lower chamber. After 12 h incubation for migration and 24 h for invasion, non‐migrating cells were removed from upper chambers. Migrated cells in lower chambers were stained with calcein AM (Life Technologies) or coomassie brilliant blue (Sigma Aldrich, St. Louis, MO, USA). Cells in six random fields were counted per chamber.
Cloning and transfection
Full‐length eIF4E 3′‐UTR was cloned into pMIR‐reporter luciferase vector (Life Technologies). Mutations in miR‐34c‐3p binding sequence of the 3′‐UTR of eIF4E were introduced using QuikChange II Site‐Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) and were verified by sequencing. The eIF4E fragment lacking 3′‐UTR was amplified from cDNA of A549 cells and subcloned into pcDNA3.1 (Life Technologies). Transfection of plasmid DNA was performed using Lipofectamine® 2000 (Life Technologies) according to the manufacturer's instructions.
Luciferase assay
Cells (5 × 104) were plated in 24‐well plates overnight. miRNA mimics or inhibitors were co‐transfected with wild‐type or mutant pMIR‐eIF4E 3′‐UTR and Renilla luciferase plasmid, into cells using Lipofectamine® 2000 reagent. Luciferase activities were measured with dual‐luciferase reporter assays (Promega) 48 h after transfection. Luciferase activity was normalized to Renilla activity.
Western blot analysis
Cells were lysed using ice‐cold RIPA buffer containing protease/phosphatase inhibitor cocktail (Boster, Wuhan, China) and protein concentration was measured using a BCA protein assay kit (Boster). Proteins in the lysate were separated on 10% SDS‐PAGE and transferred to nitrocellulose membranes (Bio‐Rad, Richmond, CA, USA) which were blocked with 5% non‐fat dry milk in Tris‐buffered saline containing 0.05% Tween‐20 for 1 h at room temperature, and then incubated overnight at 4 °C with primary antibody against eIF4E, c‐Myc, Cyclin D1, Survivin, Mcl‐1 or GAPDH (Santa Cruz Biotechnologies, Dallas, TX, USA). After washing, membranes were incubated with HRP‐conjugated secondary antibodies for 1 h and bound antibodies were visualized using enhanced chemiluminescence substrate (Boster).
Statistical analysis
Statistical analyses were performed using the SPSS 19.0 software package (SPSS, Chicago, IL, USA). One‐way ANOVA was used to investigate differences between groups. For all analyses, P‐value of less than 0.05 was considered statistically significant. All the experiments were performed in triplicate and data are presented as mean ± SD.
Results
Reduced miR‐34c‐3p expression in NSCLC tissues, patients' serum and NSCLC cell lines
To investigate expression of miR‐34c‐3p in NSCLC, we searched the GSE29248 microarray database 23. The results indicated that miR‐34c‐3p expression was significantly lower in NSCLC tissue and serum samples of NSCLS patients than those of paired adjacent normal lung tissues (P < 0.01, Fig. 1a) and serum samples from healthy controls (P < 0.05, Fig. 1b) respectively. To further confirm that miR‐34c‐3p expression was reduced in NSCLC, we analysed tissue samples collected from 15 patients with NSCLC, and found that miR‐34c‐3p was significantly lower in 14 of the 15 samples (Fig. 1c). We also analysed miR‐34c‐3p expression in five NSCLC cell lines (A549, H460, H23, H157 and H1299) and in normal lung fibroblast cell line MRC5. As shown in Fig. 1d, miR‐34c‐3p expression was lower in all five NSCLC cell lines than in MRC5. These results suggest that miR‐34c‐3p expression was down‐regulated in NSCLC cells, clinical tissue samples and patient's serum.
Figure 1.
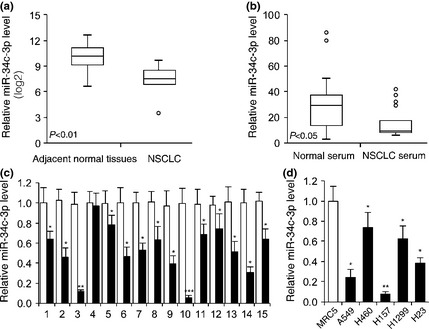
Expression of miR‐34c‐3p was down‐regulated in NSCLC tissues, patients' serum and cell lines. (a) Relative miR‐34c‐3p expression in NSCLC tissues compared to normal tissues (GSE29248, P < 0.01, n = 6 of NSCLC tissues and adjacent normal tissues). (b) Relative miR‐34c‐3p expression in NSCLC patients' serum compared to normal people's serum (GSE17681, P < 0.05, n = 17 NSCLC tissues, n = 19 control samples). (c) Relative miR‐34c‐3p expression in 15 NSCLC tissues compared to adjacent normal tissues. (d) Relative miR‐34c‐3p expression in five NSCLC cell lines compared to normal lung fibroblast cell line MRC5 cells. n = 4. miR‐34c‐3p expression was normalized to U6 levels. *P < 0.05, **P < 0.01, ***P < 0.001.
miR‐34c‐3p inhibited NSCLC cell proliferation, migration and invasion
To examine the role of miR‐34c‐3p in development of NSCLC, A549 and H157 cells were transfected with equal amounts of miR‐NC (miRNA negative control), miR‐34c‐3p, miR‐in‐NC (miRNA inhibitor negative control) or miR‐34c‐3p‐in (miR‐34c‐3p inhibitor). Expression of miR‐34c‐3p was significantly increased by miR‐34c‐3p transfection (Fig. 2a), whereas miR‐34c‐3p‐in transfection reduced endogenous miR‐34c‐3p expression in both A549 and H157 cells (Fig. 2b). Results of the cell proliferation assays revealed that both A549 and H157 cells, when transfected with miR‐34c‐3p, proliferated significantly slower than those transfected with miR‐NC (Fig. 2c). In contrast, cells transfected with miR‐34c‐3p‐in proliferated significantly faster than miR‐in‐NC‐transfected cells (Fig. 2d). Results of cell cycle analysis revealed that overexpression of miR‐34c‐3p led to approximately 15% increase in number of cells in G0/G1 phase (Fig. 2e), whereas inhibition of miR‐34c‐3p with miR‐34c‐3p‐in promoted cell cycle progression with increased number of cells in S and G2/M phases (Fig. 2f).
Figure 2.
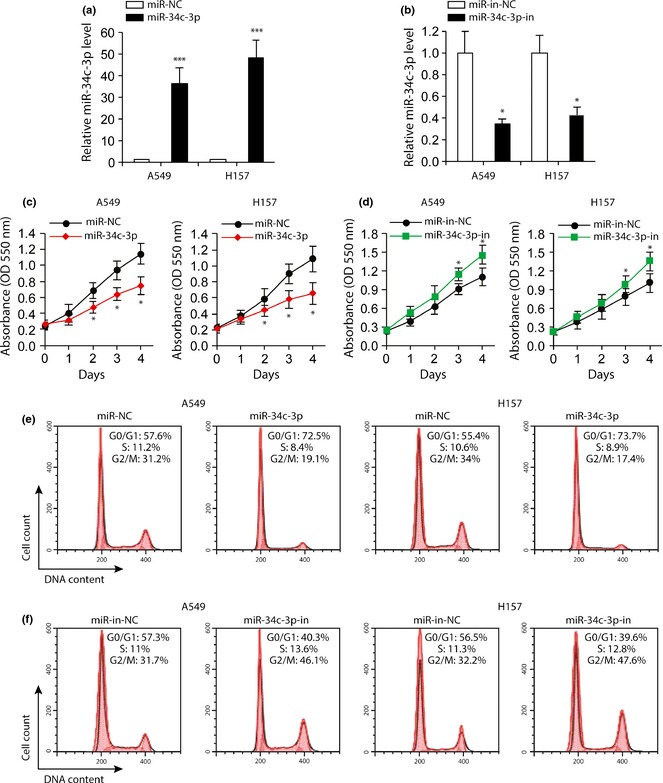
Effects of miR‐34c‐3p and miR‐34c‐3p‐in on NSCLC cell proliferation. (a) miR‐NC (negative control) or miR‐34c‐3p was transfected into A549 and H157 cells. Relative expression of miR‐34c‐3p was accessed by RT‐PCR after 48 h of transfection. n = 3. (b) Relative miR‐34c‐3p levels in A549 and H157 cells following 48 h transfection with miR‐in‐NC or miR‐34c‐3p‐in. (c and d) MTT cell viability assay of A549 and H157 cells transfected with miR‐NC or miR‐34c‐3p (c), miR‐in‐NC or anti‐miR‐34c‐3p (d) at indicated time points. n = 5. (e and f) Cell cycle analysis of A549 and H157 cells transfected with miR‐NC or miR‐34c‐3p (e), miR‐in‐NC or miR‐34c‐3p‐in (f) 2 days after transfection. n = 4. *P < 0.05, ***P < 0.001 compared to miR‐NC‐ or miR‐in‐NC‐transfected cells.
To examine whether miR‐34c‐3p could regulate NSCLC cell migration and invasion, we performed a transwell assay. Overexpression of miR‐34c‐3p suppressed A549 and H157 cell migration (Fig. 3a), whereas inhibition of miR‐34c‐3p promoted migration (Fig. 3b). Furthermore, overexpression of miR‐34c‐3p inhibited NSCLC cell invasion (Fig. 3c), whereas miR‐34c‐3p‐in significantly enhanced it (Fig. 3d). Taken together, these results suggest that miR‐34c‐3p inhibited NSCLC cell proliferation, migration and invasion.
Figure 3.
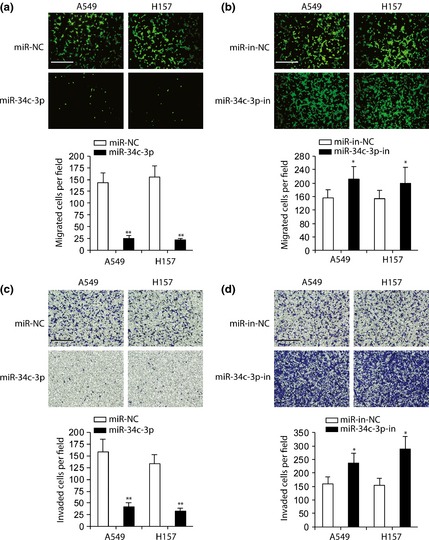
Inhibitory role of miR‐34c‐3p on NSCLC cell migration and invasion. (a–b) Migration assay in A549 and H157 cells transfected with miR‐NC or miR‐34c‐3p (a), miR‐in‐NC or miR‐34c‐3p‐in (b). Numbers of migrated cells per field were counted (bottom panel). (c and d) Invasion assay in A549 and H157 cells transfected with miR‐NC or miR‐34c‐3p (c), miR‐in‐NC or miR‐34c‐3p‐in (d); invaded cells per field were quantified (bottom panel). Scale bar: 500 μm. n = 4. *P < 0.05, **P < 0.01, compared to miR‐NC‐ or miR‐in‐NC‐transfected cells.
miR‐34c‐3p directly targeted eIF4E in NSCLC cells
To understand molecular mechanisms by which miR‐34c‐3p would inhibit NSCLC cell proliferation, migration and invasion, we searched for miR‐34c‐3p targets using miRanda, TargetScan and DIANA tools (29, 30, 31.) and results identified eIF4E, as a putative target of miR‐34c‐3p predicted in 19 vertebrate sequences (Fig. 4a). To determine whether eIF4E was a direct target of miR‐34c‐3p, we cloned wild‐type and mutant eIF4E 3′‐UTR (mutations in miR‐34c‐3p binding sequence) into a luciferase reporter plasmid. A549 and H157 cells co‐transfected with plasmid containing wild‐type eIF4E 3′‐UTR and miR‐34c‐3p had significantly less luciferase activity than their controls (Fig. 4b). Mutation of the potential miR‐34c‐3p binding sites in the eIF4E 3′‐UTR abolished this effect (Fig. 4b). In contrast, miR‐34c‐3p‐in promoted eIF4E 3′‐UTR wild‐type luciferase activity, which was abrogated by mutating the tentative miR‐34c‐3p binding sites in eIF4E (Fig. 4c). Quantitative PCR and western blotting revealed that ectopic expression of miR‐34c‐3p inhibited both mRNA and protein expression of eIF4E in NSCLC cells (Fig. 4d and 4e). As shown in Fig. 4f and 4g, inhibition of endogenous miR‐34c‐3p promoted eIF4E mRNA and protein expression in A549 and H157 cells. Taken together, these results suggested that miR‐34c‐3p directly targeted eIF4E in NSCLC cells.
Figure 4.
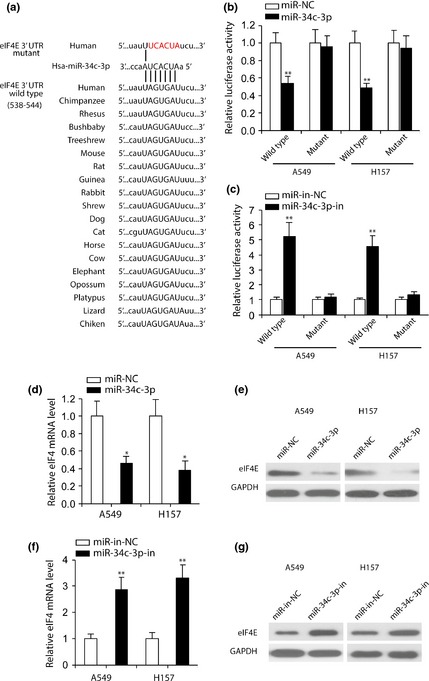
miR‐34c‐3p directly targeted eIF 4E in NSCLC cells. (a) Sequences of miR‐34c‐3p binding sites within eIF4E 3′UTR in vertebrates. Wild‐type eIF4E 3′UTR was fused into pMIR luciferase reporter plasmid. eIF4E 3′UTR mutant represents the reporter constructs containing mutated nucleotides (red). (b and c) Relative luciferase activities of plasmids carrying wild‐type or mutant eIF4E 3′UTR in A549 and H157 cells co‐transfected with miR‐NC versus miR‐34c‐3p (b) and miR‐in‐NC versus miR‐34c‐3p‐in (c). n = 4. (d and f) RT‐PCR analysis of eIF4E mRNA levels in NSCLC cells transfected with miR‐NC or miR‐34c‐3p (d), miR‐in‐NC or miR‐34c‐3p‐in (f). Expression of eIF4E was normalized to GAPDH. n = 4. (e and g) western blot analysis of eIF4E expression in A549 and H157 cells transfected with miR‐34c‐3p compared to miR‐NC (e) and miR‐34c‐3p‐in compared to miR‐in‐NC (g). GAPDH was used as a loading control. *P < 0.05, **P < 0.01 compared to miR‐NC‐ or miR‐in‐NC‐transfected cells.
miR‐34c‐3p/eIF4E‐mediated NSCLC cell tumourigenesis
To examine whether miR‐34c‐3p played an inhibitory role in NSCLC progression by targeting eIF4E, we co‐transfected A549 cells with eIF4E plasmid and miR‐34c‐3p. Western blot analysis indicated that miR‐34c‐3p inhibited eIF4E overexpression (Fig. 5a). Additionally, overexpression of eIF4E significantly enhanced NSCLC cell proliferation, and partially reversed inhibition of cell proliferation caused by miR‐34c‐3p (Fig. 5b and 5c).
Figure 5.
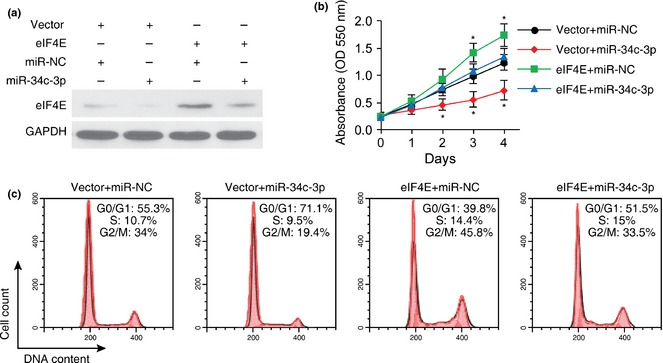
Overexpression of eIF 4E abrogated the inhibitory role of miR‐34c‐3p on A549 cell proliferation. (a) Western blot analysis of eIF4E expression in A549 cells co‐transfected with pcDNA3.1 vector + miR‐NC, pcDNA3.1 vector + miR‐34c‐3p, pcDNA3.1‐eIF4E + miR‐NC or pcDNA3.1‐eIF4E + miR‐34c‐3p. (b and c) MTT assay (b) and cell cycle analyses (c) of A549 cells in indicated groups. n = 4. *P < 0.05 compared to pcDNA3.1 vector + miR‐NC transfected cells.
We next performed transwell assays to examine effects of eIF4E on miR‐34c‐3p‐mediated inhibition of migration and invasion of NSCLC cells. As can be seen in Fig. 6a and 6b, transfection of A549 cells with eIF4E and miR‐NC promoted migration and invasion, and eIF4E reversed inhibition of cell migration and invasion caused by miR‐34c‐3p. To analyse effects of miR‐34c‐3p on expression of genes downstream of eIF4E (c‐myc, Cyclin D1, Survivin and Mcl‐1) 13, we transfected miR‐34c‐3p into A549 and H157 cells and performed western blot analysis. Results indicated that miR‐34c‐3p down‐regulated expression of c‐myc, Cyclin D1, survivin and Mcl‐1 in NSCLC cells (Fig. 7). Taken together, these results suggest that eIF4E is involved in miR‐34c‐3p‐induced inhibition of cell migration, proliferation and invasion of NSCLC cells and that miR‐34c‐3p exerted anti‐tumour effects likely to be by regulating expression of c‐myc, Cyclin D1, survivin and Mcl‐1 downstream of eIF4E.
Figure 6.
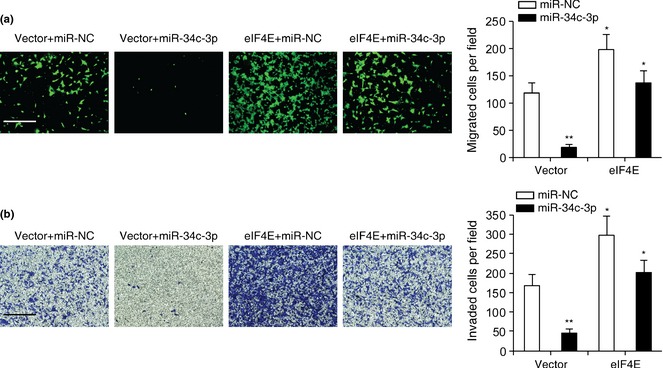
Overexpression of eIF 4E abrogated the inhibitory role of miR‐34c‐3p on A549 cell migration and invasion. (a and b) Migration (a) and invasion (b) assay in A549 cells in indicated groups. Scale bar: 500 μm. n = 4. *P < 0.05, **P < 0.01 compared to pcDNA3.1 vector and miR‐NC transfection.
Figure 7.
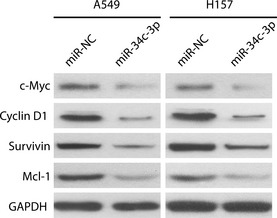
miR‐34c‐3p down‐regulated eIF 4E‐dependent downstream gene expression. A549 and H157 cell were transfected with miR‐NC or miR‐34c‐3p, and protein levels of c‐Myc, Cyclin D1, survivin and Mcl‐1 were compared using western blot analysis 48 h later. GAPDH was used as a loading control.
Discussion
In this study, we found that miR‐34c‐3p directly targeted and inhibited expression of eIF4E and played a significant role in regulating proliferation, migration and invasion of NSCLC cells. Elevated expression of eIF4E and reduced expression of several miRNAs in NSCLC have been reported previously 12, 13, 32, 33. However, dysregulated mechanism(s) responsible for up‐regulated eIF4E expression in NSCLC had not been investigated previously.
We found that miR‐34c‐3p expression was significantly lower in NSCLC cells and tissue samples than in normal lung fibroblasts and paired normal tissue, respectively. Overexpression of miR‐34c‐3p reduced eIF4E expression and caused significant reduction in NSCLC cell proliferation, migration and invasion. Inhibition of miR‐34c‐3p resulted in increased eIF4E expression along with rapid cell cycle progression, and increased cell proliferation, migration and invasion. These results suggest that miR34c‐3p functions as a tumour suppressor by limiting eIF4E expression, and that restoring expression of miR‐34c‐3p could be useful for treatment of NSCLS.
eIF4E is overexpressed in a variety of tumours and correlates with disease progression in NSCLC patients 14. Overexpression of eIF4E alone has been shown to be sufficient for transformation and tumourigenesis in cultured fibroblasts and epithelial cells 34, 35, 36. eIF4E is a mRNA cap‐binding protein, whose activity is regulated through phosphorylation by mitogen‐ and stress‐activated protein kinase Mnk1 or by binding to eukaryotic initiation factor 4E binding proteins (4E‐BPs). Phosphorylation by Mnk1 at Ser 209 enhances eIF4E's cap‐binding activity and stimulates protein synthesis 37. By binding with eIF4E, 4E‐BPs negatively regulates eIF4E activity by limiting availability of eIF4E to complex with eIF4G. Phosphorylation of 4E‐BP1 by mTOR releases eIF4E and increases its activity38. Overexpression of 4E‐BP1 or inhibition of its phosphorylation by rapamycin treatment inhibits cell proliferation and increases the susceptibility of the cells to undergo apoptosis.
Although regulation of eIF4E activity has been extensively studied, little is known about the mechanisms that regulate eIF4E expression. In fibroblasts, serum and growth factors increase eIF4E mRNA expression 39. Further, it has been reported that c‐myc, a potent regulator of cell proliferation, increases eIF4E expression 40. eIF4E itself has been found to promote expression of c‐myc, suggesting a feedback mechanism of regulation. Some studies have suggested that miRNAs regulate expression of eIF4E. Chen et al. have reported that miR‐145 inhibited NSCLC cell proliferation by targeting the c‐myc/eIF4E pathway 41. These authors observed a significant reduction of miR‐145 levels in tumour tissues NSCLC cell lines. Recently, Jiang et al. reported that down‐regulation of miR‐768‐3p enhanced expression of eIF4E and protein synthesis in melanoma cells 42.
It has been reported that p53 transcriptionally targets the miR‐34 family to regulate c‐Myc, CDK6 and c‐MET expression, apoptosis and cell cycle progression 43, 44, 45. miR‐34 genes are expressed in a tissue‐specific manner (although miR‐34a is ubiquitously expressed with highest expression in the brain), miR‐34b and miR‐34c are predominantly expressed in lungs. miR‐34c is a precursor that generates two mature miRNAs, namely miR‐34c‐5p and miR‐34c‐3p. Reduced expression of miR‐34 has been found in several tumour types, including NSCLC 9, 10.
It has been shown that combination therapy using miR‐34c and let‐7 suppresses tumour growth in mouse models of NSCLS 46. In contrast, Catuagno et al. found that miR‐34c‐5p induces resistance to apoptosis in lung cancer 47. Although miR‐34c has been identified as a tumour suppressor, its cellular targets and functions remained largely unknown.
As our analysis using miRanda, TargetScan and DIANA determined eIF4E to be a potential target for miR‐34c‐3p, we further analysed effects of overexpression and inhibition of miR‐34c‐3p on eIF4E‐mediated cell proliferation and tumour progression. Overexpression of miR‐34c‐3p significantly reduced eIF4E mRNA and protein expression (Fig. 4d and 4e), and reduced luciferase activity in eIF4E 3′‐UTR‐luciferase transfected cells (Fig. 4b). Inhibition of endogenous miR‐34c‐3p, on the other hand, enhanced luciferase activity (Fig. 4c) and promoted eIF4E mRNA and protein expression (Fig. 4f and 4g). Consistent with reduced eIF4E expression, cell cycle progression, cell proliferation, migration and invasion were reduced in cells overexpressing of miR‐34c‐3p. Additionally, our results showed that when expressed together, miR34c‐3p inhibited expression of eIF4E and partially reversed eIF4E‐induced changes in cell cycle progression, proliferation, migration and invasion (Figs. 5 and 6). Negative control miR‐NC and the inhibitor miR‐34c‐3p‐in failed to alter effects of overexpression of eIF4E, suggesting that miR‐34c‐3p specifically targeted eIF4E to suppress NSCLC cell proliferation, migration and invasion. It is worth mentioning that, for results in Figs. 5 and 6, total level of eIF4E consisted of endogenous and ectopic expression of the gene. Its ectopic expression was from eIF4E plasmid which does not contain 3′UTR and cannot be targeted by miR‐34c‐3p. However, intact endogenous eIF4E can be regulated by miR‐34‐3p. Thus, the difference between eIF4E + miR‐NC and eIF4E + miR‐34C‐3p group in terms of eIF4E protein expression (Fig. 5a), cell proliferation (Fig. 5b), cell cycle (Fig. 5c), cell migration (Fig. 6a) and cell invasion (Fig. 6b) was from down‐regulation of endogenous eIF4E expression by miR‐34c‐3p. Results from Figs. 5 and 6 demonstrate that overexpression of eIF4E rescued the inhibitory role of miR‐34‐3p in NSCLC progression. In other words, miR‐34c‐3p inhibited NSCLC progression by targeting endogenous eIF4E which can be rescued by ectopic expression of eIF4E without 3′UTR.
Mutations in p53 are commonly found in lung cancers, with approximately 75% frequency seen in NSCLC 48. Some studies have found that transcriptional activation of the miR34 family by p53 occurs during DNA damage and apoptosis 49. Therefore, it is likely that reduced expression of miR‐34c‐3p in NSCLC is a direct reflection of changes in p53 activity during tumour progression.
In summary, we found that miR‐34c‐3p negatively regulated eIF4E expression, and that miR‐34c‐3p expression was down‐regulated in NSCLC. Down‐regulated miR‐34c‐3p expression allows for aberrant eIF4E expression, which promotes oncogene expression, cell cycle progression, proliferation, migration and invasion. Our observations provide new insight into NSCLC biology and potentially a new avenue for NSCLC therapy.
References
- 1. He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. [DOI] [PubMed] [Google Scholar]
- 2. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- 3. Friedlander MR, Lizano E, Houben AJ, Bezdan D, Banez‐Coronel M, Kudla G et al (2014) Evidence for the biogenesis of more than 1,000 novel human microRNAs. Genome Biol. 15, R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grange C, Collino F, Tapparo M, Camussi G (2014) Oncogenic micro‐RNAs and Renal Cell Carcinoma. Front Oncol. 4, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma L, Teruya‐Feldstein J, Weinberg RA (2007) Tumour invasion and metastasis initiated by microRNA‐10b in breast cancer. Nature 449, 682–688. [DOI] [PubMed] [Google Scholar]
- 6. Jansson MD, Lund AH (2012) MicroRNA and cancer. Mol. Oncol. 6, 590–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD et al (2008) Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu Z, Wu Y, Tian Y, Sun X, Liu J, Ren H et al (2013) Differential effects of miR‐34c‐3p and miR‐34c‐5p on the proliferation, apoptosis and invasion of glioma cells. Oncol. Lett. 6, 1447–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang S, Li Y, Gao J, Zhang T, Li S, Luo A et al (2013) MicroRNA‐34 suppresses breast cancer invasion and metastasis by directly targeting Fra‐1. Oncogene 32, 4294–4303. [DOI] [PubMed] [Google Scholar]
- 10. Garofalo M, Jeon YJ, Nuovo GJ, Middleton J, Secchiero P, Joshi P et al (2013) MiR‐34a/c‐Dependent PDGFR‐alpha/beta Downregulation Inhibits Tumorigenesis and Enhances TRAIL‐Induced Apoptosis in Lung Cancer. PLoS ONE 8, e67581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bahlis NJ, McCafferty‐Grad J, Jordan‐McMurry I, Neil J, Reis I, Kharfan‐Dabaja M et al (2002) Feasibility and correlates of arsenic trioxide combined with ascorbic acid‐mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin. Cancer Res. 8, 3658–3668. [PubMed] [Google Scholar]
- 12. Herbst RS, Heymach JV, Lippman SM (2008) Lung cancer. N. Engl. J. Med. 359, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y, Fan S, Koo J, Yue P, Chen ZG, Owonikoko TK et al (2012) Elevated expression of eukaryotic translation initiation factor 4E is associated with proliferation, invasion and acquired resistance to erlotinib in lung cancer. Cancer Biol. Ther. 13, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang R, Geng J, Wang JH, Chu XY, Geng HC, Chen LB (2009) Overexpression of eukaryotic initiation factor 4E (eIF4E) and its clinical significance in lung adenocarcinoma. Lung Cancer 66, 237–244. [DOI] [PubMed] [Google Scholar]
- 15. Hu A, Sun M, Yan D, Chen K (2014) Clinical significance of mTOR and eIF4E expression in invasive ductal carcinoma. Tumori 100, 541–546. [DOI] [PubMed] [Google Scholar]
- 16. Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khoury T, Alrawi S, Ramnath N, Li Q, Grimm M, Black J et al (2009) Eukaryotic initiation factor‐4E and cyclin D1 expression associated with patient survival in lung cancer. Clin. Lung Cancer 10, 58–66. [DOI] [PubMed] [Google Scholar]
- 18. Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA et al (2011) Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 71, 1849–1857. [DOI] [PubMed] [Google Scholar]
- 19. Raught B, Gingras AC (1999) eIF4E activity is regulated at multiple levels. Int. J. Biochem. Cell Biol. 31, 43–57. [DOI] [PubMed] [Google Scholar]
- 20. Yoshizawa A, Fukuoka J, Shimizu S, Shilo K, Franks TJ, Hewitt SM et al (2010) Overexpression of phospho‐eIF4E is associated with survival through AKT pathway in non‐small cell lung cancer. Clin. Cancer Res. 16, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fukaya T, Iwakawa HO, Tomari Y (2014) MicroRNAs block assembly of eIF4F translation initiation complex in Drosophila. Mol. Cell 56, 67–78. [DOI] [PubMed] [Google Scholar]
- 22. Fukao A, Mishima Y, Takizawa N, Oka S, Imataka H, Pelletier J et al (2014) MicroRNAs trigger dissociation of eIF4AI and eIF4AII from target mRNAs in humans. Mol. Cell 56, 79–89. [DOI] [PubMed] [Google Scholar]
- 23. Ma L, Huang Y, Zhu W, Zhou S, Zhou J, Zeng F et al (2011) An integrated analysis of miRNA and mRNA expressions in non‐small cell lung cancers. PLoS ONE 6, e26502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang L, Huang Q, Zhang S, Zhang Q, Chang J, Qiu X et al (2010) Hsa‐miR‐125a‐3p and hsa‐miR‐125a‐5p are downregulated in non‐small cell lung cancer and have inverse effects on invasion and migration of lung cancer cells. BMC Cancer 10, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eilertsen M, Andersen S, Al‐Saad S, Richardsen E, Stenvold H, Hald SM et al (2014) Positive prognostic impact of miR‐210 in non‐small cell lung cancer. Lung Cancer 83, 272–278. [DOI] [PubMed] [Google Scholar]
- 26. Zha W, Cao L, Shen Y, Huang M (2013) Roles of Mir‐144‐ZFX pathway in growth regulation of non‐small‐cell lung cancer. PLoS ONE 8, e74175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu XG, Zhu WY, Huang YY, Ma LN, Zhou SQ, Wang YK et al (2012) High expression of serum miR‐21 and tumor miR‐200c associated with poor prognosis in patients with lung cancer. Med. Oncol. 29, 618–626. [DOI] [PubMed] [Google Scholar]
- 28. Peltier HJ, Latham GJ (2008) Normalization of microRNA expression levels in quantitative RT‐PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 14, 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS (2003) MicroRNA targets in Drosophila. Genome Biol. 5, R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- 31. Maragkakis M, Vergoulis T, Alexiou P, Reczko M, Plomaritou K, Gousis M et al (2011) DIANA‐microT Web server upgrade supports Fly and Worm miRNA target prediction and bibliographic miRNA to disease association. Nucleic Acids Res. 39, W145–W148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M et al (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189–198. [DOI] [PubMed] [Google Scholar]
- 33. Wang J, Tian X, Han R, Zhang X, Wang X, Shen H et al (2014) Downregulation of miR‐486‐5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene 33, 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lazaris‐Karatzas A, Sonenberg N (1992) The mRNA 5' cap‐binding protein, eIF‐4E, cooperates with v‐myc or E1A in the transformation of primary rodent fibroblasts. Mol. Cell. Biol. 12, 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Benedetti A, Graff JR (2004) eIF‐4E expression and its role in malignancies and metastases. Oncogene 23, 3189–3199. [DOI] [PubMed] [Google Scholar]
- 36. Zimmer SG, DeBenedetti A, Graff JR (2000) Translational control of malignancy: the mRNA cap‐binding protein, eIF‐4E, as a central regulator of tumor formation, growth, invasion and metastasis. Anticancer Res. 20, 1343–1351. [PubMed] [Google Scholar]
- 37. Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA (1999) Phosphorylation of the cap‐binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol. 19, 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Showkat M, Beigh MA, Bhat BB, Batool A, Andrabi KI (2014) Phosphorylation dynamics of eukaryotic initiation factor 4E binding protein 1 (4E‐BP1) is discordant with its potential to interact with eukaryotic initiation factor 4E (eIF4E). Cell. Signal. 26, 2117–2121. [DOI] [PubMed] [Google Scholar]
- 39. Rosenwald IB, Hutzler MJ, Wang S, Savas L, Fraire AE (2001) Expression of eukaryotic translation initiation factors 4E and 2alpha is increased frequently in bronchioloalveolar but not in squamous cell carcinomas of the lung. Cancer 92, 2164–2171. [DOI] [PubMed] [Google Scholar]
- 40. Rosenwald IB, Rhoads DB, Callanan LD, Isselbacher KJ, Schmidt EV (1993) Increased expression of eukaryotic translation initiation factors eIF‐4E and eIF‐2 alpha in response to growth induction by c‐myc. Proc. Natl Acad. Sci. USA 90, 6175–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Z, Zeng H, Guo Y, Liu P, Pan H, Deng A et al (2010) miRNA‐145 inhibits non‐small cell lung cancer cell proliferation by targeting c‐Myc. J. Exp. Clin. Cancer Res. 29, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang CC, Croft A, Tseng HY, Guo ST, Jin L, Hersey P et al (2014) Repression of microRNA‐768‐3p by MEK/ERK signalling contributes to enhanced mRNA translation in human melanoma. Oncogene 33, 2577–2588. [DOI] [PubMed] [Google Scholar]
- 43. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al (2007) A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V et al (2014) Mir‐34: a new weapon against cancer? Mol. Ther. Nucleic Acids 3, e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Corney DC, Flesken‐Nikitin A, Godwin AK, Wang W, Nikitin AY (2007) MicroRNA‐34b and MicroRNA‐34c are targets of p53 and cooperate in control of cell proliferation and adhesion‐independent growth. Cancer Res. 67, 8433–8438. [DOI] [PubMed] [Google Scholar]
- 46. Kasinski AL, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E et al (2014) A combinatorial microRNA therapeutics approach to suppressing non‐small cell lung cancer. Oncogene 34, 3547–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Catuogno S, Cerchia L, Romano G, Pognonec P, Condorelli G, de Franciscis V (2013) miR‐34c may protect lung cancer cells from paclitaxel‐induced apoptosis. Oncogene 32, 341–351. [DOI] [PubMed] [Google Scholar]
- 48. Mogi A, Kuwano H (2011) TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 583929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hermeking H (2010) The miR‐34 family in cancer and apoptosis. Cell Death Differ. 17, 193–199. [DOI] [PubMed] [Google Scholar]